

Abstract
High-valent iron-oxo species are thought to be intermediates in the catalytic cycles of oxygenases and peroxidases. An attractive route to these iron-oxo intermediates involves laser flash-quench oxidation of ferric hemes, as demonstrated by our work on the ferryl (compound II) and ferryl porphyrin radical cation (compound I) intermediates of horseradish peroxidase. Extension of this work to include cytochrome P450-BM3 (CYP102A1) has required covalent attachment of a RuII photosensitizer to a nonnative cysteine near the heme (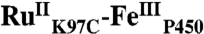 ), in order to promote electron transfer from the FeIII porphyrin to photogenerated RuIII. The
), in order to promote electron transfer from the FeIII porphyrin to photogenerated RuIII. The 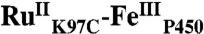 conjugate was structurally characterized by X-ray crystallography (2.4 Å resolution; Ru-Fe distance, 24 Å). Flash-quench oxidation of the ferric-aquo heme produces an FeIV-hydroxide species (compound II) within 2 ms. Difference spectra for three singly oxidized P450-BM3 intermediates were obtained from kinetics modeling of the transient absorption data in combination with generalized singular value decomposition analysis and multiexponential fitting.
conjugate was structurally characterized by X-ray crystallography (2.4 Å resolution; Ru-Fe distance, 24 Å). Flash-quench oxidation of the ferric-aquo heme produces an FeIV-hydroxide species (compound II) within 2 ms. Difference spectra for three singly oxidized P450-BM3 intermediates were obtained from kinetics modeling of the transient absorption data in combination with generalized singular value decomposition analysis and multiexponential fitting.
Keywords: ruthenium bipyridine, enzyme catalysis
The cytochromes P450 constitute a superfamily of thiolate-ligated heme enzymes so named because the Soret absorption band in their CO-bound derivatives peaks near 450 nm. These monooxygenases catalyze a dazzling array of regio- and stereospecific oxidation reactions, including the hydroxylation of aliphatic and aromatic hydrocarbons and the epoxidation of alkenes (1, 2). P450s take two reducing equivalents from NAD(P)H and deliver one atom from dioxygen to the organic substrate; the other oxygen atom is released as water. The consensus mechanism for P450 catalysis (Fig. 1) implicates a ferryl porphyrin radical cation [compound I (CI): Fig. 1, intermediate 6] as the active oxygenating agent (3), but this elusive species has not been observed in P450 under single-turnover or steady-state catalytic conditions. In the postulated mechanism, CI is proposed to abstract a hydrogen atom from the substrate to form transient FeIV-hydroxide complex (compound II, CII), followed by radical recombination to produce oxygenated product (4, 5). Mechanistic studies of P450 catalysis in cryogenic matrices have suggested that the barrier to formation of CI (5 → 6) is higher than that for its reaction with substrate (6 → 7 → 1) (6). Consequently, recent efforts have focused on alternate routes to P450 CI that bypass the hydroperoxide intermediate (5). One approach employs generation of CII using peroxynitrite, followed by laser flash photolysis to yield CI (7). This technique has been used in studies of the spectra and reactivity of the putative CI species, but the interpretation of the results remains open to question.
Fig. 1.

Schematic representation of the consensus P450 catalytic cycle and peroxide shunt pathway.
In earlier work, we employed [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) in a bimolecular flash-quench photochemical oxidation protocol to generate CII and CI in horseradish peroxidase (HRP) and the heme octapeptide from cyctochrome c (MP8) (8, 9). This approach was unsuccessful with P450, however, owing to the deep burial of the heme inside the polypeptide matrix of the enzyme. We have circumvented this problem by covalently attaching a ruthenium-diimine photosensitizer to a nonnative cysteine residue on the P450 surface.
The structurally characterized soluble heme domain of P450-BM3 from Bacillus megaterium serves as a model for the mammalian proteins (10). To ensure a unique attachment of the ruthenium-diimine photosensitizer to the P450, we have used site-directed mutagenesis to remove two native cysteine residues (C62A, C156S) and replace a surface lysine with cysteine at position 97 (K97C); this triple mutant has been overexpressed in Escherichia coli. The complex [Ru(bpy)2(IA-phen)]2+ (IA-phen = 5-iodoacetamido-1,10-phenanthroline) was covalently coupled to Cys97 to give the conjugate 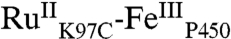 . This conjugate was characterized by mass spectrometry, UV-visible and luminescence spectroscopies, and X-ray crystallography. In our laser flash-quench experiments, this species undergoes rapid photooxidation of the resting ferric-aquo complex to form CII.
. This conjugate was characterized by mass spectrometry, UV-visible and luminescence spectroscopies, and X-ray crystallography. In our laser flash-quench experiments, this species undergoes rapid photooxidation of the resting ferric-aquo complex to form CII.
Results and Discussion
X-Ray Crystal Structure Analysis
We have determined the X-ray crystal structure of 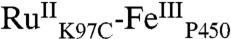 to a resolution of 2.4 Å (Table S1). Two monomers were found in the asymmetric unit; the rmsd between the Cα atom positions in the two monomers is 0.34 Å, confirming that the two polypeptides have nearly identical conformations. The structure of
to a resolution of 2.4 Å (Table S1). Two monomers were found in the asymmetric unit; the rmsd between the Cα atom positions in the two monomers is 0.34 Å, confirming that the two polypeptides have nearly identical conformations. The structure of 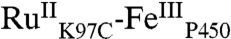 more closely resembles that of the substrate-bound P450-BM3 enzyme (rmsd of 0.5 Å for the structure 2UWH; ref. 11) than the substrate-free protein (rmsd of 1.3 Å for the structure 2BMH; ref. 12). Interestingly, the substrate channel is occupied by two unidentified electron density peaks; the
more closely resembles that of the substrate-bound P450-BM3 enzyme (rmsd of 0.5 Å for the structure 2UWH; ref. 11) than the substrate-free protein (rmsd of 1.3 Å for the structure 2BMH; ref. 12). Interestingly, the substrate channel is occupied by two unidentified electron density peaks; the 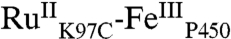 structure likely represents the substrate-bound conformational state. The Ru-photosensitizer is well defined only in one monomer of the crystal structure, owing to π-stacking of the bipyridine ligands with aromatic residues on adjacent crystal units. The Fe-Ru distance between the heme and Ru-photosensitizer is 24 Å (Fig. 2). The Ru-photosensitizer in the second monomer, which lacks the π-stacking interactions with neighboring protein molecules, is highly disordered, possibly due to flexibility of the cysteine-acetamide linkage. These observations indicate a degree of conformational freedom for the Ru-photosensitizer. The 24-Å Ru-Fe distance is likely near the maximum separation in the distribution of conformations sampled by the Ru complex in dilute
structure likely represents the substrate-bound conformational state. The Ru-photosensitizer is well defined only in one monomer of the crystal structure, owing to π-stacking of the bipyridine ligands with aromatic residues on adjacent crystal units. The Fe-Ru distance between the heme and Ru-photosensitizer is 24 Å (Fig. 2). The Ru-photosensitizer in the second monomer, which lacks the π-stacking interactions with neighboring protein molecules, is highly disordered, possibly due to flexibility of the cysteine-acetamide linkage. These observations indicate a degree of conformational freedom for the Ru-photosensitizer. The 24-Å Ru-Fe distance is likely near the maximum separation in the distribution of conformations sampled by the Ru complex in dilute 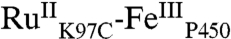 solutions since the ligands of the photosensitizer may form favorable hydrophobic or π-stacking interactions with amino acid residues on the protein surface, decreasing the Fe-Ru separation.
solutions since the ligands of the photosensitizer may form favorable hydrophobic or π-stacking interactions with amino acid residues on the protein surface, decreasing the Fe-Ru separation.
Fig. 2.

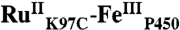 heme region based on an X-ray crystal structure analysis showing covalent attachment of [Ru(bpy)2(A-phen)]2+ at the nonnative cysteine residue (C97) with d(Ru-Fe) = 24 Å and the hydrogen bonding interaction between the tryptophan residue at position 96 and one of the propionics.
heme region based on an X-ray crystal structure analysis showing covalent attachment of [Ru(bpy)2(A-phen)]2+ at the nonnative cysteine residue (C97) with d(Ru-Fe) = 24 Å and the hydrogen bonding interaction between the tryptophan residue at position 96 and one of the propionics.
Laser Flash-Quench Experiments
As expected, the Ru-diimine sensitizer in 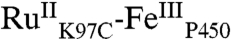 luminesces upon 480-nm excitation: The spectrum closely resembles those of [Ru(bpy)2(IA-phen)]2+ and [Ru(bpy)3]2+. The luminescence decay in
luminesces upon 480-nm excitation: The spectrum closely resembles those of [Ru(bpy)2(IA-phen)]2+ and [Ru(bpy)3]2+. The luminescence decay in  (
(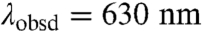 ) is biexponential, whereas τ(∗[Ru(bpy)2(IA-phen)]2+ is monoexponential [τ1
) is biexponential, whereas τ(∗[Ru(bpy)2(IA-phen)]2+ is monoexponential [τ1 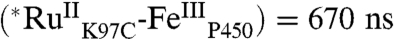 , τ2
, τ2 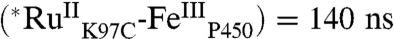 ; τ( ∗ [Ru(bpy)2(IA-phen)]2+) = 720 ns], possibly due to multiple conformations of the tethered photosensitizer (Fig. S1). Stern–Volmer analysis of
; τ( ∗ [Ru(bpy)2(IA-phen)]2+) = 720 ns], possibly due to multiple conformations of the tethered photosensitizer (Fig. S1). Stern–Volmer analysis of 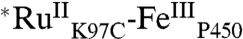 luminescence decay in the presence of an exogenous electron-transfer quencher ([Ru(NH3)6]3+) produces a quenching rate constant (kq) of 1.4 × 109 M-1 s-1 (Fig. S2).
luminescence decay in the presence of an exogenous electron-transfer quencher ([Ru(NH3)6]3+) produces a quenching rate constant (kq) of 1.4 × 109 M-1 s-1 (Fig. S2).
We used transient absorption (TA) spectroscopy to characterize the intermediates formed following excitation of 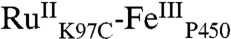 ; measurements in the heme Soret region (390–440 nm) provide the largest TA signals. The Ru-diimine complex also contributes substantially to absorbance in this region, enabling us to monitor changes in both photosensitizer and heme oxidation states (Fig. S3). TA traces of
; measurements in the heme Soret region (390–440 nm) provide the largest TA signals. The Ru-diimine complex also contributes substantially to absorbance in this region, enabling us to monitor changes in both photosensitizer and heme oxidation states (Fig. S3). TA traces of 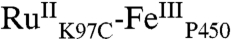 following excitation in the absence of exogenous quencher (pH 8,
following excitation in the absence of exogenous quencher (pH 8, 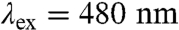 , 10 ns pulse) reveals bleaching (ΔAbs < 0) at wavelengths corresponding to RuII absorption (390–440 nm), consistent with the well-characterized behavior of metal-to-ligand charge-transfer (MLCT) excited Ru-diimine complexes (13). The transient signal returns to baseline at the same rate as luminescence decays and we find no evidence for transient species associated with changes in heme Soret absorption.
, 10 ns pulse) reveals bleaching (ΔAbs < 0) at wavelengths corresponding to RuII absorption (390–440 nm), consistent with the well-characterized behavior of metal-to-ligand charge-transfer (MLCT) excited Ru-diimine complexes (13). The transient signal returns to baseline at the same rate as luminescence decays and we find no evidence for transient species associated with changes in heme Soret absorption.
In the presence of quencher (17 mM [Ru(NH3)6]3+), the TA kinetics become substantially more complex, revealing the presence of multiple intermediates that form and disappear over the microsecond to second time range (Fig. 3). A generalized singular value decomposition analysis of the data (14) indicates that as many as five distinct kinetics phases contribute to the recovery of the TA signal (Fig. S4). Global least-squares fitting of the TA data recorded at six different wavelengths to a sum of five exponentials with amplitude coefficients ρ1-5 produces the observed rate constants γ1-5 listed in Table 1. We find analogous trends in the TA data recorded at pH 6 and 7.
Fig. 3.

Single-wavelength transient absorption kinetics for 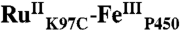 with quencher, pH 8. Circled numbers correspond to the beginning of each phase: (A) 420 nm
with quencher, pH 8. Circled numbers correspond to the beginning of each phase: (A) 420 nm 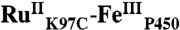 (blue) and [Ru(bpy)2(IA-phen)]2+ (yellow); (B) 390 nm; (C) 440 nm.
(blue) and [Ru(bpy)2(IA-phen)]2+ (yellow); (B) 390 nm; (C) 440 nm.
Table 1.
Rate constants extracted from the global fitting of 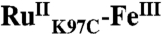 P450 transient absorption data
P450 transient absorption data
| pH |
γ1, s-1 |
γ2, s-1 |
γ3, s-1 |
γ4, s-1 |
γ5, s-1 |
| 6 | 2.5(3) × 107 | 1.5(5) × 106 | 2.0(3) × 105 | 2(1) × 104 | 6(1) × 101 |
| 7 | 2.4(3) × 107 | 1.5(6) × 106 | 1.5(3) × 105 | 1.2(3) × 104 | 7(2) × 101 |
| 8 | 3.0(4) × 107 | 2(5) × 106 | 1.0(3) × 105 | 4(1) × 103 | 3(1) × 101 |
Data from six different wavelengths were fit to the following function: ΔAbs(λ,t) = ρ1(λ) exp(-γ1t) + ρ2(λ) exp(-γ2t) + ρ3(λ) exp(-γ3t) + ρ4(λ) exp(-γ4t) + ρ5(λ) exp(-γ5t). The rate constant γ1 was fixed to the value obtained from single-exponential fits to the luminescence decay kinetics.
Flash-Quench Electron Transfer
The five distinct phases observed in the TA data suggest that six distinct species are formed following excitation of the Ru photosensitizer. The first transient species is easily identified as 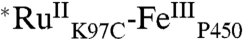 , and the second,
, and the second, 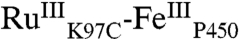 , is formed in the quenching reaction with [Ru(NH3)6]3+. The final species formed is
, is formed in the quenching reaction with [Ru(NH3)6]3+. The final species formed is 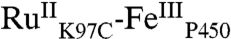 , because all TA signals return to baseline. Three transient species remain to be identified; we anticipate that these are associated with oxidation of the P450 heme center (P450OX1-3) (Scheme 1).
, because all TA signals return to baseline. Three transient species remain to be identified; we anticipate that these are associated with oxidation of the P450 heme center (P450OX1-3) (Scheme 1).
Scheme 1.

In our studies of flash-quench CII generation in HRP and MP8, we found that ferryl-heme formation was preceded by a porphyrin oxidation step; subsequent internal rearrangement and deprotonation led to the FeIV(O)P product. Based on this reasoning, we have developed a sequential kinetics model for the 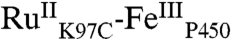 TA data (Fig. 4). We have solved the rate law for this model, allowing us to express the observed rate constants (γ1-5) and amplitude coefficients (ρ1-5) in terms of nine elementary rate constants (k0, k1, k2, k3, k4, k5, k6, k-6, k7) and the initial
TA data (Fig. 4). We have solved the rate law for this model, allowing us to express the observed rate constants (γ1-5) and amplitude coefficients (ρ1-5) in terms of nine elementary rate constants (k0, k1, k2, k3, k4, k5, k6, k-6, k7) and the initial 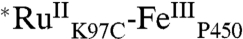 concentration.
concentration.
Fig. 4.

Diagram of the model for photochemical heme oxidation in 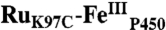 used in the analysis of transient absorption kinetics data.
used in the analysis of transient absorption kinetics data.
The parameters in the kinetics model cannot be determined by the data alone: Values for k1, k3, k5, and Keq = k6/k-6 must be supplied in order to determine absolute molar difference spectra for the six intermediate species. The known spectra of *RuII- and RuIII-diimine constrain the possible values of k1 and k3. The balance between k5 and k7 has no effect on the relative difference spectra extracted from the data, so k5 was set equal to k7. The equilibrium constant Keq was optimized to provide the best agreement between the transient difference spectra recorded at the three different pH values (Fig. 5).
Fig. 5.

Dependence of transient absorption kinetics on pH (red, pH 6; green, 7; blue, 8): (A) 390 nm, (B) 440 nm.
The difference spectra of 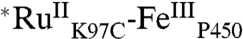 and
and 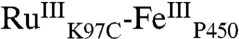 extracted from the kinetics analysis exhibit bleaching at 430 nm of the RuII MLCT absorption band (Fig. S5). Of primary interest are the difference spectra corresponding to intermediates P450OX1-3 (Fig. S5). The spectra of P450OX1 and P450OX2 are quite similar, characterized by a bleach of the Soret absorption band (centered at 420 nm). The spectrum of P450OX2 displays somewhat more absorbance at 390 nm than that of P450OX1 but otherwise closely resembles that of P450OX1. Reasoning by analogy to our results on the oxidation of HRP and MP8, and the similarity of their difference spectra, we suggest that P450OX1 and P450OX2 correspond to porphyrin radical cations:
extracted from the kinetics analysis exhibit bleaching at 430 nm of the RuII MLCT absorption band (Fig. S5). Of primary interest are the difference spectra corresponding to intermediates P450OX1-3 (Fig. S5). The spectra of P450OX1 and P450OX2 are quite similar, characterized by a bleach of the Soret absorption band (centered at 420 nm). The spectrum of P450OX2 displays somewhat more absorbance at 390 nm than that of P450OX1 but otherwise closely resembles that of P450OX1. Reasoning by analogy to our results on the oxidation of HRP and MP8, and the similarity of their difference spectra, we suggest that P450OX1 and P450OX2 correspond to porphyrin radical cations: 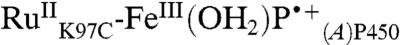 and
and 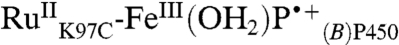 . These difference spectra are consistent with the difference spectrum of CPO CI, which is characterized as an FeIV(O)P•+ center (15). Flash-quench-triggered oxidation of HRP and MP8 produces a similarly bleached, six-coordinate ferric porphyrin radical cation (FeIIIP•+) (8). The blue shift in absorption for these porphyrin radical intermediates is also consistent with synthetic models of Fe(III)-porphyrin cation radicals (16).
. These difference spectra are consistent with the difference spectrum of CPO CI, which is characterized as an FeIV(O)P•+ center (15). Flash-quench-triggered oxidation of HRP and MP8 produces a similarly bleached, six-coordinate ferric porphyrin radical cation (FeIIIP•+) (8). The blue shift in absorption for these porphyrin radical intermediates is also consistent with synthetic models of Fe(III)-porphyrin cation radicals (16).
The specific rate of 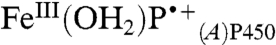 formation in our conjugate (Table S2) is comparable to that found for reconstituted myoglobin containing a heme tethered directly to
formation in our conjugate (Table S2) is comparable to that found for reconstituted myoglobin containing a heme tethered directly to 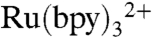 (17). This observation suggests that a favorable pathway couples the P450 porphyrin to RuIII, possibly involving the Trp96-heme propionate hydrogen bond (18). The conversion of
(17). This observation suggests that a favorable pathway couples the P450 porphyrin to RuIII, possibly involving the Trp96-heme propionate hydrogen bond (18). The conversion of 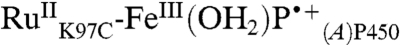 to
to 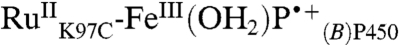 may be a consequence of changes in polypeptide or solvent conformation in the P450 heme pocket (19).
may be a consequence of changes in polypeptide or solvent conformation in the P450 heme pocket (19).
The apparent equilibrium constant between P450OX2 and P450OX3 varies with pH (Keq: 0.8, pH 6; 2.9, pH 7; 10, pH 8) suggesting that a proton is lost in the formation of P450OX3. Moreover, the difference spectrum for this species indicates a red-shifted Soret absorption band analogous to that reported for the FeIV(OH)P center in CPO CII (20). Hence, we suggest that P450OX3 is 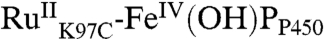 . Internal charge transfer in
. Internal charge transfer in 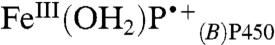 is accompanied by rapid loss of a proton (possibly to water), producing FeIV(OH)PP450. On the basis of results for HRP and MP8, we expect this heme rearrangement to be pH dependent. The formation of flash-quench-generated CII in HRP was slower (kobs of 4.1 s-1) due to rate-limiting water ligation. CII formation in P450 proceeds on the millisecond timescale because the sixth coordination site of the ferric heme is occupied by a water molecule.
is accompanied by rapid loss of a proton (possibly to water), producing FeIV(OH)PP450. On the basis of results for HRP and MP8, we expect this heme rearrangement to be pH dependent. The formation of flash-quench-generated CII in HRP was slower (kobs of 4.1 s-1) due to rate-limiting water ligation. CII formation in P450 proceeds on the millisecond timescale because the sixth coordination site of the ferric heme is occupied by a water molecule.
All of these intermediates are short-lived; transient absorbances return to baseline within 500 ms, indicating recovery of 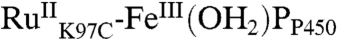 . We have modeled this process as recovery from both the ferryl species [k7,
. We have modeled this process as recovery from both the ferryl species [k7, 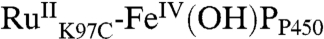 ] and its porphyrin radical cation precursor [k5,
] and its porphyrin radical cation precursor [k5, 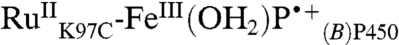 ] (Fig. 4), but it is not possible to determine the two rate constants because equilibration between
] (Fig. 4), but it is not possible to determine the two rate constants because equilibration between 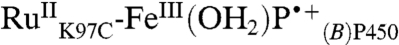 and
and 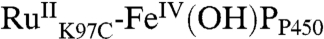 is faster than the ground-state recovery process. The precise nature of the ground-state recovery process remains unclear. It should be described by second-order reaction kinetics, but the experimental data are modeled better by a simple exponential process. Nevertheless, [Ru(NH3)6]2+ seems to be involved in the recovery reaction because, in the presence of [Ru(NH3)6]3+ as quencher, transient absorption data are consistent over the course of multiple hours, and the Soret absorption band appears relatively unaffected after many rounds of flash-quench excitation. However, similar measurements with an irreversible quencher ([Co(NH3)5Cl]3+) induces bleaching of the Soret band and rapid sample degradation.
is faster than the ground-state recovery process. The precise nature of the ground-state recovery process remains unclear. It should be described by second-order reaction kinetics, but the experimental data are modeled better by a simple exponential process. Nevertheless, [Ru(NH3)6]2+ seems to be involved in the recovery reaction because, in the presence of [Ru(NH3)6]3+ as quencher, transient absorption data are consistent over the course of multiple hours, and the Soret absorption band appears relatively unaffected after many rounds of flash-quench excitation. However, similar measurements with an irreversible quencher ([Co(NH3)5Cl]3+) induces bleaching of the Soret band and rapid sample degradation.
Concluding Remarks
We have developed a flash-quench method to oxidize the buried resting state ferric-aquo compound of the P450-BM3 heme domain to CII without the use of reactive oxygen species (O2, H2O2). The catalytic cycle runs in reverse by photochemically splitting water at the heme site. It is likely that the observed ferryl species is protonated over a pH range of 6–8, as consistent with the current view of chloroperoxidase and P450 CII. The finding that internal oxidation of the iron center is rate limiting has allowed us to observe porphyrin radical cation intermediates. As porphyrin oxidation occurs on the microsecond timescale, we can reasonably expect a second round of flash-quench on photochemically generated CII to produce CI.
Materials and Methods
Protein Expression.
A plasmid containing the C62A/C156S double mutant of the P450-BM3 heme domain in the vector pCWori+ was obtained courtesy of Andrew Udit (Occidental College, Los Angeles, CA). A cysteine residue for photosensitizer coupling was introduced at residue 97 using a QuikChange Site-Directed Mutagenesis kit (Qiagen). All primers were purchased from Operon. Mutant cytochromes were expressed with an N-terminal His6 tag in Escherichia coli BL21(DE3) cells and purified according to literature procedure (21). Proteins were characterized by SDS-PAGE and MS analysis.
Photosensitizer Synthesis and Coupling to P450-BM3.
Ruthenium(II) bisbipyridine 5-acetamido-1,10-phenanthroline ([Ru(bpy)2(IA-phen)]2+) was synthesized according to the published procedure (22) with the following modification: In lieu of final precipitation as the PF6 salt, the compound was redissolved in water without further purification. Approximately twofold excess of [Ru(bpy)2(IA-phen)]2+ was added to a 20 μM solution of C62A/C156S/K97C P450-BM3 in 20 mM Tris buffer (pH 8), and shaken in the dark at 4 °C. Labeling progress was monitored by MALDI mass spectrometry; no further increase in the peak at 54.2 kDa (corresponding to the predicted mass of 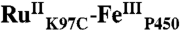 ) was observed after 2 h. Excess [Ru(bpy)2(IA-phen)]2+ was removed during concentration in 30 kDa filters, followed by desalting on an FPLC HiPrep column. Photosensitizer-labeled (
) was observed after 2 h. Excess [Ru(bpy)2(IA-phen)]2+ was removed during concentration in 30 kDa filters, followed by desalting on an FPLC HiPrep column. Photosensitizer-labeled (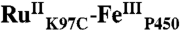 ) and unlabeled proteins were separated using an anion exchange MonoQ column. The labeled protein was characterized by CO binding, and purity was confirmed by SDS-PAGE and mass spectrometry. The conjugate
) and unlabeled proteins were separated using an anion exchange MonoQ column. The labeled protein was characterized by CO binding, and purity was confirmed by SDS-PAGE and mass spectrometry. The conjugate 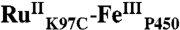 demonstrated activity in the hydroxylation of lauric acid via the peroxide shunt (21).
demonstrated activity in the hydroxylation of lauric acid via the peroxide shunt (21).
Crystal Structure Determination.
Crystals of 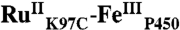 were obtained by the sitting-drop vapor diffusion method: 27 mg/mL
were obtained by the sitting-drop vapor diffusion method: 27 mg/mL 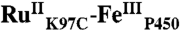 in 10 mM potassium phosphate, pH 8.4, were mixed with a crystallization well solution of 2 M (NH4)2SO4 (wt/vol) in a 1∶1 ratio (vol/vol). Crystals formed over a period of 2 d at 4 °C, and were flash-frozen directly from the crystallization solution. X-ray diffraction data were collected at 100 K using beamline 7-1 at the Stanford Synchrotron Radiation Laboratory. Detailed description of the structure determination is provided in the SI Text. Statistics for data collection and refinement are shown in Table S1. Atomic coordinates and structure factors were deposited in the Protein Data Bank under the entry 3NPL.
in 10 mM potassium phosphate, pH 8.4, were mixed with a crystallization well solution of 2 M (NH4)2SO4 (wt/vol) in a 1∶1 ratio (vol/vol). Crystals formed over a period of 2 d at 4 °C, and were flash-frozen directly from the crystallization solution. X-ray diffraction data were collected at 100 K using beamline 7-1 at the Stanford Synchrotron Radiation Laboratory. Detailed description of the structure determination is provided in the SI Text. Statistics for data collection and refinement are shown in Table S1. Atomic coordinates and structure factors were deposited in the Protein Data Bank under the entry 3NPL.
Laser Flash-Quench.
Samples consisting of [Ru(bpy)2(IA-phen)]2+ or 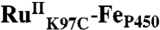 (∼10 μM) with and without quencher (17 mM ruthenium(III)-hexaammine trichloride) were prepared in buffered solution (pH 6, 20 mM sodium acetate; pH 7, 20 mM sodium acetate; pH 8, 50 mM sodium borate or 50 mM Tris). Deoxygenation was achieved via 30 gentle pump-backfill cycles with argon. Samples were excited with 10 ns laser pulses at 480 nm, delivered by an optical parametric oscillator pumped by the third harmonic from a Spectra Physics Q-switched Nd:YAG laser. Luminescence decays were monitored at 630 nm. Single wavelength TA kinetics were monitored every 10 nm from 390–440 nm, averaging ∼500 shots per wavelength. Data from five separate timescales (2 μs, 40 μs, 400 μs, 10 ms, and 500 ms) were collected and spliced together to produce full kinetics traces.
(∼10 μM) with and without quencher (17 mM ruthenium(III)-hexaammine trichloride) were prepared in buffered solution (pH 6, 20 mM sodium acetate; pH 7, 20 mM sodium acetate; pH 8, 50 mM sodium borate or 50 mM Tris). Deoxygenation was achieved via 30 gentle pump-backfill cycles with argon. Samples were excited with 10 ns laser pulses at 480 nm, delivered by an optical parametric oscillator pumped by the third harmonic from a Spectra Physics Q-switched Nd:YAG laser. Luminescence decays were monitored at 630 nm. Single wavelength TA kinetics were monitored every 10 nm from 390–440 nm, averaging ∼500 shots per wavelength. Data from five separate timescales (2 μs, 40 μs, 400 μs, 10 ms, and 500 ms) were collected and spliced together to produce full kinetics traces.
Supplementary Material
Acknowledgments.
This research was supported by the National Institutes of Health (DK019038) and The Arnold and Mabel Beckman Foundation. L.C. thanks Dr. Phoebe Glazer for helpful discussions.
Footnotes
The authors declare no conflict of interest.
Data deposition: The crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3NPL and Research Collaboratory for Structural Bioinformatics (RCSB) ID code RCSB060120).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012381107/-/DCSupplemental.
References
- 1.Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 2.Ortiz de Montellano P. Cytochrome P450: Structure, Mechanism and Biochemistry. 3rd Ed. New York: Kluwer/Plenum; 2005. p. 689. [Google Scholar]
- 3.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 4.Groves JT. The bioinorganic chemistry of iron in oxygenases and supramolecular assemblies. Proc Natl Acad Sci USA. 2003;100:3569–3574. doi: 10.1073/pnas.0830019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green MT. C-H bond activation in heme proteins: The role of thiolate ligation in cytochrome P450. Curr Opin Chem Biol. 2009;13:84–88. doi: 10.1016/j.cbpa.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 6.Davydov R, et al. Hydroxylation of camphor by reduced oxy-cytochrome P450cam: Mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J Am Chem Soc. 2001;123:1403–1415. doi: 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- 7.Wang Q, Sheng X, Horner JH, Newcomb M. Quantitative production of compound I from a cytochrome P450 enzyme at low temperatures. Kinetics, activation parameters, and kinetic isotope effects for oxidation of benzyl alcohol. J Am Chem Soc. 2009;131:10629–10636. doi: 10.1021/ja9031105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berglund J, Pascher T, Winkler JR, Gray HB. Photoinduced oxidation of horseradish peroxidase. J Am Chem Soc. 1997;119:2464–2469. [Google Scholar]
- 9.Low D, Winkler J, Gray H. Photoinduced oxidation of microperoxidase-8: Generation of ferryl and cation-radical porphyrins. J Am Chem Soc. 1996;118:117–120. [Google Scholar]
- 10.Munro AW, et al. P450 BM3: The very model of a model flavocytochrome. Trends Biochem Sci. 2002;27:250–255. doi: 10.1016/s0968-0004(02)02086-8. [DOI] [PubMed] [Google Scholar]
- 11.Huang W, et al. Filling a hole in cytochrome P450 BM3 improves substrate binding and catalytic efficiency. J Mol Biol. 2007;373:633–651. doi: 10.1016/j.jmb.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Poulos T. Modeling protein-substrate interactions in the heme domain of cytochrome P450(BM-3) Acta Crystallogr, Sect D: Biol Crystallogr. 1995;51:21–32. doi: 10.1107/S0907444994009194. [DOI] [PubMed] [Google Scholar]
- 13.Creutz C, Chou M, Netzel TL, Okumura M. Lifetimes, spectra, and quenching of the excited states of polypyridine complexes of iron(II), ruthenium(II), and osmium(II) J Am Chem Soc. 1980;102:1309–1319. [Google Scholar]
- 14.Hansen PC. Regularization tools: A Matlab package for analysis and solution of discrete ill-posed problems. Numer Algorithms. 1994;6:1–35. [Google Scholar]
- 15.Rittle J, Younker JM, Green MT. Cytochrome P450: The active oxidant and its spectrum. Inorg Chem. 2010;49:3610–3617. doi: 10.1021/ic902062d. [DOI] [PubMed] [Google Scholar]
- 16.Ikezaki A, Tukada H, Nakamura M. Control of electronic structure of a six-coordinate iron(III) porphyrin radical by means of axial ligands. Chem Commun. 2008:2257–2259. doi: 10.1039/b800674a. [DOI] [PubMed] [Google Scholar]
- 17.Hamachi I, Tsukiji S, Shinkai S, Oishi S. Direct observation of the ferric-porphyrin cation radical as an intermediate in the phototriggered oxidation of ferric- to ferryl-heme tethered to Ru(bpy)3Ru(bpy)3 in reconstituted materials. J Am Chem Soc. 1999;121:5500–5506. [Google Scholar]
- 18.Shih C, et al. Tryptophan-accelerated electron flow through proteins. Science. 2008;320:1760–1762. doi: 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- 19.Fishelovitch D, Shaik S, Wolfson HJ, Nussinov R. How does the reductase help to regulate the catalytic cycle of cytochrome P450 3A4 using the conserved water channel. J Phys Chem B. 2010;114:5964–5970. doi: 10.1021/jp101894k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green MT, Dawson JH, Gray HB. Oxoiron(IV) in chloroperoxidase compound II is basic: Implications for P450 chemistry. Science. 2004;304:1653–1656. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- 21.Cirino PC, Arnold FH. Regioselectivity and activity of cytochrome P450 BM-3 and mutant F87A in reactions driven by hydrogen peroxide. Adv Synth Catal. 2002;344:932–937. [Google Scholar]
- 22.Castellano FN, Dattelbaum JD, Lakowicz JR. Long-lifetime Ru(II) complexes as labeling reagents for sulfhydryl groups. Anal Biochem. 1998;255:165–170. doi: 10.1006/abio.1997.2468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.