

Abstract
A series of N-acyl O-amino derivatives of seco-CBI-indole2 are reported and examined as prototypical members of a unique class of reductively activated (cleaved) prodrugs of the duocarmycin and CC-1065 family of antitumor agents. These prodrugs were designed to be potentially preferentially activated in hypoxic tumor environments, which carry an intrinsically higher concentration of “reducing” nucleophiles (e.g., thiols) capable of activating such derivatives by nucleophilic cleavage of a weak N–O bond. A remarkable range of stabilities and a resulting direct correlation with in vitro/in vivo biological potencies was observed for these prodrugs even enlisting subtle variations in the electronic and steric environment around the weak N–O bond. An in vivo evaluation of several of the prodrugs demonstrates that some approach the potency and exceed the efficacy of the free drug itself (CBI-indole2), suggesting the prodrugs may offer an additional advantage related to a controlled or targeted release.
Introduction
CC-1065 (1)1a and duocarmycin SA (2)1b represent the parent members of a class of antitumor agents that derive their biological activity from their ability to selectively alkylate duplex DNA (Figure 1).1–3 The study of the natural products, their synthetic unnatural enantiomers,4 and key analogues has defined many of the fundamental features that control the alkylation selectivity, efficiency, and catalysis,5 ultimately providing a detailed understanding of the relationship between structure, reactivity, and biological activity.3–5
Figure 1.

Natural products
Among the most widely studied duocarmycin analogue is CBI6 (1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one), Figure 2. This simplified alkylation subunit is not only synthetically more accessible, but it exhibits an enhanced chemical stability (4×) and biological potency (4×) relative to the alkylation subunit found in 1, approaching the stability and potency of duocarmycin SA (2) and yatakemycin derivatives. Derivatives bearing this modified alkylation subunit have been shown to exhibit efficacious antitumor activity in animal models at doses that reflect this potency.7 Thus, it is on this scaffold that new design concepts are often evaluated.
Figure 2.

Prodrug design
A unique feature of this class of molecules is the observation that the phenol synthetic precursors possess indistinguishable biological properties (DNA alkylation efficiency and selectivity, in vitro cytotoxic activity, in vivo antitumor activity) in comparison to the cyclopropane derivatives themselves. These phenol precursors undergo in situ Ar-3′ spirocyclization with the displacement of an appropriate leaving group to afford the cyclopropane found in the natural products (Figure 2). This reliable behavior of the precursor phenols has provided the basis on which the development of prodrugs has been conducted.8 Thus, protection of the phenol, which prevents spirocyclization to the cyclopropane, provides precursors that are inactive, yet readily yield the active products upon deprotection. Development of this protection and release strategy has been pursued, where phenol release in vivo is coupled with features that may allow for selective tumor activation or delivery.9 Despite the attractiveness of such an approach, only a surprisingly small number of such studies have been disclosed.10–12
We recently reported an initial study examining the potential for N-acyl O-amino derivatives of such phenols to serve as duocarmycin prodrugs subject to reductive activation, with the expectation being that these compounds might be capable of preferential activation in hypoxic tumors.12 At this initial stage of the studies, it was not known whether such derivatives possessed the required reactivity for effective chemical cleavage and release of the free drug or the necessary stability for their synthesis and subsequent storage. Consequently, it is remarkable that the initial studies not only established that such derivatives possessed the requisite combination of chemical stability and cleavage reactivity, but that one of the initially explored derivatives (5) demonstrated effective in vivo release of the free drug leading to more efficacious antitumor activity than the free drug itself and with a potency that approached that of the free drug.12 Alternative approaches to incorporate reductive activation into this class of molecules have been disclosed, but target an intrinsic enzyme activity that differentiates normal versus tumor cells.11 However, in many cases (e.g., mitomycin) on which the design principles are based, it may not be the enzymatic reduction itself, but rather the re-oxidation of the drug in normal cells (vs hypoxic tumor environments) that protects them from damage. Systems utilized for reductive activation have been reviewed,13 and such agents include mitomycin C and other aziridoquinones, nitro-aromatics, N-oxides, various metal complexes, azides, and di- and trisulfides. Those most relevant to the design disclosed herein are a class of natural products (FR900482 and its related congeners)14 as well as a dehydromonocrotaline progenitor15 incorporating hydroxylamine hemiketals that are irreversibly activated by reductive cleavage of a N–O bond. The approach outlined herein is not designed for reversible or enzymatic reductive activation, but rather for irreversible cleavage of a weak N–O bond by reducing nucleophiles (Figure 2).12 The key question was whether one can take advantage of the fact that hypoxic tumor environments contain a higher concentration of reducing nucleophiles (i.e., glutathione) over normal cells capable of activating such agents.16 Further contributing to the properties of the prodrugs and their sites of release, it has been reported that intracellular glutathione (thiol) concentrations are 100-fold higher than in the extracellular matrix, suggesting preferential intracellular versus plasma release of such drugs.16,17
Herein, we report an examination of the impact of the electronic and steric features of the functionality on the nitrogen atom of the O-aminophenols that can be utilized to tune the reactivity of the N–O bond. Thus, a range of functional groups that affect both the electronic (amines vs ureas vs carbamates vs amides vs sulfonamides) and steric (t-butyl vs methyl carbamate) environment around nitrogen were examined. In addition, the introduction of steric bulk directly on the nitrogen atom was explored (N-methylation). It has been shown that the addition of alkyl groups adjacent to the bond undergoing in vivo cleavage can increase stability, presumably by interfering with the approach of incoming nucleophiles.18
To access the desired analogs (Figure 3), a direct SN2 phenol displacement of an O-tosylated hydroxamide using the fully elaborated seco-CBI-indole2 (11) was examined and utilized to provide the desired prodrugs in a single step.12 In turn, the requisite hydroxamides were prepared in 2–4 steps from commercially available materials.
Figure 3.
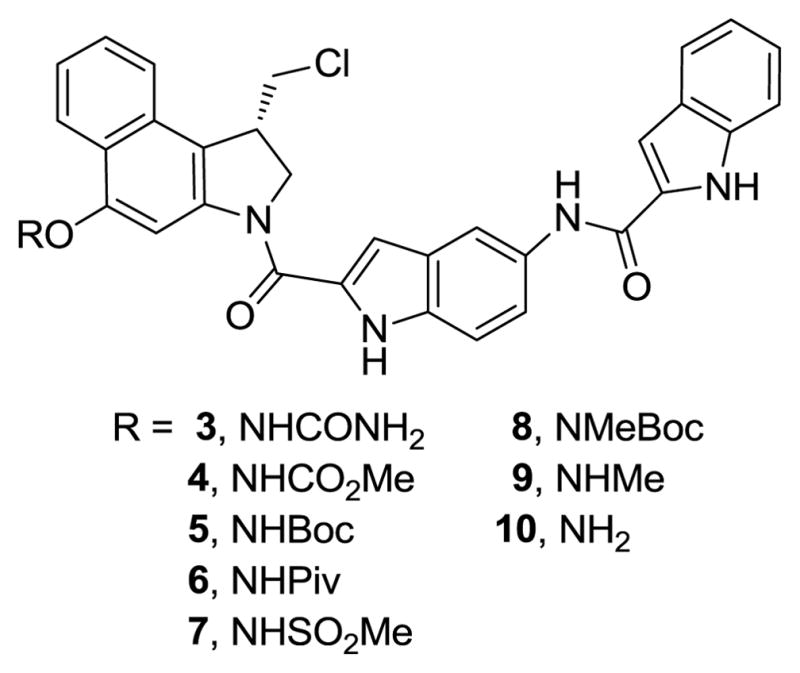
N–O prodrugs
Chemistry
Synthesis
The starting seco-CBI-indole2 (11) was synthesized by a 10-step sequence in 28% overall yield.19 Deprotonation of 11 with LiHMDS in ether:1,4-dioxane (1:1) at 0 °C, followed by the addition of TsONHBoc20 and maintaining the reaction mixture at room temperature (0.1 M) for 4 h resulted in consistent yields of 35–42% for compound 5 (Scheme 1). Remarkably, use of the lithium phenolate generated at 0 °C and the judicious choice of a nonpolar, aprotic solvent12 prevents the competitive facile spirocyclization from occurring. Moreover, the O-amination reaction can be effectively conducted on the fully elaborated seco-CBI-indole2 without competing sites of reaction permitting a late stage introduction of the prodrug variations. In the case of 5, separation of the desired product from unreacted seco-CBI-indole2 (11) was not possible by silica gel chromatography. Therefore, deliberate spirocylization of the remaining starting material 11 in the reaction product mixture with 1:1 saturated aqueous NaHCO3:THF afforded the much more polar spirocyclized CBI-indole2 (Figure 2) which was readily separated from 5 by conventional chromatography.
Scheme 1.

Applying these conditions to the more electron-rich urea substrate (TsONHCONH2) provided only trace amounts of product 3. After extensive optimization, small amounts of urea 3 were isolated by increasing the scale and concentration of the reaction to 0.2 M. Similarly, methyl carbamate 4 was formed in trace amounts using the original reaction conditions (TsONHCO2Me). In this case, raising the reaction temperature to 50 °C and increasing the concentration to 0.2 M provided carbamate 4. The N-trimethyacetyl prodrug 6 was formed in much higher conversions (TsONHCOtBu), but the relative instability of its N–O bond prevented the use of chromatography for purification. However, precipitation of the product from THF using distilled hexanes afforded the desired prodrug 6 in 65% isolated yield. Although unrecognized at the time the synthesis was conducted, the yield of the O-amidation reactions were found to be inversely correlated with the relative stability of the products and directly correlated with the relative reactivity of the cleavable N–O bond.
As anticipated from the stability of 6, initial efforts on the synthesis of the more electron-withdrawing methanesulfonyl prodrug 7 (Scheme 1) proved challenging. Not surprisingly, recovered phenol was observed in all of the attempts to prepare 7 (TsONHSO2Me) and likely reflects not its lack of formation, but rather its instability to isolation or even its detection (TLC). Since ongoing efforts conducted alongside efforts to generate 7 suggested it would possess a reactivity too high to be useful for our purposes, the synthesis of 7 was not pursued in depth. Similarly, the synthesis of N-Me-t-butyl carbamate 8 by direct N–O bond formation with TsONMeBoc failed to afford the desired product, presumably due to the steric hinderance on nitrogen. However, selective N-methylation of 5 with dimethyl sulfate and LiHMDS afforded the methylated product 8. Efforts to remove the t-butyl carbamate from 8 to provide 9 were unsuccessful, leading only to complex reaction mixtures. Direct O-amination of seco-CBI-indole2 (11) was also explored with several different aminating reagents, but failed to provide 10. Overall, the exploratory work led to the syntheses of the urea 3, methyl carbamate 4, t-butyl carbamate 5, pivoloyl amide 6, and N-Me-t-butyl carbamate 8 containing prodrugs that proved sufficient to span the range of useful reactivity.
Stability and Reactivity of the N–O Prodrugs and Release of Free Drug
The stability of the prodrugs and their ability to release the free drug 11 were examined under a wide variety of acidic, basic, and nucleophilic conditions. As expected based on electronic properties, the relative stability followed the order of 3 ~ 8 > 4 > 5 > 6 with the urea 3 and carbamate 8 proving stable to all conditions except strong acid, whereas 6 was cleaved under almost all conditions examined (Supporting Information Table S1). Although the trends in reactivity can be easily rationalized based on the electronic effect of the N-acyl group (6 > 5 > 4 > 3) or the steric effect of further N-methylation (5 > 8), the magnitude of the differences in reactivity represented, in our opinion, a remarkable range given the subtle changes in the structures. Derivatives 3, 4, 5, and 8 proved robust to acidic conditions (1:1 TFA:CH2Cl2, 4 N HCl in EtOAc) and stable to mild base in nonpolar, aprotic solvents (Et3N or DMAP, CH2Cl2), but showed diminished stability to base releasing 11 as the solvent polarity increased. All but 6 were stable to NaHCO3 in THF and 1:1 THF:H2O, but 5 underwent cleavage in NaHCO3/DMF:H2O (1:1), and both 4 and 5 were cleaved in DBU/MeCN to release 11. Similarly, all the prodrugs proved stable in MeOH, but cleavage of 4, 5, and 6 to provide 11 was observed upon treatment with NaHCO3 in MeOH (2 h, 23 °C). Most relevant to the potential source of cleavage under physiological conditions (i.e., thiols), 6 was rapidly cleaved by BnSH under all conditions examined, whereas 3, 4, 5, and 8 were stable to treatment with BnSH in THF (72 h, 23 °C) or MeOH (72 h, 23 °C), and stable to treatment with BnSH in THF even in the presence of insoluble NaHCO3 (2 h, 23 °C). However, all were cleaved to release 11 and provide CBI-indole2 upon treatment with BnSH (100 equiv) in MeOH in the presence of NaHCO3 (100 equiv), albeit at relative rates that reflect the N–O bond reactivity (6 > 5 > 4 > 38, ). The half life of the prodrugs under these thiol cleavage conditions, releasing CBI-indole2, were estimated to be: 6 (< 0.5 h), 5 (6 h), 4 (10 h), 8 (48 h) and 3 (35 h). The stability of these compounds was also assessed in pH 7.0 phosphate buffer, and no significant cleavage was observed for derivatives 3, 4, 5, and 8 (72 h), whereas 6 was completely cleaved to release CBI-indole2 with a t1/2 of ca. 6 h. Finally, 5 was observed to exhibit a robust half-life in human plasma (t1/2 = 3–6 h) with clean release of 11 and its subsequent spirocyclization to CBI-indole2.12
Additionally, we have shown that the prodrug 5 is incapable of alkylating DNA in cell free systems indicating that any activity observed in the following cell-based assay or in vivo model requires cleavage of the prodrug N–O bond with release of the free drug 11.12
Biological Properties
In Vitro Cytotoxic Activity
Both enantiomers of prodrugs 3–6 and 8 were tested for cell growth inhibition in a cytotoxic assay with L1210 cells (Figure 4). Importantly, all the prodrugs exhibited potent cytotoxic activity (> 100-fold more active than the phenol methyl ether of 11),12 and the natural (1S)-enantiomers were on average 30-fold more potent than the unnatural (1R)-enantiomers analogous to the enantiomer pairs of 11, indicating the release of the free drug under the assay conditions. The relative order of activity for the compounds correlated with their observed reactivity for release of 11 under the conditions described above. The unstable pivoloyl amide prodrug 6 was the most potent derivative (IC50 = 80 pM), exhibiting a slightly greater potency than t-butyl carbamate 5 (IC50 = 100 pM) and approaching the potency of the free drug 11 itself (IC50 = 30 pM). Urea 3 (IC50 = 1.1 nM) was ten times less potent than 5, which is a testament to its stability due to the less electron-withdrawing nature of the urea functionality. Further, 5 was found to be five times more potent than the methyl carbamate 4 (IC50 = 500 pM), indicating that the steric size of the carbamate distal from the N–O bond may subtly decrease its stability (see above), increase its rate of in situ free drug release, and increase its cytotoxic activity. The addition of the N-methyl group in 8 increased the stability of the compound considerably (above), and a resulting nine-fold decrease in cytotoxic potency was observed relative to 5. Based on these results, the productive N–O bond cleavage and free drug release proved much more sensitive to the subtle differences in the electron-withdrawing properties of the N-acyl group than initially expected. It is notable that the enantiomeric pairs of each prodrug exhibited the same distinguishable differences in cytotoxic potency as 11 (ca. 30-fold) consistent with, but not proving, simple in vitro chemical versus enzymatic reductive cleavage of the N–O bond. Most significant in these assessments was the fact that the biological activity (potency) of the prodrugs in this cellular functional assay correlates remarkably well with their corresponding reactivity for free drug release.
Figure 4.

Summary data for N–O prodrugs.
In Vivo Antitumor Activity
From the stability studies and the results of in vitro cytotoxic assays, a clear depiction of the relationship between relative N–O cleavage ability and functional activity in a cell based assay was defined. However, it was not clear how these features might translate to in vivo use of the prodrugs. Clearly they must be sufficiently reactive to effectively release the free drug, yet sufficiently stable to permit its controlled and potentially more selective release. Since the pivoloyl amide prodrug 6 was found to be unstable in pH 7.0 phosphate buffer (t1/2 = 6 h), it was not examined alongside 3–5. The key question being addressed was whether the more reactive or more stable N–O prodrugs in the remaining series (3–5) performed best in an in vivo tumor model. Consequently, the in vivo antitumor activity of 3–5 was assessed alongside the parent drug 11 in a widely employed antitumor model consisting of L1210 murine leukemia cells implanted i.p. into DBA/2J mice. A dose range (10–100 μg/kg) and a dosing schedule (administered three times i.p. on days 1, 5, and 9) found suitable for related parent drugs including CBI-indole2 was used and extended to 2-fold and 5-fold higher doses for 4 and 3, respectively.7 Although the free drug 11 exhibited a narrow range of dose dependent efficacy, exhibiting its maximal effect at 60 μg/kg and less effective efficacy at 30 μg/kg (too little drug) or 100 μg/kg (too much drug), 5 exhibited increasing efficacy up to and including the 100 μg/kg dose and it matched or exceeded the efficacy of the free drug 11 at each dose. Herein, higher doses of 5 were not examined in this comparison, but the results of two independent examinations of 5 in which the studies were conducted for as long as 365 days12 complement the observations made herein and more clearly define the enhanced efficacy of 5 over 11. In all cases, the dose at which a maximal response was achieved proved similar for 5 and the free drug 11, but with 5 showing improved efficacy. Prodrugs 3 and 4 were also active, albeit only at higher doses. In both instances, the initial examination was conducted likely without reaching their optimal dosing. However, importantly the studies reveal that each of the prodrugs release the free drug 11 in the in vivo model (reductive activation), that the relative potency in vivo parallels the relative cleavage ability of the prodrug N–O bond and the relative in vitro cytotoxic activity, that even the subtle structural changes examined herein likely span the useful range of relative reactivities, and that either the rate of release or the site of release can lead to enhancements in the efficacy of the drug. The t-butyl carbamate prodrug 5 exhibited the most robust antitumor activity, providing 6/10 long term survivors at the highest dose examined (100 μg/kg). Especially notable is that the in vivo study was conducted for 120 days with meaningful numbers of long term survivors (untreated controls avg. life span = 22 days) suggesting cures with 5 even without a dosing optimization. Finally, it is worth highlighting that the compounds are extraordinarily potent, requiring less than 1 mg of sample to conduct the in vivo antitumor testing and indicating that clinical amounts of such agents could easily be supplied by chemical synthesis.
Conclusions
A unique series of N-acyl O-aminophenol prodrugs of CBI-indole2 were explored. These prodrugs, subject to reductive activation through nucleophilic cleavage of a weak N–O bond, were found to exhibit predictable and tunable N–O cleavage properties subject to both electronic and steric control of their relative reactivity, and to exhibit a remarkable range of reactivity given the subtle nature of the structural changes examined. All were found to effectively release the free drug both in functional cellular assays and in vivo antitumor models, and the most effective of the derivatives examined (5) exhibited potent and efficacious antitumor activity approaching the potency of the free drug 11 and surpassing its in vivo efficacy. Further characterization of 5 is in progress and will be reported in due course. Remarkable in these studies is that the small structural changes in moving from the urea to carbamate to amide based prodrug proved sufficient to result in large differences in both the stability and in vitro/in vivo potency of these agents. Coupled with the addition of steric bulk on or adjacent to the N–O bond, a useful range of stabilities and biological potencies was observed for these duocarmycin prodrugs.
Experimental Section
General
Reagents and solvents were purchased reagent-grade and used without further purification. THF was freshly distilled from sodium benzophenone ketyl. All reactions were performed in oven-dried glassware under an Argon atmosphere. Evaporation and concentration in vacuo was performed at 20 °C and atmospheric pressure. Further drying of the new compounds was carried out at ca. 5 millibar. TLC: pre-coated SiO2 60 F254 glass plates from EMD, visualization by UV light (254 or 366 nm). Analytical reverse-phase HPLC was performed using a Waters HPLC on a Waters Nova-Pac® HR C18 6 μm, 60 Å column (25 × 100 mm) using a gradient of MeCN/water with 0.07% TFA at flow rate of 5 mL/min and with UV detection at λ = 254 and 280 nm. NMR (1H or 13C): Varian Inova-400, Bruker DRX-500 and Bruker DRX-600 NMR spectrophotometers at 298 K. Residual solvent peaks were used as an internal reference. Coupling constants (J) (H,H) are given in Hz. Coupling patterns are designated as singlet (s), doublet (d), triplet (t), quadruplet (q), quintuplet (qt), multiplet (m), or broad signal (br). IR spectra: Thermo Scientific Nicolet 380 FT-IR spectrophotometer measured neat. High resolution mass spectral data were acquired on an Agilent Technologies High Resolution LC/MSD-TOF and the detected masses are given as m/z with M representing the molecular ion. The purity of compounds was determined as >95% as determined by HPLC analysis.
N-(2-(1-(Chloromethyl)-5-(ureidooxy)-2,3-dihydro-1H-benzo[e]indole-3-carbonyl)-1H-indol-5-yl)-1H-indole-2-carboxamide (Urea Prodrug 3)
A solution of seco-CBI-indole2 (11, 73.0 mg, 0.137 mmol) in 1:1 ether:dioxane (0.8 mL) at 0 °C and treated with LiHMDS (0.410 mL, 0.410 mmol, 1.0 M in THF). The solution was stirred for 30 min, after which 18 (94.2 mg, 0.410 mmol) was added and the solution was warmed to room temperature. The reaction was stirred for 4 h, diluted with ethyl acetate, washed with H2O and saturated aqueous NaCl, and dried (MgSO4). The solution was concentrated under reduced pressure and the resulting residue was purified by PTLC (1:1 THF:toluene, eluted 2×) to afford 3 as a pale yellow solid (4.1 mg, 5.1%): 1H NMR (DMSO-d6, 600 MHz) δ 11.92 (s, 1H), 11.73 (s, 1H), 10.20 (s, 1H), 8.26 (s, 1H), 7.93 (d, J = 7.8 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.66 (t, J = 7.2 Hz, 1H), 7.63 (d, J = 9.0 Hz, 1H), 7.50 (t, J = 8.4 Hz, 1H), 7.50 (d, J = 11 Hz, 1H), 7.48 (d, J = 11 Hz, 1H), 7.47 (s, 1H), 7.43 (s, 1H), 7.25 (s, 1H), 7.22 (t, J = 7.8 Hz, 1H), 7.07 (t, J = 7.2 Hz, 1H), 6.93 (s, 1H), 5.49 (s, 2H), 4.77 (dd, J = 10, 4.8 Hz, 1H), 4.48 (d, J = 10 Hz, 1H), 4.23 (m, 1H), 3.80 (dd, J = 13, 3.6 Hz, 1H), 3.52 (dd, J = 14, 3.6 Hz, 1H); 13C NMR (DMSO-d6, 150 MHz) δ 162.1, 159.5, 158.3, 157.5, 138.7, 136.6, 133.8, 132.0, 131.9, 131.7, 131.0, 129.9, 128.0, 127.6, 127.03, 126.96, 126.1, 125.5, 123.5, 121.6, 120.2, 119.8, 113.0, 112.4, 112.3, 111.5, 107.5, 106.9, 103.4, 66.5, 60.9, 43.4; IR (film) νmax 3307, 2958, 2889, 1647, 1521 cm−1; ESI-TOF HRMS m/z 593.1681 (M+H+, C32H25ClN6O4 requires 593.1698).
1S-3: [α]23D +11 (c 0.20, THF), natural enantiomer.
1R-3: [α]23D −10 (c 0.11, THF), unnatural enantiomer.
Methyl(3-(5-(1H-indole-2-carboxamido)-1H-indole-2-carbonyl)-1-(chloromethyl)-2,3-dihydro-1H-benzo[e]indol-5-yl)oxycarbamate (Methylcarbamate Prodrug 4)
A solution of seco-CBI-indole2 (11, 76.1 mg, 0.142 mmol) in 1:1 ether:dioxane (2.5 mL) at 0 °C was treated with LiHMDS (0.427 mL, 0.427 mmol, 1.0 M in THF). The solution was stirred for 30 min, after which 17 (105 mg, 0.427 mmol) was added and the solution was heated at 50 °C for 1 h. The reaction mixture was diluted with ethyl acetate, washed with H2O and saturated aqueous NaCl, and dried (MgSO4). The solution was concentrated under reduced pressure and the resulting residue was purified by PTLC (1:1 THF:toluene, eluted 2×) to afford 4 as a pale yellow solid (8.0 mg, 9.2%): 1H NMR (DMSO-d6, 600 MHz) δ 11.92 (s, 1H), 11.73 (s, 1H), 10.20 (s, 1H), 8.42 (s, 1H), 8.27 (s, 1H), 7.92 (d, J = 7.2 Hz, 1H), 7.694 (d, J = 8.4 Hz, 1H), 7.689 (t, J = 9.0 Hz, 1H), 7.62 (d, J = 9.0 Hz, 1H), 7.52 (t, J = 7.2 Hz, 1H), 7.50 (d, J = 7.2 Hz, 1H), 7.48 (d, J = 10 Hz, 1H), 7.43 (s, 1H), 7.38 (s, 1H), 7.31 (s, 1H), 7.22 (t, J = 7.2 Hz, 1H), 7.07 (t, J = 6.6 Hz, 1H), 4.81 (dd, J = 10, 4.8 Hz, 1H), 4.47 (d, J = 10 Hz, 1H), 3.71 (m, 1H), 3.43 (dd, J = 11, 3.0 Hz, 1H), 3.37 (s, 3H), 3.33 (d, J = 10 Hz, 1H); 13C NMR (DMSO-d6, 150 MHz) δ 161.9, 159.5, 155.8, 154.2, 137.4, 136.6, 133.8, 132.4, 132.2, 131.9, 131.7, 129.9, 129.6, 128.5, 128.3, 127.0, 126.9, 126.7, 125.1, 123.5, 121.6, 120.2, 119.8, 113.1, 112.34, 112.28, 107.2, 106.9, 103.4, 66.5, 60.3, 51.2, 42.9; IR (film) νmax 3271, 2923, 2853, 1713, 1646, 1597, 1517 cm−1; ESI-TOF HRMS m/z 608.1699 (M+H+, C33H26ClN5O5 requires 608.1695).
1S-4: [α]23D −33 (c 0.44, THF), natural enantiomer.
1R-4: [α]23D +32 (c 0.31, THF), unnatural enantiomer.
tert-Butyl(3-(5-(1H-indole-2-carboxamido)-1H-indole-2-carbonyl)-1-(chloromethyl)-2,3-dihydro-1H-benzo[e]indol-5-yl)oxycarbamate (tert-Butylcarbamate Prodrug 5)
A solution of seco-CBI-indole2 (11, 106 mg, 0.191 mmol) in 1:1 ether:dioxane (6 mL) at 0 °C was treated with LiHMDS (0.594 mL, 0.594 mmol, 1.0 M in THF). The solution was stirred for 30 min, after which 13 (171 mg, 0.594 mmol) was added and the solution was warmed to room temperature and stirred for 4 h. The reaction mixture was diluted with ethyl acetate, washed with H2O and saturated aqueous NaCl, and dried (MgSO4). The solution was concentrated under reduced pressure and purified by PTLC (1:1 THF:toluene, eluted 2×) to afford 5 as a pale yellow solid (52.1 mg, 42%): 1H NMR (DMSO-d6, 500 MHz) δ 11.80 (s, 1H), 11.73 (s, 1H), 10.84 (br s, 1H), 10.20 (s, 1H), 8.28 (s, 1H), 8.26 (s, 1H), 8.06 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.5 Hz, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.63 (t, J = 7.5 Hz, 1H), 7.60 (dd, J = 9.0, 2.0 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.48 (d, J = 8.0 Hz, 1H), 7.44 (s, 1H), 7.29 (s, 1H), 7.22 (t, J = 7.5 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 4.92 (t, J = 9.5 Hz, 1H), 4.67 (dd, J = 11, 2.0 Hz, 1H), 4.44 (m, 1H), 4.11 (dd, J = 11, 3.0 Hz, 1H), 4.04 (dd, J = 11, 6.5 Hz, 1H), 1.27 (s, 9H); 13C NMR (DMSO-d6, 150 MHz) δ 160.1, 159.5, 155.5, 151.4, 141.5, 139.1, 136.7, 133.4, 131.8, 131.7, 130.8, 129.5, 128.0, 127.7, 127.1, 124.9, 124.3, 123.5, 122.4, 121.9, 121.6, 119.8, 119.4, 112.9, 112.3, 112.2, 110.4, 106.0, 103.4, 80.8, 54.9, 47.7, 41.2, 26.2; IR (film) νmax 3271, 2973, 1714, 1632, 1517 cm−1; ESI-TOF HRMS m/z 650.2167 (M+H+, C36H32ClN5O5 requires 650.2165).
1S-5: [α]23D +6.6 (c 0.58, THF), natural enantiomer.
1R-5: [α]23D −6.0 (c 0.40, THF), unnatural enantiomer.
N-(2-(1-(Chloromethyl)-5-(pivalamidooxy)-2,3-dihydro-1H-benzo[e]indole-3-carbonyl)-1H-indol-5-yl)-1H-indole-2-carboxamide (Trimethylacetamide Prodrug 6)
A solution of seco-CBI-indole2 (11, 33.9 mg, 0.0634 mmol) in 1:1 ether:dioxane (0.1 mL) at 0 °C was treated with LiHMDS (0.190 mL, 0.190 mmol, 1.0 M in THF). The solution was stirred for 30 min, after which 15 (51.6 mg, 0.190 mmol) was added and the solution was warmed to room temperature and stirred for 4 h. The reaction mixture was diluted with ethyl acetate, washed with H2O and saturated aqueous NaCl, and dried (MgSO4). The solution was concentrated and purified by recrystallization (THF:hexanes) × 4 to afford 6 as a pale yellow solid (45.5 mg, 65%): 1H NMR (DMSO-d6, 400 MHz) δ 11.87 (s, 1H), 11.72 (s, 1H), 10.17 (s, 1H), 8.25 (s, 1H), 8.09 (br s, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.583 (d, J = 8.8 Hz, 1H), 7.575 (t, J = 6.8 Hz, 1H), 7.48 (d, J = 8.4 Hz, 1H), 7.45 (t, J = 8.4 Hz, 1H), 7.44 (d, J = 8.4 Hz, 1H), 7.43 (s, 1H), 7.26 (s, 1H), 7.22 (t, J = 8.0 Hz, 1H), 7.07 (t, J = 7.6 Hz, 1H), 4.87 (t, J = 10 Hz, 1H), 4.63 (dd, J = 11, 2.0 Hz, 1H), 4.34 (m, 1H), 4.08 (dd, J = 11, 3.2 Hz, 1H), 3.97 (dd, J = 12, 7.2 Hz, 1H), 1.20 (s, 9H); 13C NMR (THF-d8, 150 MHz) δ 161.6, 160.7, 159.1, 153.6, 143.8, 138.4, 135.0, 133.7, 133.6, 132.9, 131.7, 129.4, 128.3, 126.5, 124.9, 124.6, 124.5, 123.7, 122.6, 120.9, 120.1, 118.3, 113.9, 113.1, 112.8, 106.9, 106.7, 103.2, 56.2, 50.2, 47.6, 44.3, 30.0; IR (film) νmax 3442, 3347, 3222, 2968, 1650, 1613, 1552 cm−1; ESI-TOF HRMS m/z 656.2030 (M+Na+, C36H32ClN5O4 requires 656.2035).
1S-6: [α]23D +9.0 (c 0.20, THF), natural enantiomer.
1R-6: [α]23D −8.8 (c 0.25, THF), unnatural enantiomer.
tert-Butyl(3-(5-(1H-indole-2-carboxamido)-1H-indole-2-carbonyl)-1-(chloromethyl)-2,3-dihydro-1H-benzo[e]indol-5-yl)oxy(methyl)carbamate (N-Methyl-tert-Butylcarbamate Prodrug 8)
A solution of 5 (15.2 mg, 0.0234 mmol) in THF (0.1 mL) at −78 °C was treated with LiHMDS (23 μL, 0.023 mmol, 1.0 M in THF) and was stirred for 15 min. Me2SO4 (2.2 μL, 0.023 mmol) was added and the solution was warmed to room temperature and stirred for 2 h. The solvent was removed under a stream of N2 and the residue was purified by PTLC (1:1 THF:toluene) to afford 8 as a pale yellow solid (5.5 mg, 35%): 1H NMR (DMSO-d6, 600 MHz) δ 11.79 (s, 1H), 11.74 (s, 1H), 10.19 (s, 1H), 8.28 (s, 1H), 8.25 (s, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.64 (t, J = 7.8 Hz, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.54 (t, J = 7.8 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.44 (s, 1H), 7.29 (s, 1H), 7.22 (t, J = 7.8 Hz, 1H), 7.07 (t, J = 7.2 Hz, 1H), 4.92 (t, J = 10 Hz, 1H), 4.68 (d, J = 10 Hz, 1H), 4.45 (m, 1H), 4.11 (dd, J = 11, 3.0 Hz, 1H), 4.04 (dd, J = 11, 6.6 Hz, 1H), 3.39 (s, 3H), 1.35 (s, 9H); 13C NMR (DMSO-d6, 150 MHz) δ 160.1, 159.8, 157.1, 146.9, 141.5, 136.6, 133.4, 131.8, 131.7, 130.8, 129.5, 127.7, 127.0, 125.2, 124.0, 123.48, 123.46, 122.7, 122.0, 121.6, 119.8, 112.8, 112.3, 112.2, 110.3, 106.9, 106.0, 103.3, 82.2, 66.9, 54.8, 47.7, 41.1, 26.9; IR (film) νmax 3286, 2976, 1715, 1632, 1502 cm−1; ESI-TOF HRMS m/z 686.2153 (M+Na+, C37H34ClN5O5 requires 686.2141).
1S-8: [α]23D +24 (c 0.45, THF), natural enantiomer.
1R-8: [α]23D −22 (c 0.43, THF), unnatural enantiomer.
tert-Butyl N-Hydroxycarbamate (12)
A solution of hydroxylamine hydrochloride (5.0 g, 72.0 mmol) and (BocO)2CO (15.7 g, 72.0 mmol) in 1:1 THF:H2O (160 mL) at 0 °C was treated with NaHCO3 (12.1 g, 144 mmol). The solution was stirred at 0 °C for 2 h, after which it was diluted with ethyl acetate, washed with saturated aqueous NaHCO3, H2O, and saturated aqueous NaCl, and dried (MgSO4). The solvent was removed under reduced pressure to provide 12 as a white solid (9.25 g, 96%): 1H NMR (acetone-d6, 400 MHz) δ 8.39 (s, 1H), 7.64 (s, 1H), 1.40 (s, 9H).
tert-Butyl N-Tosyloxycarbamate (13)
A solution of 12 (3.83 g, 28.8 mmol) and TsCl (5.23 g, 27.4 mmol) in THF (40 mL) at 0 °C was treated with triethylamine (4.00 mL, 28.8 mmol). The resulting suspension was stirred at 0 °C for 2 h. The solution was filtered and the filtrate was concentrated under reduced pressure. The residue was dissolved in ethyl acetate, washed with 1 N HCl, H2O, and saturated aqueous NaCl, and dried (MgSO4). The solution was concentrated and the resulting yellow solid was triturated with hexanes to provide 13 as a pure white solid (4.1 g, 52%): 1H NMR (acetone-d6, 400 MHz) δ 10.03 (s, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 2.48 (s, 3H), 1.29 (s, 9H).
tert-Butyl Pivaloyl(N-tosyloxy)carbamate (14)
A solution of 13 (1.00 g, 3.48 mmol) and trimethylacetyl chloride (2.14 mL, 17.4 mmol) in THF (10 mL) at 0 °C was treated with triethylamine. The solution was warmed to room temperature and stirred overnight (16 h). The reaction mixture was diluted with ethyl acetate, washed with H2O and saturated aqueous NaCl, and dried (MgSO4). The solution was concentrated and the residue purified by flash chromatography to provide 14 as a white solid (1.29 g, 99%): 1H NMR (acetone-d6, 400 MHz) δ 7.90 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 8.0 Hz, 2H), 2.51 (s, 3H), 1.30 (s, 9H), 1.28 (s, 9H); 13C NMR (acetone-d6, 150 MHz) δ 183.7, 175.4, 148.4, 132.9, 131.9, 131.3, 87.4, 45.1, 28.7, 27.6, 22.6; IR (film) νmax 2978, 2936, 1808, 1737 cm−1; ESI-TOF HRMS m/z 394.1294 (M+Na+, C17H25NO6S requires 394.1295).
N-(Tosyloxy)pivalamide (15)
A vial containing 14 (91 mg, 0.245 mmol) was treated with 4 N HCl in EtOAc (10 mL). The reaction mixture was stirred at room temperature for 2 h. The solvent was evaporated under a stream of N2 to provide 15 as the HCl salt (68 mg, 91%): 1H NMR (acetone-d6, 400 MHz) δ 10.88 (s, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 2.46 (s, 3H), 1.08 (s, 9H); 13C NMR (acetone-d6, 150 MHz) δ 177.8, 147.7, 133.1, 131.4, 130.5, 54.0, 28.1, 22.6; IR (film) νmax 3197, 2977, 1695 cm−1; ESI-TOF HRMS m/z 272.0941 (M+H+, C12H17NO4S requires 272.0951).
Methyl Hydroxycarbamate (16)
A solution of hydroxylamine hydrochloride (2.00 g, 28.8 mmol) in H2O (30 mL) at 0 °C was treated with KOH (4.85 g, 86.4 mmol) and the reaction mixture was stirred for 10 minutes. Dimethyl carbonate (2.42 mL, 28.8 mmol) was added dropwise, and the reaction mixture was stirred at 5 °C for 8 h. The reaction mixture was acidified with the addition of 1 N HCl and extracted with diethyl ether (3×100 mL). The ether layers were combined and concentrated under reduced pressure to provide 16 as a clear oil (308 mg, 12%): 1H NMR (acetone-d6, 400 MHz) δ 8.83 (s, 1H), 3.63 (s, 3H).
Methyl Tosyloxycarbamate (17)
A solution of 16 (308 mg, 3.38 mmol) and TsCl (537 mg, 2.82 mmol) in diethyl ether (5 mL) at 0 °C was treated with triethylamine (0.471 mL, 3.38 mmol). The resulting suspension was stirred at 0 °C for 1 h, after which the solution was diluted with ethyl acetate, washed with H2O and saturated aqueous NaCl, and dried (MgSO4). The solution was concentrated under reduced pressure to provide 17 as an off-white solid (350 mg, 51%): 1H NMR (acetone-d6, 400 MHz) δ 10.25 (br s, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 3.60 (s, 3H), 2.48 (s, 3H); 13C NMR (acetone-d6, 125 MHz) δ 158.3, 147.9, 133.0, 131.6, 131.2, 54.5, 22.6; IR (film) νmax 3222, 2964, 1730 cm−1; ESI-TOF HRMS m/z 268.0250 (M+Na+, C9H11NO5S requires 268.0250).
1-(Tosyloxy)urea (18)
A solution of hydroxyurea (2.00 g, 26.3 mmol) and TsCl (4.80 g, 25.0 mmol) in DMF (20 mL) at 0 °C was treated with triethylamine (3.67 mL, 26.3 mmol). The solution was stirred for 3 h at 0 °C, diluted with ethyl acetate, washed with H2O and saturated aqueous NaCl, and dried (MgSO4). The solution was concentrated under reduced pressure and purified by flash chromatography to provide 18 as a white solid (716 mg, 12%): 1H NMR (acetone-d6, 400 MHz) δ 9.33 (s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 6.23 (br s, 2H), 2.48 (s, 3H); 13C NMR (acetone-d6, 125 MHz) δ 160.7, 147.9, 132.9, 131.7, 131.2, 22.6; IR (film) νmax 3444, 3233, 3052, 2855, 1698 cm−1; ESI-TOF HRMS m/z 253.0250 (M+Na+, C8H10N2O4S requires 253.0253).
In Vivo Antitumor Activity
DBA/2J mice were injected intraperitoneal (i.p.) with syngeneic L1210 cells (1 × 106) on day 0. Ten mice were randomly assigned to treatment groups for compounds 3–5, 11, or vehicle at doses of 50, 100, 250, and 500 μg/kg/inj. for 3, 10, 30, 60, 100, and 200 μg/kg/inj. for 4, and 10, 30, 60, and 100 μg/kg/inj. for 5 and 11. Compounds 3–5 and 11 were formulated with 30% DMSO plus 0.7% glucose solution and were i.p. injected consecutively on days 1, 5, and 9. Animals were monitored daily and weighed three times per week following injection of tumor cells. Tumor burden in the peritoneal cavity and survivorship were determined for animals in each treatment group.
Supplementary Material
Figure 5.

In vivo antitumor activity (L1210, i.p.)
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA 41986), the Skaggs Institute for Chemical Biology, and a grant from the Kansas Biotech Authority. JPL and WMR are Skaggs Fellows.
Abbreviations
- BnSH
benzyl thiol
- CBI
1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one
- LiHMDS
lithium bis(trimethylsilyl)amide
- THF
tetrahydrofuran
- TFA
trifluoroacetic acid
- EtOAc
ethyl acetate
- DMAP
4-dimethylaminopyridine
- DMF
dimethylformamide
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
Footnotes
Supporting Information Available: A table of the stability data for prodrugs 3–6 and 8 is provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Martin DG, Biles C, Gerpheide SA, Hanka LJ, Krueger WC, McGovren JP, Mizsak SA, Neil GL, Stewart JC, Visser J. CC-1065 (NSC 298223), a Potent New Antitumor Agent. Improved Production and Isolation, Characterization and Antitumor Activity. J Antibiot. 1981;34:1119–1125. doi: 10.7164/antibiotics.34.1119. [DOI] [PubMed] [Google Scholar]; (b) Ichimura M, Ogawa T, Takahashi K, Kobayashi E, Kawamoto I, Yasuzawa T, Takahashi I, Nakano H. Duocarmycin SA, a New Antitumor Antibiotic From Streptomyces sp. J Antibiot. 1990;43:1037–1038. doi: 10.7164/antibiotics.43.1037. [DOI] [PubMed] [Google Scholar]; (c) Takahashi I, Takahashi K, Ichimura M, Morimoto M, Asano K, Kawamoto I, Tomita F, Nakano H. Duocarmycin A, a New Antitumor Antibiotic From Streptomyces. J Antibiot. 1988;41:1915–1917. doi: 10.7164/antibiotics.41.1915. [DOI] [PubMed] [Google Scholar]; (d) Igarashi Y, Futamata K, Fujita T, Sekine A, Senda H, Naoki H, Furumai T. Yatakemycin, a Novel Antifungal Antibiotic Produced by Streptomyces sp. TP-A0356. J Antibiot. 2003;56:107–113. doi: 10.7164/antibiotics.56.107. [DOI] [PubMed] [Google Scholar]
- 2.For duocarmycin SA, see: Boger DL, Johnson DS, Yun W. (+)- and ent-(−)-Duocarmycin SA and (+)- and ent-(−)-N-BOC-DSA DNA Alkylation Properties. Alkylation Site Models That Accommodate the Offset AT-Rich Adenine N3 Alkylation Selectivity of the Enantiomeric Agents. J Am Chem Soc. 1994;116:1635–1656. doi: 10.1016/s0968-0896(00)82007-6.For yatakemycin, see: Parrish JP, Kastrinsky DB, Wolkenberg SE, Igarashi Y, Boger DL. DNA Alkylation Properties of Yatakemycin. J Am Chem Soc. 2003;125:10971–10976. doi: 10.1021/ja035984h.Trzupek JD, Gottesfeld JM, Boger DL. Alkylation of Duplex DNA in Nucleosome Core Particles by Duocarmycin SA and Yatakemycin. Nat Chem Biol. 2006;2:79–82. doi: 10.1038/nchembio761.Tichenor MS, MacMillan KS, Trzupek JD, Rayl TJ, Hwang I, Boger DL. Systematic Exploration of the Structural Features of Yatakemycin Impacting DNA Alkylation and Biological Activity. J Am Chem Soc. 2007;129:10858–10869. doi: 10.1021/ja072777z.For CC-1065, see: Hurley LH, Lee CS, McGovren JP, Warpehoski MA, Mitchell MA, Kelly RC, Aristoff PA. Molecular Basis for Sequence-Specific DNA Alkylation by CC-1065. Biochemistry. 1988;27:3886–3892. doi: 10.1021/bi00410a054.Boger DL, Johnson DS, Yun W, Tarby CM. Molecular Basis for Sequence Selective DNA Alkylation by (+)- and ent-(−)-CC-1065 and Related Agents: Alkylation Site Models that Accommodate the Offset AT-Rich Adenine N3 Alkylation Selectivity. Bioorg Med Chem. 1994;2:115–135. doi: 10.1016/s0968-0896(00)82007-6.Boger DL, Coleman RS, Invergo BJ, Sakya SM, Ishizaki T, Munk SA, Zarrinmayeh H, Kitos PA, Thompson SC. Synthesis and Evaluation of Aborted and Extended CC-1065 Functional Analogs: (+)- and (−)-CPI-PDE-I1, (+)- and (−)-CPI-CDPI1, and (±)-, (+)-, and (−)-CPI-CDPI3. Preparation of Key Partial Structures and Definition of an Additional Functional Role of the CC-1065 Central and Right-Hand Subunits. J Am Chem Soc. 1990;112:4623–4640.For duocarmycin A, see: Boger DL, Ishizaki T, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. Duocarmycin-Pyrindamycin DNA Alkylation Properties and Identification, Synthesis, and Evaluation of Agents Incorporating the Pharmacophore of the Duocarmycin-Pyrindamycin Alkylation Subunit. Identification of the CC-1065 Duocarmycin Common Pharmacophore. J Am Chem Soc. 1990;112:8961–8971.Boger DL, Ishizaki T, Zarrinmayeh H. Isolation and Characterization of the Duocarmycin-Adenine DNA Adduct. J Am Chem Soc. 1991;113:6645–6649.Boger DL, Yun W, Terashima S, Fukuda Y, Nakatani K, Kitos PA, Jin Q. DNA Alkylation Properties of the Duocarmycins: (+)-Duocarmycin A, epi-(+)-Duocarmycin A, ent-(−)-Duocarmycin A and epi,ent-(−)-Duocarmycin A. Bioorg Med Chem Lett. 1992;2:759–765.Boger DL, Yun W. Reversibility of the Duocarmycin A and SA DNA Alkylation Reaction. J Am Chem Soc. 1993;115:9872–9873.
- 3.Reviews: Boger DL, Johnson DS. CC-1065 and the Duocarmycins: Understanding Their Biological Function Through Mechanistic Studies. Angew Chem Int Ed Engl. 1996;35:1438–1474.Boger DL. The Duocarmycins: Synthetic and Mechanistic Studies. Acc Chem Res. 1995;28:20–29.Boger DL, Johnson DS. CC-1065 and the Duocarmycins: Unraveling the Keys to a New Class of Naturally Derived DNA Alkylating Agents. Proc Natl Acad Sci USA. 1995;92:3642–3650. doi: 10.1073/pnas.92.9.3642.Boger DL, Garbaccio RM. Shape-Dependent Catalysis: Insights into the Source of Catalysis for the CC-1065 and Duocarmycin DNA Alkylation Reaction. Acc Chem Res. 1999;32:1043–1052.Tichenor MS, Boger DL. Yatakemycin: Total Synthesis, DNA Alkylation, and Biological Properties. Natural Prod Rep. 2008;25:220–226. doi: 10.1039/b705665f.MacMillan KS, Boger DL. Fundamental Relationships Between Structure, Reactivity, and Biological Activity for the Duocarmycins and CC-1065. J Med Chem. 2009;52:5771–5780. doi: 10.1021/jm9006214.Searcey M. Duocarmycins: Natures Prodrugs? Curr Pharm Des. 2002;8:1375–1389. doi: 10.2174/1381612023394539.Tse WC, Boger DL. Sequence-Selective DNA Recognition: Natural Products and Nature’s Lessons. Chem Biol. 2004;11:1607–1617. doi: 10.1016/j.chembiol.2003.08.012.
- 4.(a) Boger DL, Coleman RS. Total Synthesis of (+)-CC-1065 and ent-(−)-CC- J Am Chem Soc 1988. 1065;110:1321–1323. [Google Scholar]; (b) Boger DL, Coleman RS. Total Synthesis of CC-1065,, and the Precise, Functional Agents CPI-CDPI2. J Am Chem Soc. 1988;110:4796–4807. [Google Scholar]; (c) Boger DL, Machiya K. Total Synthesis of (+)-Duocarmycin SA. J Am Chem Soc. 1992;114:10056–10058. [Google Scholar]; (d) Boger DL, Machiya K, Hertzog DL, Kitos PA, Holmes D. Total Synthesis and Preliminary Evaluation of (+)- and ent-(−)-Duocarmycin SA. J Am Chem Soc. 1993;115:9025–9036. [Google Scholar]; (e) Boger DL, McKie JA, Nishi T, Ogiku T. Enantioselective Total Synthesis of (+)-Duocarmycin A, epi-(+)-Duocarmycin A, and Their Unnatural Enantiomers. J Am Chem Soc. 1996;118:2301–2302. [Google Scholar]; (f) Boger DL, McKie JA, Nishi T, Ogiku T. Total Synthesis of (+)-Duocarmycin A and epi-(+)-Duocarmycin A and Their Unnatural Enantiomers: Assessment of Chemical and Biological Properties. J Am Chem Soc. 1997;119:311–325. [Google Scholar]; (g) Tichenor MS, Kastrinsky DB, Boger DL. Total Synthesis, Structure Revision, and Absolute Configuration of (+)-Yatakemycin. J Am Chem Soc. 2004;126:8396–8398. doi: 10.1021/ja0472735. [DOI] [PubMed] [Google Scholar]; (h) Tichenor MS, Trzupek JD, Kastrinsky DB, Shiga F, Hwang I, Boger DL. Asymmetric Total Synthesis of (+)- and ent-(−)-Yatakemycin and Duocarmycin SA. Evaluation of Yatakemycin Key Partial Structures and Its Unnatural Enantiomer. J Am Chem Soc. 2006;128:15683–15696. doi: 10.1021/ja064228j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Boger DL, Hertzog DL, Bollinger B, Johnson DS, Cai H, Goldberg J, Turnbull P. Duocarmycin SA Shortened, Simplified, and Extended Agents: a Systematic Examination of the Role of the DNA Binding Subunit. J Am Chem Soc. 1997;119:4977–4986. [Google Scholar]; (b) Boger DL, Bollinger B, Hertzog DL, Johnson DS, Cai H, Mesini P, Garbaccio RM, Jin Q, Kitos PA. Reversed and Sandwiched Analogs of Duocarmycin SA: Establishment of the Origin of the Sequence-Selective Alkylation of DNA and New Insights into the Source of Catalysis. J Am Chem Soc. 1997;119:4987–4988. [Google Scholar]; (c) Boger DL, Garbaccio RM. Catalysis of the CC-1065 and Duocarmycin DNA Alkylation Reaction: DNA Binding Induced Conformational Change in the Agent Results in Activation. Bioorg Med Chem. 1997;5:263–276. doi: 10.1016/s0968-0896(96)00238-6. [DOI] [PubMed] [Google Scholar]
- 6.(a) Boger DL, Ishizaki T, Kitos PA, Suntornwat O. Synthesis of N-(tert-Butyloxycarbonyl)-CBI, CBI, CBI-CDPI1, and CBI-CDPI2: Enhanced Functional Analogs of CC-1065 Incorporating the 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) Left-Hand Subunit. J Org Chem. 1990;55:5823–5832. [Google Scholar]; (b) Boger DL, Ishizaki T, Wysocki RJ, Jr, Munk SA, Kitos PA, Suntornwat O. Total Synthesis and Evaluation of (±)-N-(tert-Butoxycarbonyl)-CBI, (±)-CBI-CDPI1, and (±)-CBI-CDPI2: CC-1065 Functional Agents Incorporating the Equivalent 1,2,9,9a-Tetrahydrocyclopropa[1,2-c]benz[1,2-e]indol-4-one (CBI) Left-Hand Subunit. J Am Chem Soc. 1989;111:6461–6463. [Google Scholar]; (c) Boger DL, Ishizaki T. Resolution of a CBI Precursor and Incorporation into the Synthesis of (+)-CBI, (+)-CBI-CDPI1, (+)-CBI-CDPI2: Enhanced Functional Analogs of (+)-CC-1065. A Critical Appraisal of a Proposed Relationship Between Electrophile Reactivity, DNA Binding Properties, and Cytotoxic Potency. Tetrahedron Lett. 1990;31:793–796. [Google Scholar]; (d) Boger DL, Ishizaki T, Zarrinmayeh H, Kitos PA, Suntornwat O. A Potent, Simple Derivative of an Analog of the CC-1065 Alkylation Subunit. Bioorg Med Chem Lett. 1991;1:55–58. [Google Scholar]; (e) Boger DL, Munk SA. DNA Alkylation Properties of Enhanced Functional Analogs of CC-1065 Incorporating the 1,2,9,9a-Tetrahydrocyclopropa[1,2-c]benz[1,2-e]indol-4-one (CBI) Alkylation Subunit. J Am Chem Soc. 1992;114:5487–5496. [Google Scholar]; (f) Boger DL, Yun W. Role of the CC-1065 and Duocarmycin N2 Substituent: Validation of a Direct Relationship between Solvolysis Chemical Stability and in vitro Biological Potency. J Am Chem Soc. 1994;116:5523–5524. [Google Scholar]; (g) Boger DL, Yun W, Cai H, Han N. CBI-CDPBO1 and CBI-CDPBI1: CC-1065 Analogs Containing Deep-Seated Modifications in the DNA Binding Subunit. Bioorg Med Chem. 1995;3:761–775. doi: 10.1016/0968-0896(95)00066-p. [DOI] [PubMed] [Google Scholar]; (h) Boger DL, Yun W. CBI-TMI: Synthesis and Evaluation of a Key Analog of the Duocarmycins. Validation of a Direct Relationship between Chemical Solvolytic Stability and Cytotoxic Potency and Confirmation of the Structural Features Responsible for the Distinguishing Behavior of Enantiomeric Pairs of Agents. J Am Chem Soc. 1994;116:7996–8006. [Google Scholar]
- 7.Boger DL, Yun W, Han N. 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) Analogs of CC-1065 and the Duocarmycins: Synthesis and Evaluation. Bioorg Med Chem. 1995;3:1429–1453. doi: 10.1016/0968-0896(95)00130-9. [DOI] [PubMed] [Google Scholar]
- 8.For Carzelesin, see: Aristoff PA. CC-1065 Analogs: Sequence Specific DNA-Alkylating Antitumor Agents. Adv Med Chem. 1993;2:67–110.For KW-2189, see: Kobayashi E, Okamoto A, Asada M, Okabe M, Nagamura S, Asai A, Saito H, Gomi K, Hirata T. Characteristics of Antitumor Activity of KW-2189, a Novel Water-Soluble Derivative of Duocarmycin, Against Murine and Human Tumors. Cancer Res. 1994;54:2404–2410.For CBI, see: Boger DL, Boyce CW, Garbaccio RM, Searcey M, Jin Q. CBI Prodrug Analogs of CC-1065 and the Duocarmycins. Synthesis. 1999:1505–1509.Tietze LF, Schuster HJ, Schmuck K, Schuberth I, Alves F. Duocarmycin-based Prodrugs for Cancer Prodrug Monotherapy. Bioorg Med Chem. 2008;16:6312–6318. doi: 10.1016/j.bmc.2008.05.009.Li LS, Sinha SC. Studies Toward the Duocarmycin Prodrugs for the Antibody Prodrug Therapy Approach. Tetrahedron Lett. 2009;50:2932–2935. doi: 10.1016/j.tetlet.2009.03.205.
- 9.Wolkenberg SE, Boger DL. Mechanisms of in Situ Activation for DNA-Targeting Antitumor Agents. Chem Rev. 2002;102:2477–2496. doi: 10.1021/cr010046q.Reviews on reductive activation: Papadopoulou MV, Bloomer WD. Exploiting Hypoxia in Solid Tumors with DNA-Targeted Bioreductive Drugs. Drugs Future. 2004;29:807–819.Jaffar M, Stratford IJ. Bioreductive Drugs: Selectivity Towards Hypoxic Tissue. Exp Opin Ther Pat. 1999;9:1371–1380.
- 10.(a) Chari RV, Jackel KA, Bourret LA, Derr SJ, Tadayoni BM, Mattocks KM, Shah SA, Liu C, Blattler WA, Goldmacher VS. Enhancement of the Selectivity and Antitumor Efficacy of a CC-1065 Analog Through Immunoconjugate Formation. Cancer Res. 1995;55:4079–4084. [PubMed] [Google Scholar]; (b) Lillo AM, Sun C, Gao C, Ditzel H, Parrish J, Gauss CM, Moss J, Felding–Habermann B, Wirsching P, Boger DL, Janda KD. A Human Single-Chain Antibody Specific for Integrin α3β1 Capable of Cell Internalization and Delivery of Antitumor Agents. Chem Biol. 2004;11:897–906. doi: 10.1016/j.chembiol.2004.04.018. [DOI] [PubMed] [Google Scholar]; (c) Tietze LF, Herzig T, Feuerstein T, Schuberth I. Synthesis and Biological Evaluation of Novel Analogues and Prodrugs of the Cytotoxic Antibiotic CC-1065 for Selective Cancer Therapy. Eur J Org Chem. 2002;10:1634–1645. [Google Scholar]; (d) Tietze LF, Major F, Schuberth I. Antitumor Agents: Development of Highly Potent Glycosidic Duocarmycin Analogs for Selective Cancer Therapy. Angew Chem Int Ed. 2006;45:6574–6577. doi: 10.1002/anie.200600936. [DOI] [PubMed] [Google Scholar]; (e) Wang Y, Yuan H, Wright SC, Wang H, Larrick JW. Synthesis and Preliminary Cytotoxicity Study of Glucuronide Derivatives of CC-1065 Analogues. Bioorg Med Chem. 2003;11:1569–1575. doi: 10.1016/s0968-0896(02)00603-x. [DOI] [PubMed] [Google Scholar]; (f) Kline T, Torgov MY, Mendelsohn BA, Cerveny CG, Senter PD. Novel Antitumor Prodrugs Designed for Activation by Matrix Metalloproteinases-2 and -9. Mol Pharm. 2004;1:9–22. doi: 10.1021/mp0340183. [DOI] [PubMed] [Google Scholar]
- 11.(a) Hay MP, Anderson RF, Ferry DM, Wilson WR, Denny WA. Synthesis and Evaluation of Nitroheterocyclic Carbamate Prodrugs for Use with Nitroreductase-Mediated Gene-Directed Enzyme Prodrug Therapy. J Med Chem. 2003;46:5533–5545. doi: 10.1021/jm030308b. [DOI] [PubMed] [Google Scholar]; (b) Hay MP, Sykes BM, Denny WA, Wilson WR. A 2-Nitroimidazole Carbamate Prodrug of 5-Amino-1-(chloromethyl)-3-[(5,6,7-trimethoxyindol-2-yl)carbonyl]-1,2-dihydro-3H-benz[e]indole (Amino-seco-CBI-TMI) for Use With ADEPT and GDEPT. Bioorg Med Chem Lett. 1999;9:2237–2242. doi: 10.1016/s0960-894x(99)00381-9. [DOI] [PubMed] [Google Scholar]; (c) Townes H, Summerville K, Purnell B, Hooker M, Madsen E, Hudson S, Lee M. Investigation of a Novel Reductively-Activatable Anticancer Prodrug of seco-CBI-TMI, an Analog of Duocarmycin SA. Med Chem Res. 2002;11:248–253. [Google Scholar]; (d) Boger DL, Garbaccio RM. A Novel Class of CC-1065 and Duocarmycin Analogues Subject to Mitomycin-Related Reductive Activation. J Org Chem. 1999;69:8350–8362. doi: 10.1021/jo991301y. [DOI] [PubMed] [Google Scholar]
- 12.Jin W, Trzupek JD, Rayl TJ, Broward MA, Vielhauer GA, Weir SJ, Hwang I, Boger DL. A Unique Class of Duocarmycin and CC-1065 Analogues Subject to Reductive Activation. J Am Chem Soc. 2007;129:15391–15397. doi: 10.1021/ja075398e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denny WA. Hypoxia-Selective Cytotoxins. In: Foye WO, editor. Cancer Chemotherapeutic Agents. American Chemical Society; Washington DC: 1995. pp. 483–500. [Google Scholar]
- 14.(a) Paz MM, Hopkins PB. DNA-DNA Interstrand Crosslinking by FR66979: Intermediates in the Activation Cascade. J Am Chem Soc. 1997;119:5999–6005. [Google Scholar]; (b) Williams RM, Rajski SR, Rollins SB. FR 900482, a Close Cousin of Mitomycin C that Exploits Mitosene-based DNA Crosslinking. Chem Biol. 1997;4:127–137. doi: 10.1016/s1074-5521(97)90256-8. [DOI] [PubMed] [Google Scholar]
- 15.Tepe JJ, Kosogof C, Williams RM. DNA Interstrand Cross-link Formation by Reductive Activation of Dehydropyrrolizidine Progenitors. Tetrahedron. 2002;58:3553–3559. [Google Scholar]
- 16.(a) Meister A, Anderson ME. Glutathione. Ann Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]; (b) Perry RR, Mazetta JA, Levin M, Barranco SC. Glutathione Levels and Variability in Breast Tumors and Normal Tissue. Cancer. 1993;72:783–787. doi: 10.1002/1097-0142(19930801)72:3<783::aid-cncr2820720325>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; (c) Oberli-Schrämmli AE, Joncourt F, Stadler M, Altermatt HJ, Buser K, Ris HB, Schmid U, Cerny T. Parallel Assessment of Glutathione-based Detoxifying Enzymes, O6-Alkylguanine-DNA Alkyltransferase and P-Glycoprotein as Indicators of Drug Resistance in Tumor and Normal Lung of Patients with Lung Cancer. Int J Cancer. 1994;59:629–636. doi: 10.1002/ijc.2910590509. [DOI] [PubMed] [Google Scholar]; (d) Cook JA, Pass HI, Iype SN, Friedman N, DeGraff W, Russo A, Mitchell JB. Cellular Glutathione and Thiol Measurements from Surgically Resected Human Lung Tumor and Normal Lung Tissue. Cancer Res. 1991;51:4287–4294. [PubMed] [Google Scholar]
- 17.Meister A. Glutathione Deficiency Produced by Inhibition of its Synthesis, and its Reversal; Applications in Research and Therapy. Pharmacol Ther. 1991;51:155–194. doi: 10.1016/0163-7258(91)90076-x. [DOI] [PubMed] [Google Scholar]
- 18.Chari RVJ. Targeted Cancer Therapy: Conferring Specificity to Cytotoxic Drugs. Acc Chem Res. 2008;41:98–107. doi: 10.1021/ar700108g. [DOI] [PubMed] [Google Scholar]
- 19.(a) Boger DL, Ishizaki T, Sakya SM, Munk SA, Kitos PA, Jin Q, Besterman JM. Synthesis and Preliminary Evaluation of (+)-CBI-indole2: An Enhanced Functional Analog of (+)-CC-1065. Bioorg Med Chem Lett. 1991;1:115–120. [Google Scholar]; (b) Boger DL, Boyce CW, Garbaccio RM, Searcey M. Synthesis of CC-1065 and Duocarmycin Analogs via Intramolecular Aryl Radical Cyclization of a Tethered Vinyl Chloride. Tetrahedron Lett. 1998;29:2227–2230. [Google Scholar]; (c) Kastrinksy DB, Boger DL. Effective Asymmetric Synthesis of 1,2,9,9a-Tetrahydrocyclopropa[c]benzo[e]indol-4-one (CBI) J Org Chem. 2004;69:2284–2289. doi: 10.1021/jo035465x. [DOI] [PubMed] [Google Scholar]; (d) Boger DL, McKie JA, Boyce CW. Asymmetric Synthesis of the 1,2,9,9a-Tetrahydrocyclopropa[c]benzo[e]indol-4-one (CBI) Alkylation Subunit of the CC-1065 and Duocarmycin Analogs. Synlett. 1997;5:515–517. [Google Scholar]; (e) Boger DL, McKie JA. An Efficient Synthesis of 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one CBI: an Enhanced and Simplified Analog of the CC-1065 and Duocarmycin Alkylation Subunits. J Org Chem. 1995;60:1271–1275. [Google Scholar]; (f) Boger DL, Yun W, Teegarden BR. An Improved Synthesis of 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI): a Simplified Analog of the CC-1065 Alkylation Subunit. J Org Chem. 1992;57:2873–2876. [Google Scholar]; (g) Boger DL, Wysocki RJ, Ishizaki T. Synthesis of N-(Phenylsulfonyl)-CI, N-Boc-CI, CI-CDPI1 and CI-CDPI2: CC-1065 Analogs Incorporating the Parent 1,2,7,7a-Tetrahydrocycloprop[1,2-c]indol-4-one (CI) Left-hand Subunit. J Am Chem Soc. 1990;112:5230–5240. [Google Scholar]; (h) Boger DL, Boyce CW, Garbaccio RM, Goldberg JA. CC-1065 and the Duocarmycins: Synthetic Studies. Chem Rev. 1997;97:787–828. doi: 10.1021/cr960095g. [DOI] [PubMed] [Google Scholar]
- 20.Greck C, Bischoff L, Girard A, Hajicek J, Genet JP. Electrophilic Amination of Carbanions with Metalated t-Butyl-N-Tosyloxycarbamate. Bull Soc Chim Fr. 1994;131:429–433. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.