

Abstract
Cyclooxygenase-2 is frequently over-expressed and associated with a poor prognosis in breast cancer. The COX-2 product prostaglandin E2 (PGE2) elicits cellular responses through four G-protein-coupled receptors designated EP1–4 coupled to distinct intracellular signaling pathways. EP4, expressed on malignant breast cells, promotes metastasis, but a role for EP1 in metastasis has not been investigated. Using a murine model of metastatic breast cancer, we now show that pharmacologic antagonism of EP1 with SC19220 or AH6809 promoted lung colonization of mammary tumor cells by 3.7–5.4 fold. Likewise, reducing EP1 gene expression by shRNA also increased metastatic capacity relative to cells transfected with non-silencing vector but did not affect the size of transplanted tumors. Examination of invasive ductal carcinomas by immunohistochemistry shows that EP1 was detected in both the cytoplasm and nucleus of benign ducts as well as malignant cells in some samples, but was absent or limited to either the nucleus or cytoplasm in other malignant samples. Overall survival for women with tumors that were negative for nuclear EP1 was significantly worse than for women with EP1 expression (p=0.008). There was no difference in survival for women with differences in cytoplasmic EP1 expression (p=0.46). Comparing EP1 mRNA in breast tumors from African-American and European-American women revealed that many more AA breast tumors lacked detectable EP1 mRNA (p=0.04). These studies support the hypothesis that EP1 functions as a metastasis suppressor and that loss of nuclear EP1 is associated with poorer overall survival and may contribute to disparities in outcome in different populations.
Introduction
In tumors, the cyclooxygenase -2 (COX-2) enzyme is often highly expressed and is associated with poor survival in breast and other malignancies (1). The principle COX-2 product is prostaglandin E2. Cellular effects of PGE2 are mediated through a family of G-protein-coupled receptors designated EP1,2,3 and EP4 (2). Despite structural and sequence similarities among the four EP receptors, they are coupled to different intracellular signaling pathways. Ligand binding of EP1 is associated with Gq protein, calcium flux and subsequent PKC activation, whereas EP2 and EP4 are coupled to Gs, PKA/adenyl cyclase and mediate elevations in intracellular cAMP. EP4 also activates ERK1 and ERK2 by way of PI3K (3). Each of the EP receptors has been implicated in primary carcinogenesis (4–8), however, little is known regarding the role of individual EPs in the process of tumor metastasis. We and others have shown that EP4 functions to promote metastasis in breast, lung and colon cancer (9–12), but other EPs, including EP1, had not been investigated. A study by Thorat et al (13) reported that nuclear expression of EP1 was associated with lymph-node negative breast cancer and we had reported some years ago that EP antagonists, then of unknown specificity, promoted metastasis (14), however, no study had directly examined the role of EP1 in metastasis. We now report that pharmacologic antagonism or gene silencing of EP1 promotes metastasis leading us to propose that EP1 is a novel metastasis suppressor. Our studies also show that expression of EP1 in the nucleus of malignant cells is associated with significantly better overall survival in women with early stage disease and hint at a contribution of EP1 to breast cancer disparities.
Materials and Methods
Cells
Line 66.1 and 410 tumor cells were derived from a spontaneously occurring mammary adenocarcinoma in a Balb/cfC3H mouse (15). Line 410.4 was derived from a pulmonary tumor of a mouse implanted with line 410 tumor cells. Both lines 66.1 and 410.4 are highly tumorigenic and metastatic following s.c. or i.v. injection into syngeneic Balb/c mice. Cells are grown in DMEM, supplemented with 10% fetal bovine serum (Gemini Bioproducts), 1.5 mg/mL sodium bicarbonate, 2 mmol/L L-glutamine, 100 umol/L nonessential amino acids, 100 units/mL penicillin and 100 units/mL streptomycin in a 10% CO2 atmosphere.
Construction and characterization of EP1 gene-silenced cell lines
Line 66.1 tumor cells were double-transfected with a control vector or plasmid expressing EP1 shRNA (SureSilencing, SABiosciences, Frederick, MD) and with a TRC shRNA construct targeting EP1 or control vector pLKO.1 (Thermo Scientific Open Biosystems, Huntsville, AL) and selected in puromycin (Sigma Chem. Co.). Multiple stable transfectants were analyzed for expression of EP1 mRNA by standard reverse transcription-PCR protocols using EP1 or glyceraldehyde-3-phosphate dehydrogenase-specific primers and by western blotting with antibodies to EP1 or β-actin.
Quantitative Real-Time RT-PCR
Total RNA extracted from cells, reverse transcribed and quantitative PCR amplification performed using iQ™ SYBR Green Supermix (Bio-Rad, Hercules, CA) and the following gene-specific primers: 5′-CACCCAGGCTCCCCAATAC-3′ (sense) and 5′-GCGACGAACAACAGGAAGG-3′-3′ (anti-sense) for mouse EP1 and 5′-GCCTTCCGTGTTCCTACC-3′ (sense) and 5′-GCCTGCTTCACCACCTTC-3′ (anti-sense) for mouse GAPDH.
Mice
Balb/cByJ female mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice were housed, cared for, and used in strict accordance with the U.S. Department of Agriculture regulations and the NIH Health Guide for the Care and Use of Laboratory Animals. The University of Maryland School of Medicine Animal Facility is fully accredited by the American Association for the Accreditation of Laboratory Animal Care.
In Vivo Studies
Local tumor growth and spontaneous metastases were evaluated by injecting tumor cells s.c. proximal to the left abdominal mammary gland of syngeneic female mice. Tumor diameters were measured with a caliper twice weekly and mice were euthanized on an individual basis when the s.c. tumor measured 16 mm in diameter or earlier if moribund. The lungs were weighed and surface tumor colonies were quantified in a blinded fashion under a dissecting microscope. Lung colonization was evaluated by injecting 2–3 × 105 viable tumor cells i.v. into the lateral tail vein of syngeneic female mice. Parental tumor cells were cultured in the presence of the EP1 antagonists SC19220, AH6809 or vehicle at the indicated concentrations for 48 hrs, washed and injected into mice. All mice were euthanized on day 18–22 posttransplantation or earlier if moribund. Lungs were examined for tumor colonies as above.
Immunohistochemistry
A tissue microarray was prepared from formalin-fixed, paraffin-embedded tissue specimens acquired through the Breast Cancer Prognostic Study of the Karmanos Cancer Institute, Detroit, MI and described previously (16). This was a prospective study of 1,306 women with newly diagnosed breast cancer from 1975 to 1983. Clinical and pathologic data were collected from all study subjects. Follow-up data collection from the women continued until 1998. An analysis of all lesions (55) that were classified as invasive ductal carcinoma were used for the current study. For women who died of breast cancer, the mean time to death was 6.1 yr and for those alive at last follow-up, the mean follow-up time was 12.4 y. Immunohistochemistry was done using monoclonal EP1 antibody clone 5F12 described in detail in (17) or isotype control antibody. This antibody identifies a protein of the appropriate m.w. in human or murine tissue or cells transduced with the EP1 gene, but no band is visible in lysates prepared from tissues of EP1−/− mice. Staining was performed on a DAKO automatic stainer using diaminobenzidine as the chromagen and detection with EnVision+ detection system. Staining intensity was scored semiquantitatively separately for the cytoplasm and nucleus, using a scale from 0 to 3; 0, no staining; 1, equivocally positive; 2, definitely positive; 3, intensely positive. Specimens were scored in a blinded fashion by two investigators (N.K. and O. I).
Statistical Methods
For studies in mice, the distribution of the number of metastases was compared between the prespecified treatment groups using the nonparametric exact Wilcoxon rank-sum test for groups with a relatively small sample size (fewer than 10 mice). The general linear model approach was applied to estimate and compare average number of lung metastases across larger treatment groups. The square root transformation of the metastasis data was used to decrease variability and to ensure approximate normality. All tests were exact, two-sided and done at the 0.05 level of significance. In the retrospective cohort study, survival time was defined as time from study entry to death. The censored follow-up time for patients without death information was the date of last contact. EP1 expression for each patient was defined to be the minimum EP1 score of the patients’ multiple tissue microarray cores. Overall survival functions were estimated by Kaplan-Meier method for 55 patients grouped by staining intensity of nuclear or cytoplasmic EP1. The hazard ratios (HR) with the corresponding confidence intervals (CI) were estimated using the Cox proportional hazard models. The association of EP1 level and presence of positive lymph nodes was assessed using the Jonckheere-Terpstra test for doubly ordered rxc contingency table.
Microarray analyses
The preparation and analysis of the gene microarray was described in detail previously (18). Total RNA was extracted from thirty-six fresh frozen breast cancers obtained at the University of Maryland Medical System between 1994 and 1999, from eighteen African-American and eighteen European-American women. Twenty patients had stage II disease, and 16 were diagnosed with stage III disease. RNA was amplified from 20–50 ug of total RNA with a T7-based protocol and labeled using random primers and Superscript reverse transcriptase. Microarray slides containing 8,064 sequence verified human cDNA clones were prepared and gene expression analyzed as described. The Lawrence Livermore Laboratory cDNA library was used as a clone source (Invitrogen, Carlsbad, CA). Data points representing gene expression ratios were log-transformed. To explore the relationship of race to EP1 gene expression, the general linear model was used.
Results
To examine the role of EP1 in modulating tumor behavior, we employed a syngeneic murine model of metastatic breast cancer. Lines 410, 66.1 and 410.4 were derived from a spontaneously-occurring mammary tumor; the latter two lines are highly metastatic in syngeneic mice. By RT-PCR and western blotting we first showed that all three cell lines express EP1 as well as EP2-EP4 (Figure 1a, b). Others have reported that EP1 is expressed in the nucleus of malignant cells (13,19). By confocal microscopy, EP1 protein was detected in both the cytoplasm and nucleus of murine mammary tumor cells (Figure 1c).
Figure 1.

(A) RNA isolated from murine mammary tumor cell lines 410, 410.4 and 66.1 and analyzed by RT-PCR using probes specific for murine EP1, EP2, EP3 or EP4. (B) lyates of the same cell lines immunoblotted with antibodies specific for each EP receptor subtype. (C) Line 66.1 cells examined by confocal microscopy using DAPI (upper panel), antibody to EP1 (middle panel) or the merged images (lower panel).
A role for EP1 in modulation of metastatic behavior has not been elucidated. To begin to determine a functional role for EP1 in metastasis, we asked if pharmacologic antagonists of EP1 (SC19220) or EP1/EP2 (AH6809), could modulate lung-colonizing ability of metastatic cell lines 66.1 and 410.4. In figure 2a, line 66.1 cells were cultured in the presence of SC19220 or AH6809 (1.0 ug/ml) for 48 hrs, washed and 2 × 105 viable tumor cells injected into the lateral tail vein of syngeneic female Balb/cByJ mice. On day 18, all mice were euthanized and surface lung tumor colonies were quantified. Pretreatment of line 66.1 tumor cells with either SC19220 or AH6809 increased the number of lung tumor colonies by 3.7- or 5.4-fold, respectively. These results were confirmed using mammary tumor cell line 410.4 pretreated with three different concentrations of SC19220 (1.0, 0.5, 0.1 ug/ml) (Figure 2b). Even though vehicle-treated 410.4 cells are highly metastatic, EP1 antagonism was able to further increase the number of lung tumor colonies. The observation that EP1 antagonism enhances metastatic potential has been replicated in seven independent experiments.
Figure 2.

(A) Line 66.1 cells cultured in SC19220 (1.0 ug/ml) or AH6809 (1.0 ug/ml) for 48 hours, washed and 2 × 105 viable cells injected i.v. into Balb/cByJ mice. On day 18, all mice were euthanized and surface lung tumor colonies were quantified. 10–13 mice/group. (B) Line 410.4 cells cultured in SC19220 (1.0 0.5, 0.1 ug/ml) for 48 hours and 3 × 105 cells injected and tumor colonies quantified on day 22. 8–10 mice/group.
To confirm that blocking EP1 increases metastatic potential, 66.1 cells were engineered with shRNA directed to murine EP1 to stably silence EP1 expression. Line 66.1 cells were transduced with non-silencing shRNA vector (66.1Ve) or two shRNA sequences targeting EP1 (66.1shEP1) and four stable clones were derived with reduced EP1 expression as determined by quantitative RT-PCR (Figure 3a). To determine if changes in EP1 expression affect lung colonizing ability, 66.1Ve or 66.1shEP1 clones were injected i.v. into mice and surface lung tumor colonies were analyzed on day +21. Reduced expression of EP1 was associated with increased capacity to form lung tumor colonies in all four clones (Figure 3b). To determine if EP1 expression levels affect local tumor growth, 66.1Ve or three clones expressing reduced levels of EP1 (66.1shEP1C1, 66.1shEP1C2, 66.1shEP1C3) were implanted proximal to the mammary fat pad of mice and tumor growth monitored. Regardless of the EP1 expression levels, tumor size was not significantly different in any group (Figure 3c). Individual animals were euthanized and lung metastases quantified as each tumor achieved an average diameter of 16 mm or mice appeared moribund. Pulmonary metastases were significantly increased (p<0.002) in mice bearing implants of two of three EP1 silenced clones, (66.1shEP1C2, 66.1shEP1C3) in comparison to mice bearing implants of 66.1Ve (Figure 3d). These data using a spontaneous metastasis model are consistent with the data obtained using pharmacologic EP antagonists or a lung colonization model in which reduced EP1 function resulted in more lung tumor colonies.
Figure 3.


(A) Line 66.1 cells transduced with two shRNA sequences targeting EP1, four stable clones were derived and EP1 levels determined by quantitative RT-PCR. (B) Two × 105 of the indicated cell lines injected i.v. into Balb/cByJ female mice and on day +21, surface lung colonies were enumerated. (C) Three × 105 of 66.1Ve or three EP1 shRNA –expressing clones implanted proximal to the mammary gland of mice (ten mice/group) and tumor growth monitored by caliper. Tumor size reported as the average of the longest diameter and the perpendicular diameter. (D) When tumors achieve an average diameter of 16 mm, mice were euthanized on an individual basis and lung tumor colonies quantified.
We have reported previously that mammary tumor cells also express EP4 but that this receptor plays a very different role in metastasis. Either pharmacologic antagonism or genetic silencing of EP4 leads to reduced, rather than heightened metastatic capacity (9–11). In the current study, we have pooled the data from 3–6 additional independent experiments to further illustrate this point. Pharmacologic antagonism of EP4 with AH23848 inhibited lung colonization of 66.1 cells by an average of 38 ± 4% in three independent experiments. Likewise, gene silencing of EP4 reduced metastatic potential by an average of 60 ± 6% in six separate experiments. Thus, EP1 and EP4 have dramatically different roles in tumor metastasis. EP4 promotes metastasis. Based on the ability of EP1 to inhibit metastasis in the absence of effects on the primary tumor size, we propose that EP1 acts selectively as a metastasis suppressor.
Limited information regarding the expression or function of EP1 in clinical breast cancer is available. The only reported finding is that EP1 is detected in both the cytoplasm and nucleus of primary human breast tumors by immunohistochemistry and that EP1 expression in the nucleus of malignant cells correlates with good prognostic markers of progesterone expression and lymph node negative status (13). We investigated the relationship between EP1 expression levels and survival in 55 invasive ductal carcinomas from women who had participated in the Breast Cancer Prognostic Study, a prospective study of 1,306 women newly diagnosed with breast cancer from 1975 to 1983 carried out at the Karmanos Cancer Institute, Detroit, MI. A tissue microarray was prepared that contained one to three tumor samples from each specimen. The tissue array was stained with antibody to human EP1 or isotype-control antibody. Two blinded investigators evaluated the degree of cytoplasmic and nuclear epithelial EP1 staining on a scale of 0 (no positive cells) to 3+ (>75% positive cells). We saw marked heterogeneity both in the degree of staining and in the subcellular localization. Figure 4A shows representative samples of the different patterns observed. EP1 protein is indicated by brown staining, nuclei are identified by blue counterstain. Figure 4Ai is an invasive ductal carcinoma with no detectable EP1 in either the nucleus or cytoplasm; Figure 4Aii shows benign ducts with prominent brown cytoplasmic as well as brown nuclear EP1 staining obscuring the blue nucleus (panel ii); an IDC displaying primarily nuclear EP1 but very weak cytoplasmic EP1 (panel iii) and an IDC specimen in which no nuclear EP1 was detected, but moderate levels of cytoplasmic EP1 were seen (panel iv). Ten of 55 specimens revealed no EP1 staining in either subcellular compartment; nine patients had tumors in which no nuclear EP1 was detectable and 5 exhibited exclusively nuclear EP1. The remainder had detectable EP1 (1+, 2+ or 3+) in both subcellular compartments.
Figure 4.

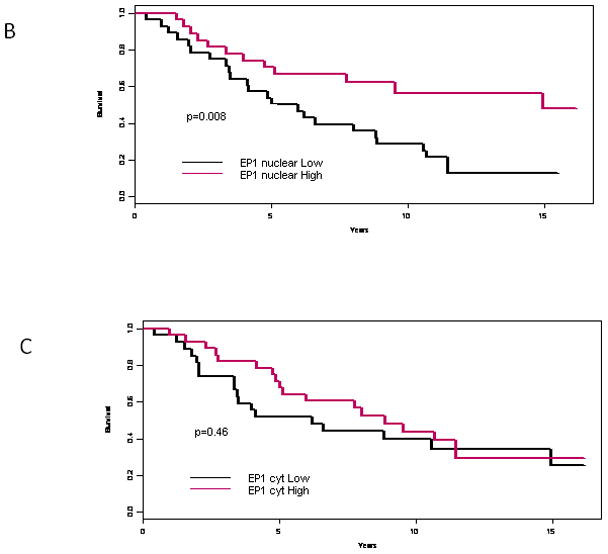
A tissue array of primary breast tumors was examined by immunohistochemstry using antibody specific to EP1 or isotype control. (A) (i) Negative EP1 staining in an invasive ductal carcinoma; (ii) benign ducts exhibiting strong nuclear and cytoplasmic EP1 reactivity; (iii) IDC with strong nuclear staining, but weak cytoplasmic staining; (iv) IDC with moderate cytoplasmic EP1 staining and negligible nuclear EP1. (B) Kaplan-Meier survival analysis for women with no nuclear EP1 detected versus EP1 of 1+, 2+ or 3+. (C) Cytoplasmic EP1 versus survival by Kaplan-Meier analysis.
Based on our prediction that the absence of EP1 would be associated with the worst prognosis, we compared the minimum EP1 intensity for each patient versus overall survival. We considered the expression levels of nuclear or cytoplasmic staining separately. Since 3+ staining was relatively rare, we grouped the intensity values as negative (0) versus positive (1,2 or 3+) staining. Figure 4B shows the impact of nuclear EP1 on overall survival estimated by the Kaplan-Meier approach. Survival for women with EP1-negative tumors is significantly worse than for women with any nuclear EP1 expression (p=0.008). The hazard ratio of death for women with positive EP1 expression is only 40% of those without EP (95% CI:0.20–0.81). In contrast, overall survival for women was not affected by the level of cytoplasmic EP1 (p value=0.46, Figure 4C). Thus, detection of even a minimal level of nuclear EP1 in breast tumors was predictive of significantly better survival. This finding is consistent with a metastasis suppressor role for EP1 revealed in the murine model shown here as well as the clinical study cited earlier in which EP1 expression was associated with lymph node negative status (13). We also observed a possible trend towards low EP1 and node-positive disease, however this relationship was not statistically significant (p=0.25).
Although breast cancer incidence is lower in African-American women in comparison to European-American populations, overall survival in the former group is worse (20,21). Even when variables such as access to medical care are eliminated, differences in clinical outcome persist, suggesting that breast cancer is inherently more aggressive in AA women. To begin to determine the basis for the observation that breast cancers are often more aggressive in AA women, we examined expression of ~8,000 genes in breast tumors from 18 AA and 18 EA women (18). We now report that expression levels of COX-2, EP2, EP3 or EP4 were not detectably different in these two populations. In contrast, EP1 mRNA was detected significantly less frequently (negative value in comparison to reference standard) in AA versus EA tumors (Figure 5). In stage II or III disease, 30% and 25% of AA tumors, respectively, were positive for EP1 mRNA. In contrast, 60% and 88% of EA stage II and III tumors were EP1 positive. This difference in the two populations was significant (p=0.04). Microarray data must be interpreted conservatively, however, these findings hint at one factor that should be investigated further and that could contribute to the overall worse outcome in AA women with breast cancer.
Figure 5.

RNA was isolated from frozen sections of breast tumors from eighteen African-American and 18 European-American women and probed on an ~8,000 gene chip microarray. Gene expression ratios are plotted for individual patients as EP1 detected at levels greater than the gene reference standard (indicated as zero) or below the reference standard. Numbers in bold indicate the number of patients in each subcategory that are positive or negative for EP1 mRNA expression.
DISCUSSION
In tumors, the principal COX-2 product is prostaglandin E2. Cellular effects of PGE2 are mediated through a family of G-protein-coupled receptors designated EP1,2,3 and EP4 (2). Despite structural and sequence similarities among the four EP receptors, they have different binding affinities for PGE2 (Ki of 20, 12, 0.85, 1.9 nM for EP1–4, respectively), and are coupled to different intracellular signaling pathways (3). Because EP1 is uniquely coupled to a distinct signaling pathway relative to other EPs, it is not surprising that this receptor functions differently in cancer as well.
The precise role of each EP in malignancy is emerging. In colon carcinogenesis, all four EP receptors have been implicated (5–8). EP1 antagonism prevents chemically-induced breast carcinogenesis in a murine model (22). Transgenic mice expressing COX-2 in the mammary gland develop mammary tumors, however, this effect was abrogated in EP2−/− mice implicating the EP2 receptor in mammary carcinogenesis (23). The aromatase enzyme, which catalyzes estrogen biosynthesis, is positively regulated by PGE2; EP1 and EP2 (expressed on stromal cells) are implicated in some studies (24) whereas EP2 and EP4 are key to the induction of the aromatase promoter in other cells (25). The EP1 receptor is implicated in UVB-induced inflammation and skin tumorigenesis (26) but other skin cancer models support a role for EP2 (27). In contrast to the apparent tumor-promoting role of EP1 in several models of primary tumorigenesis, we saw no effect of EP1 silencing on expansion of transplanted mammary tumors. There are many possible explanations for these differences. We directly silenced EP1 on the malignant epithelial cells, but it may be that EP1 expressed on other cells is the critical target in primary skin or mammary gland carcinogenesis. It is also possible that the role for EP1 is very different in early stage tumorigenesis versus tumor expansion or in very late stage metastasis.
In addition to affecting early events in tumor formation, we and others have shown that EP receptors also play important roles in late stage breast tumor progression. Breast and colon tumor cells migrate in response to PGE2 and this response is mediated through EP4(28,29). Using pharmacologic antagonists or genetic inhibition of EP4, we showed that EP4 promotes metastasis whereas EP3 antagonists do not affect tumor dissemination (9,11). The protective effect of EP4 blockade was dependent on functioning Natural Killer cells in the host. Other laboratories have also shown that EP4 silencing inhibits metastasis in murine models of lung and colon cancer (12). PGE2 induces angiogenesis in a model of prostate cancer through the EP4 and EP2 receptors (30). Cell lines derived from inflammatory breast cancer are stimulated to proliferate and invade via EP4 activation (31).
While a consensus is developing that EP4 supports tumor growth and progression, very little is known regarding a function for EP1. In the current study, we asked whether EP1 had the same prometastatic role as EP4. Using both pharmacologic and genetic approaches, we now show that inhibition of EP1 signaling or expression increases rather than decreases metastatic capacity. We were the first to suggest a role for EP receptors in metastasis and had reported some years ago that the EP1 antagonist SC19220 promoted metastasis (14), however, it was not then known that more than one EP receptor exists or that SC19220 would selectively antagonize EP1. To our knowledge, no other studies have examined a role for EP1 in tumor metastasis. Both SC19220 and AH6809 are widely used to examine the physiology of EP receptors but conclusions regarding the role of individual EP receptors cannot be based on pharmacologic inhibitors alone. While SC19220 is relatively selective for EP1 on human tissues, AH6809 inhibits both EP1 and EP2 and it is not clear that the receptor specificity of these antagonists is the same in murine tissues (32). Despite the limitations of pharmacologic antagonists, it is quite clear from our previously published studies using two selective EP4 antagonists as well as EP4 gene silencing, that EP1 and EP4 play very different roles in tumor biology. Since our gene silencing data are consistent with the results obtained using pharmacologic approaches, we think this data supports a role for EP1 as a negative regulator of metastatic potential. Because changes in EP1 expression or function did not affect the expansion of the primary tumor, we propose the novel hypothesis that EP1 functions as a suppressor of metastasis.
It is clear that the absolute levels of EP1 alone do not determine function. Similar amounts of EP1 protein were detected in whole cell lysates of murine tumor cell lines regardless of metastatic capacity. The subcellular location also is likely to determine function. A recent study compared EP1 expression by immunohistochemistry in normal human breast tissue and primary breast cancers (13). EP1 was detected in both the cytoplasm and nuclear membrane in malignant cells. Nuclear EP1 expression was inversely correlated with lymph node involvement and positively correlated with progesterone receptors. Thus, nuclear expression of EP1 was correlated with better prognostic markers. Like that study, we observed cytoplasmic and nuclear EP1 in primary breast cancer. We have extended those findings to now show that the absence of nuclear EP1 is associated with significantly worse overall survival. These data strongly support a protective role for EP1. EP1 has also been detected in non-melanoma skin cancers (17,33). Interestingly, well-differentiated squamous cell carcinomas have more detectable EP1 than less differentiated lesions (17), consistent with a protective role for EP1.
G-protein-coupled receptors (GPCRs), including EP receptors, belong to a family of receptors comprised of 7 transmembrane spanning domains. There is now considerable evidence derived from immunofluorescence, electron microscopy and radio-ligand binding studies, that perinuclear or nuclear GPCRs, including EP1 and EP3, are functional and these functions may be distinct from the cell surface receptor function (34–36). For example, plasma membrane-expressed EP3 functions in signal transduction, whereas nuclear-EP3 is more directly associated with transcription of a distinct set of genes. Most importantly, our data and that of Thorat el al (13) report that nuclear expression of EP1, but not cytoplasmic EP1, is associated with lymph node-negative disease and better long-term survival in breast cancer, further supporting a distinct role for nuclear-expressed EP1. Functional EP1 has been detected in the nucleus of cholangiocarcinoma cells and is associated with stat3 and EGFR signaling (19) and nuclear calcium signaling (36). Thus, it is intriguing that nuclear EP1 is most closely associated with differences in survival. We have also detected EP1 on normal, immortalized murine and human mammary epithelial cells so it is not a property unique to malignant cells.
As part of an initial effort to understand possible biologic bases for differences in outcome for African-American versus European-American women with breast cancer, we had analyzed the expression of approximately 8,000 genes by gene chip (18). In the current study, we show that the majority of tumors from EA women (72%) have detectable levels of EP1 mRNA, whereas only 28% of AA women had detectable EP1 mRNA. While gene array data must be interpreted with caution, this marked difference suggests that EP1 should be examined further as a possible contributor to differences in outcome in these two populations. A larger study, including more annotated specimens from AA women will be required to explore this relationship further.
We do not know the molecular mechanism by which EP1 could suppress metastasis. In skin, EP1 supports differentiation (17) and a similar function in breast could result in fewer metastases. Both direct effects on EP1-mediated downstream effectors as well as indirect effects on other EP receptors are possible. One proposed mechanism is by shifting PGE2 binding to EP2 and EP4 (37). EP4 promotes metastasis and it is possible that antagonism of EP1 could enhance EP4-mediated signaling events leading to the observed increased metastatic potential. It is also possible that, due to the lower binding affinity of EP1 for PGE2, that this receptor is rarely activated in the tumor milieu and that the potentially protective effects of EP1 on metastasis are not enabled. Teasing out the role of each EP in the context of multiple EPs will be challenging. Future studies will examine the molecular function of cytoplasmic versus nuclear EP1 in breast cancer cells in order to determine the mechanism by which EP1 suppresses metastasis.
References
- 1.Ristimaki A, Sivula A, Lundin M, Salminen T, Haglund C, Joensuu H, Isola J. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002;62:632–635. [PubMed] [Google Scholar]
- 2.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures properties and functions. Physiological Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 3.Fujino H, Regan JW. Prostanoid receptors and phosphatidylinositol 3-kinase: a pathway to cancer? Trends in Pharmacol Sci. 2003;24:335–340. doi: 10.1016/S0165-6147(03)00162-7. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe T, Satoh H, Togoh M, Taniguchi S, Hashimoto Y, Kurokawa K. Positive and negative regulation of cell proliferation through prostaglandin receptors in NIH-3T3 cells. J Cell Physiol. 1996;69:401–409. doi: 10.1002/(SICI)1097-4652(199611)169:2<401::AID-JCP20>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 5.Mutoh M, Watanabe K, Kitamura T, Shoji Y, Takahashi M, Kawamori T, Tani K, Kobayashi M, Maruyama T, Kobayashi K, Ohuchida S, Sugimoto Y, Narumiya S, Sugimura T, Wakabayashi K. Involvement of prostaglandin E receptor subtype EP4 in colon carcinogenesis. Cancer Res. 2002;62:28–32. [PubMed] [Google Scholar]
- 6.Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M, Taketo MM. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med. 2001;9:1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe K, Kawamori T, Nakatsugi S, Ohta T, Ohuchida S, Yamamoto H, Maruyama T, Kondo K, Ushikubi F, Narumiya S, Sugimura T, Wakabayashi K. Role of the prostaglandin E receptor subtype EP1 in colon carcinogenesis. Cancer Res. 1999;59:5093–5096. [PubMed] [Google Scholar]
- 8.Kitamura T, Itoh M, Noda T, Tani K, Kobayashi M, Maruyama T, Kobayashi K, Ohuchida S, Sugimura T, Wakabayashi K. Combined effects of prostaglandin E receptor subtype EP1 and subtype EP4 antagonists on intestinal tumorigenesis in adenomatous polyposis coli gene knockout mice. Cancer Sci. 2003;94:618–621. doi: 10.1111/j.1349-7006.2003.tb01492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma X, Kundu N, Rifat S, Walser T, Fulton AM. Prostaglandin E receptor EP4 antagonism inhibits breast cancer metastasis. Cancer Res. 2006;66:2923–2927. doi: 10.1158/0008-5472.CAN-05-4348. [DOI] [PubMed] [Google Scholar]
- 10.Fulton AM, Ma X, Kundu N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res. 2006;66:9794–7. doi: 10.1158/0008-5472.CAN-06-2067. [DOI] [PubMed] [Google Scholar]
- 11.Kundu N, Ma X, Holt D, Goloubeva O, Ostrand-Rosenberg S, Fulton AM. Antagonism of the prostaglandin E receptor EP4 inhibits metastasis and enhances NK function. Breast Cancer Res Treatment. 2009;117:235–42. doi: 10.1007/s10549-008-0180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang L, Huang Y, Porta R, Yanagisawa K, Gonzalex A, Segi E, Johnson DH, Narumiya S, Carbone DH. Host and direct antitumor effects and profound reduction in tumor metastasis with selective EP4 receptor antagonism. Cancer Res. 2006;66:9665–72. doi: 10.1158/0008-5472.CAN-06-1271. [DOI] [PubMed] [Google Scholar]
- 13.Thorat MA, Morimiya A, Mehrotra S, Konger R, Badve S. Prostanoid receptor EP1 expression in breast cancer. Mod Pathol. 2008;21:15–21. doi: 10.1038/modpathol.3800970. [DOI] [PubMed] [Google Scholar]
- 14.Fulton AM, Zhang S-Z, Chong YC. Role of the prostaglandin E receptor in mammary tumor metastasis. Cancer Res. 1991;51:2047–2050. [PubMed] [Google Scholar]
- 15.Miller FR, Miller BE, Heppner GH. Characterization of metastatic heterogeneity among subpopulations of a single mouse mammary tumor: heterogeneity in phenotypic stability. Invasion Metastasis. 1983;3:22–31. [PubMed] [Google Scholar]
- 16.Ma X, Norsworthy K, Kundu N, Rodgers WH, Gimotty PA, Goloubeva O, Lipsky M, Li Y, Holt D, Fulton AM. CXCR3 expression is associated with poor survival in breast cancer and promotes metastasis in a murine model. Mol Cancer Ther. 2009;8:490–8. doi: 10.1158/1535-7163.MCT-08-0485. [DOI] [PubMed] [Google Scholar]
- 17.Konger RL, Billings SD, Prall NC, Katona TM, DaSilva SC, Kennedy CRJ, Badve S, Perkins SM, LaCelle PT. The EP1 subtype of prostaglandin E2 receptor: role in keratinocyte differentiation and expression in non-melanoma skin cancer. Prostagl Leukotriene and Essential Fatty Acids. 2009;81:279–90. doi: 10.1016/j.plefa.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selaru FM, Yin J, Olaru A, Mori Y, Xu Y, Epstein S, Sato F, Deacu E, Wang S, Sterian A, Fulton AM, Abraham JM, Shibata D, Baquet C, Stass SA, Meltzer SJ. An unsupervised approach to identify molecular phenotypic components influencing breast cancer features. Cancer Res. 2004;64:1584–1588. doi: 10.1158/0008-5472.can-03-3208. [DOI] [PubMed] [Google Scholar]
- 19.Han C, Demetris AJ, Stolz DB, Xu L, Lim K, Wu T. Modulation of Stat3 activation by the cytosolic phospholipase A2α and cyclooxygenase-2-controlled prostaglandin E2 signaling pathway. J Biol Chem. 2006;281:24831–24846. doi: 10.1074/jbc.M602201200. [DOI] [PubMed] [Google Scholar]
- 20.Simon MS, Severson RK. Racial differences in breast cancer survival: the interaction of socioeconomic status and tumor biology. Am J Obstet Gynecol. 1997;176:S233–239. doi: 10.1016/s0002-9378(97)70381-8. [DOI] [PubMed] [Google Scholar]
- 21.Smigal C, Jemal A, Ward E, Cokkinides V, Smith R, Howe HL, Thun M. Trends in breast cancer by race and ethnicity: update 2006. Ca Cancer J Clin. 2006;56:168–183. doi: 10.3322/canjclin.56.3.168. [DOI] [PubMed] [Google Scholar]
- 22.Kawamori T, Uchiya N, Nakatsugi S, Watanabe K, Ohuchida S, Yamamoto H, Maruyama T, Kondo K, Sugimura T, Wakabayashi K. Chemopreventive effects of ONO-8711, a selective prostaglandin E receptor EP1 antagonist, on breast cancer development. Carcinogenesis. 2001;22:2001–4. doi: 10.1093/carcin/22.12.2001. [DOI] [PubMed] [Google Scholar]
- 23.Chang S-H, Liu CH, Conway R, Han DK, Nithipatikom K, Trifan OC, Lane TF, Hla T. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase-2-induced breast cancer progression. Proc Natl Acad Sci. 2004;101:591–596. doi: 10.1073/pnas.2535911100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards JA, Brueggemeier RW. Prostaglandin E2 regulates aromatase activity and expression in human adipose stromal cells via two distinct receptor subtypes. J Clin Endocrinol Metab. 2003;88:2810–2816. doi: 10.1210/jc.2002-021475. [DOI] [PubMed] [Google Scholar]
- 25.Subbaramaiah K, Hudis C, Chang SH, Hla T, Dannenberg AJ. EP2 and EP4 receptors regulate aromatase expression in human adipocytes and breast cancer cells. Evidence of a BRCA1 and p300 exchange. J Biol Chem. 2008;283:3433–44. doi: 10.1074/jbc.M705409200. [DOI] [PubMed] [Google Scholar]
- 26.Tober KL, Wilgus TA, Kusewitt DF, Thomas-Ahner JM, Maruyama T, Oberyszyn TM. Importance of the EP1 receptor in cutaneous UVB-induced inflammation and tumor development. J Inv Derm. 2006;126:205–11. doi: 10.1038/sj.jid.5700014. [DOI] [PubMed] [Google Scholar]
- 27.Sung YM, He G, Fischer SM. Lack of expression of the EP2 but not the EP3 receptor for prostaglandin E2 results in suppression of skin tumor development. Cancer Res. 2005;65:9304–11. doi: 10.1158/0008-5472.CAN-05-1015. [DOI] [PubMed] [Google Scholar]
- 28.Timoshenko AV, Xu G, Chakrabarti S, Lala PK, Chakraborty C. Role of prostaglandin E2 receptors in migration of murine and human breast cancer cells. Expt Cell Res. 2003;289:265–274. doi: 10.1016/s0014-4827(03)00269-6. [DOI] [PubMed] [Google Scholar]
- 29.Buchanan FG, Wang D, Bargiacchi F, Dubois RN, Prostaglandin E. 2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem. 2003;278:35451–35457. doi: 10.1074/jbc.M302474200. [DOI] [PubMed] [Google Scholar]
- 30.Jain S, Chakraborty G, Raja R, Kale S, Kundu GC. Prostaglandin E2 regulates tumor angiogenesis in prostate cancer. Cancer Res. 2008;68:7750–9. doi: 10.1158/0008-5472.CAN-07-6689. [DOI] [PubMed] [Google Scholar]
- 31.Robertson FM, Simeone A-M, Mazumdar A, Shah AH, McMurray JS, Ghosh S, Cristofanilli M. Molecular and pharmacological blockade of the EP4 receptor selectively inhibits both proliferation and invasion of human inflammatory breast cancer cells. J Exp Ther Oncol. 2008;7:299–312. [PubMed] [Google Scholar]
- 32.Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Brit J Pharmacol. 1997;122:217–224. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JL, Kim A, Kopelovich L, Bickers DR, Athar M. Differential expression of E prostanoid receptors in murine and human non-melanoma skin cancer. J Invest Dermatol. 2005;125:818–825. doi: 10.1111/j.0022-202X.2005.23829.x. [DOI] [PubMed] [Google Scholar]
- 34.Gobeil F, Fortier A, Zhu T, Bossolasco M, Leduc M, Grandbois M, Heveker N, Bkaily G, Chemtob S, Barbaz D. G-protein-coupled receptors signaling at the cell nucleus: an emerging paradigm. Can J Physiol Pharmacol. 2006;84:287–297. doi: 10.1139/y05-127. [DOI] [PubMed] [Google Scholar]
- 35.Zhu T, Gobeil F, Vazquez-Tello A, Leduc M, Rihakova L, Bossolasco M, Bkaily G, Peri K, Varma DR, Orvoine R, Chemtob S. Intracrine signaling through lipid mediators and their cognate nuclear G-protein-coupled receptors: a paradigm based on PGE2, PAF and LPA1 receptors. Can J Physiol Pharmacol. 2006;84:377–391. doi: 10.1139/y05-147. [DOI] [PubMed] [Google Scholar]
- 36.Bhattacharya M, Peri KG, Almazan G, Ribeiro-da-Silva A, Shichi H, Durocher Y, Abramovitz M, Hou X, Varma DR, Chemtob S. Nuclear localization of prostaglandin E2 receptors. Proc Natl Acad Sci USA. 1998;95:15792–7. doi: 10.1073/pnas.95.26.15792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dore S. GPCR antagonists as an alternative to COX-2 inhibitors: a case for the PGE2 EP1 receptor. TRENDS in Pharmacol Sci. 2006;27:458–60. doi: 10.1016/j.tips.2006.07.001. [DOI] [PubMed] [Google Scholar]