

Abstract
AP1 (jun/fos) factors comprise a family of transcriptional regulators (c-jun, junB, junD, c-fos, FosB, Fra-1 and Fra-2) that are key controllers of epidermal keratinocyte survival and differentiation, and are important drivers of cancer development. Understanding the role of these factors in epidermis is complicated by the fact that each member is expressed in defined cell layers during epidermal differentiation, and because AP1 factors regulate competing processes (i.e., proliferation, apoptosis and differentiation). We have proposed that AP1 factors function differently in basal versus suprabasal epidermis. To test this, we inactivated suprabasal AP1 factor function in mouse epidermis by targeted expression of dominant-negative c-jun (TAM67) which inactivates function of all AP1 factors. This produces increased basal keratinocyte proliferation, delayed differentiation, and extensive hyperkeratosis. These findings contrast with previous studies showing that basal layer AP1 factor inactivation does not perturb resting epidermis. It is interesting that in spite of extensive keratinocyte hyperproliferation, susceptibility to carcinogen-dependent tumor induction is markedly attenuated. These novel observations strongly suggest that AP1 factors have distinct roles in the basal versus suprabasal epidermis, confirm that AP1 factor function is required for normal terminal differentiation, and suggest that AP1 factors play a different role in normal epidermis versus in cancer progression.
Keywords: TAM67, c-jun, keratinocyte differentiation, epidermis, skin cancer
Introduction
The activator protein one (AP1) transcription factors, which include jun (c-jun, junB, junD) and fos (c-fos, FosB, Fra-1, Fra-2), are important modulators of cell function that control diverse biological processes (Angel et al., 2001; Karin et al., 1997; Shaulian and Karin 2002; Shaulian and Karin 2001). They form jun-jun and jun-fos homo- and heterodimers that subsequently interact with specific AP1 factor consensus DNA binding sites to regulate gene expression. AP1 factor function in skin has been intensively studied, and it is difficult to imagine a more important set of regulatory proteins in epidermis. AP1 factors are implicated in control of keratinocyte proliferation (Mizuno et al., 2006; Shi and Isseroff 2005; Takahashi et al., 2002), differentiation (Adhikary et al., 2004; Eckert et al., 2003; Eckert et al., 2004), apoptosis (Efimova et al., 2004; Raj et al., 2006), and transformation (Saez et al., 1995; Kahn et al., 1993; She et al., 2002; Rutberg et al., 2000). Because multiple AP1 family members are expressed in epidermis and because specific AP1 proteins may display context-dependent function in different epidermal cell layers, investigators have searched for appropriate model systems to understand their role. Several laboratories have used in vivo systems (Florin et al., 2006; Saez et al., 1995; Zenz et al., 2005; Passegue et al., 2002; Schreiber et al., 2000; Fleischmann et al., 2000; Zenz et al., 2005). For example, JunB knockout mice display delayed wound healing (Florin et al., 2006), challenge of v-H-ras positive mice with DMBA/TPA results in malignant tumor formation which is attenuated in the absence of c-fos (Saez et al., 1995), and junB/c-jun double knockout results in a psoriasis phenotype (Zenz et al., 2005). Mouse studies also suggest that AP1 factors can compliment the function of other AP1 factors. For example Fra-1 can substitute for c-fos (Fleischmann et al., 2000) and JunB can substitute for c-jun (Passegue et al., 2002). Taken together, these studies suggest an essential role of AP1 factors in epidermis. However, knockout models are not useful in assessing function in specific layers, since these approaches generally inactivate in all layers.
We have hypothesized that AP1 factors perform different functions in the basal (proliferating) versus suprabasal (differentiating) epidermis (Eckert et al., 2004). However, testing this hypothesis is complicated by the fact that virtually all of the AP1 family members are expressed, at some level, in both the basal and suprabasal compartments (Welter and Eckert 1995; Angel et al., 2001; Mehic et al., 2005). In an effort to understand the role of AP1 factors in epidermal carcinogenesis, Colburn and colleagues have utilized the K14 promoter to target TAM67 expression to the keratinocyte basal layer (Brown et al., 1994). TAM67 is a dominant-negative form of c-jun that interacts broadly with all AP1 factors to inhibit transactivation (Brown et al., 1994). In these interesting studies, basal layer AP1 factor inactivation did not produce obvious changes in keratinocyte proliferation or epidermal or dermal appearance; however, the mice were less susceptible to cancer progression (Young et al., 1999; Thompson et al., 2002; Cooper et al., 2003).
An important goal of the present study was to assess the role of AP1 factors in the suprabasal epidermis. To achieve this, we generated mice in which TAM67 is expressed in the suprabasal epidermal layers. We utilize an inducible system that makes it possible to reversibly activate TAM67 expression in the suprabasal epidermis by administration of doxycycline in the drinking water. Our experiments indicate that suprabasal TAM67 expression produces a thickened, hyperproliferative, parakeratotic epidermis, but that in spite of this extensive hyperproliferation, the epidermis is resistant to DMBA/TPA-dependent tumor formation.
Results
Impact of suprabasal AP1 factor inactivation on epidermal morphology
To assess the impact of TAM67 expression on epidermal function, TG/-TAM67 (TAM67) and TG/-TAM67-TG/-rTA (TAM67-rTA) mice were treated for twelve days in the absence or presence of doxycycline (Dox) supplemented drinking water. Dox treatment of TAM67-rTA mice results in an approximate 3-fold increase in epidermal thickness (19 ± 5 to 57 ± 8 μm, mean ± SEM, n = 10 animals/group) (Fig. 1A). Moreover, this effect requires the presence of TAM67 and rTA, since no phenotype is produced in TAM67 mice following Dox treatment. As shown in Fig. 1B, immunoassay reveals that TAM67-FLAG expression is observed in epidermis of Dox-treated TAM67-rTA mice and is associated with the epidermal phenotype. Our goal was to selectively inactivate AP1 factor function in the suprabasal epidermis. This requires that TAM67 be expressed exclusively in the upper epidermal layers. To confirm TAM67-FLAG suprabasal expression, sections derived from Dox-treated bi-transgenic mice were incubated with peroxidase-conjugated or fluorescene-tagged FLAG-specific antibody. Fig. 1C shows that both methods detect TAM67-FLAG in the suprabasal epidermal layers and, as expected of a nuclear transcription factor, the signal is detected in the nucleus. No TAM67-FLAG expression is detected in sections derived from TAM67-rTA mice that were not treated with Dox.
Fig. 1.

Suprabasal TAM67 expression produces epidermal hyperproliferation and hyperkeratosis. A TAM67 expression alters epidermal phenotype. TAM67 and TAM67-rTA (bi-transgenic) mice were treated with drinking water (-Dox) or drinking water supplemented with 2 mg/ml doxycycline (+Dox) for 12 d. The dorsal epidermis was harvested on day twelve and sections were prepared and stained with H&E. The epidermis is indicated (Epi). B TAM67-FLAG is expressed in Dox-treated TAM67-rTA mice. Total cell extracts, prepared from dorsal epidermis, were electrophoresed and immunoblotted with HRP-conjugated mouse monoclonal anti-FLAG M2 (Sigma, A8592) and signal was detected using chemiluminescence detection reagents. Migration of the TAM67-FLAG fusion protein is indicated by an arrow. Gel loading was normalized based on β-actin level (not shown). C TAM67-FLAG is detected in the nucleus of suprabasal keratinocytes. TAM67-rTA mice were treated with or without doxycycline and frozen sections were incubated with mouse monoclonal anti-FLAG (Sigma, F1804) followed by HRP-conjugated sheep anti-mouse IgG (Amersham, NA931). The sections were not counterstained. As a second method of detection, paraffin-embedded sections were stained with FITC-conjugated anti-FLAG (Sigma, F1804). These are 1 μm confocal optical sections. The epidermis (Epi) and dermis are indicated and separated by a dotted line. The arrows identify nuclear TAM67-FLAG staining.
Suprabasal TAM67 expression increases basal layer keratinocyte proliferation
The increase in epidermal thickness could be due to increased survival of cells in the suprabasal layers, increased adhesiveness of cells in the stratified layers or increased proliferation of basal cells. We first tested whether suprabasal AP1 factor inactivation influences basal keratinocyte proliferation. To test this we monitored BrdU incorporation and Ki67 level. TAM67-expressing mice were treated with BrdU for 2 h and tissue sections were prepared for detection of BrdU and Ki67. As shown in Fig. 2A, the number of BrdU-positive and Ki67-positive nuclei is five-fold higher in TAM67-positive epidermis (+Dox), suggesting that increased basal and suprabasal layer proliferation contributes to the phenotype.
Fig. 2.

Differentiation and proliferation in suprabasal TAM67 epidermis. A Suprabasal TAM67 expression increases keratinocyte proliferation. TAM67-rTA mice were treated with or without doxycycline for 12 d. The mice were shaved on day ten, injected with 100 μg BrdU per gram body weight on day twelve, and euthanized 2 h later. Paraffin-embedded sections were prepared for detection of BrdU (peroxidase staining) and Ki67 (Fluorescence staining). Arrows indicate BrdU- or Ki67-positive nuclei. The number of BrdU-positive and Ki67-positive nuclei were counted and expressed per unit length of epidermis in five random sections derived each of three mice per treatment group. This analysis indicated five times more BrdU-positive cells per unit length of epidermis in Dox treated versus not-treated TAM67-rTA mice. B Suprabasal TAM67 expression delays keratinocyte differentiation. Epidermis from TAM67-rTA mice, treated for 12 d with or without doxycycline, was embedded in paraffin and sections were stained to detect the indicated epitopes. These are 1 μm confocal optical sections. C TAM67 impact on marker protein level. Two TAM67-rTA littermates were treated with (+Dox) and two without (-Dox) doxycycline for 12 d and epidermal extracts were prepared and electrophoresed for detection of the indicated proteins.
To further assess the impact of suprabasal TAM67 expression on cell proliferation and differentiation, we monitored expression of epidermal differentiation markers. Keratin K1, involucrin, filaggrin and loricrin are typically expressed in differentiated suprabasal keratinocytes (Eckert et al., 1997). In TAM67-positive epidermis, K1, involucrin and filaggrin exhibit normal suprabasal expression, suggesting that keratinocytes continue to undergo differentiation (Fig. 2B). However, loricrin, which is normally present as a discrete band in the uppermost epidermis, is detected diffusely in multiple suprabasal layers in TAM67-positive epidermis (Fig. 2B). In addition, absence of suprabasal AP1 factor function is associated with reduced involucrin and loricrin level (Fig. 2C); suggesting that suprabasal AP1 factor function is necessary to maintain expression of these genes.
Keratins K5 and K14 are typically expressed in basal keratinocytes; however, in TAM67-positive mice expression is observed in both basal and suprabasal layers. The presence of these proteins in the suprabasal epidermis is consistent with the idea that the cells are delayed in differentiation. Moreover, we detect high level expression of keratin K6, a keratin that is not present in normal epidermis, but is detected in hyperproliferative epidermis (Sun et al., 1983).
Suprabasal TAM67 expression blocks tumor formation
The above results suggest that reducing suprabasal epidermis AP1 factor function increases proliferation and delays differentiation. Because of this phenotype, we predicted that these animals should be more susceptible to carcinogenesis. To test this hypothesis, mice were shaved and then treated with a single topical application of DMBA followed by twice weekly treatment with TPA for 22 weeks. DMBA treatment alone did not cause tumor production in any mice. DMBA/TPA treatment, in contrast, did result in tumor formation. In both the TAM67-positive and negative groups, tumor formation was observed at week seven. Thus, reducing suprabasal AP1 factor function does not delay the onset of tumor formation. However, the number of tumors per mouse is markedly altered by TAM67 expression. Mice positive for suprabasal TAM67 expression produced an average of 1-2 tumors per mouse at 22 weeks. In contrast, TAM67-negative mice produced 12 tumors per mouse. In addition, at 22 weeks of treatment the average tumor size was 9.5 ± 0.5 mm3 in TAM67-positivie epidermis as compared to 2.1 ± 0.1 mm3 in TAM67-negative epidermis (Fig. 3B). All of the tumors were benign papillomas (Fig. 3C) and stained positively for TAM67-FLAG indicating that reduced tumor formation is associated with TAM67 expression. These findings clearly show that tumor formation is inhibited when AP1 factor function is reduced in the suprabasal epidermis.
Fig. 3.
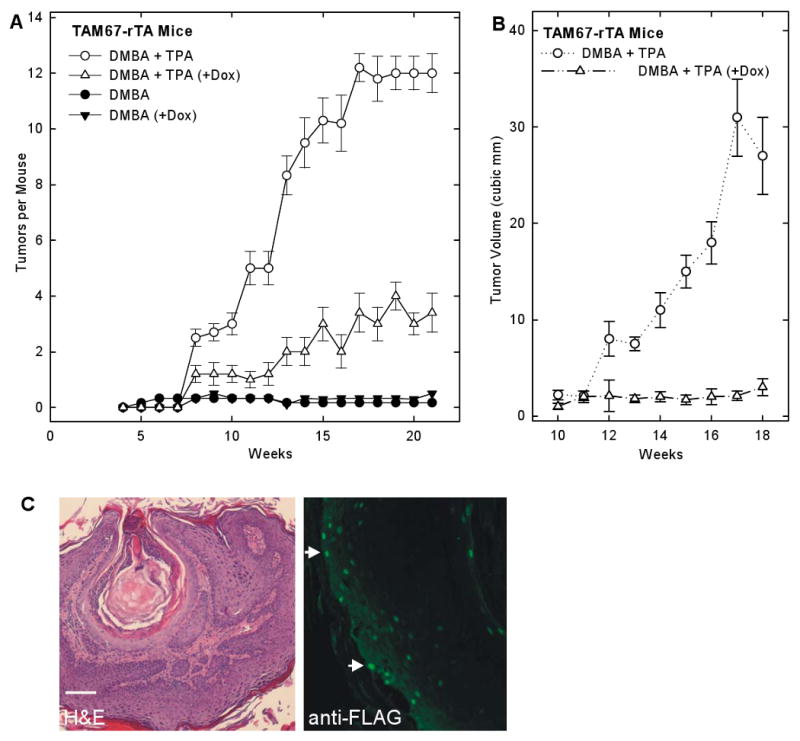
Tumor formation in suprabasal TAM67 expressing epidermis. A Reduced tumor formation in mice expressing suprabasal TAM67. TAM67-rTA mice were treated with or without a single topical application of 100 μg DMBA followed 1 week later by treatment with or without doxycycline and TPA for 22 weeks as outlined in Materials and Methods. Mice were observed weekly for tumor onset, number and size. Each treatment group included ten animals. The values are mean ± SEM. At 10 - 21 weeks, there were significantly more tumors in the DMBA + TPA group as compared to the DMBA + TPA (+Dox) group, p < 0.5 as compared using the t-test. B Reduced tumor size in suprabasal TAM67 mice. Tumor size was monitored using a caliper and the formula tumor volume = (length × Width2)/2. Only the DMBA/TPA treated (+/- Dox) groups are shown. Each treatment group includes ten animals and the values are mean ± SEM. Tumor size at 13 - 18 weeks was significantly greater in the DMBA/TPA group as compared to the DMBA + TPA (+Dox) group, p < 0.5 as compared using the t-test. C DMBA/TPA treatment produces benign papillomas. Tumors, derived from DMBA/TPA-treated TAM67-rTA mice, were collected and sectioned for H&E staining and immunofluorescence detection of TAM67-FLAG using anti-FLAG. The fluorescence picture shows a confocal 1 μm optical section. TAM67-FLAG is localized in tumor cell nuclei. Optical scanning of the tissue revealed that most tumor cells are TAM67-FLAG positive. The arrows indicate nuclear staining.
TAM67 versus TPA - a similar phenotype
Topical TPA treatment is a classical method of inducing epidermal hyperproliferation and is associated with suprabasal expression of keratins K5 and K6 (Toftgard et al., 1985; Molloy and Laskin 1987). An attenuated response to TPA could explain the lack of tumor induction in the TAM67 expressing epidermis in the DMBA-TPA carcinogenesis protocol. We therefore compared the pattern of K5 and K6 expression in TAM67-expressing mice with that observed following acute TPA-treatment of TAM67-negative mice. Fig. 4A shows that acute TPA treatment results in suprabasal K5 and K6 expression in a pattern that is similar to that observed in TAM67-expressing epidermis. This indicates that suprabasal TAM67 expression produces a hyperproliferative keratin expression profile similar to that observed with TPA treatment. We next assessed the impact of TPA treatment on cell proliferation as monitored by presence of Ki67 cell proliferation marker. Suprabasal TAM67 expression is associated with a five-fold increase in the number of Ki67 positive cells (Fig. 2A). Most of these cells are observed in the basal layer. As shown in Fig. 4B, this number is substantially increased when TAM67-positive epidermis is treated acutely with TPA. The number of basal layer Ki67 positive cells is increased and there are a substantially greater number of suprabasal Ki67-positive cells. Thus, TAM67-positive mice are extremely responsive to acute treatment with TPA.
Fig. 4.
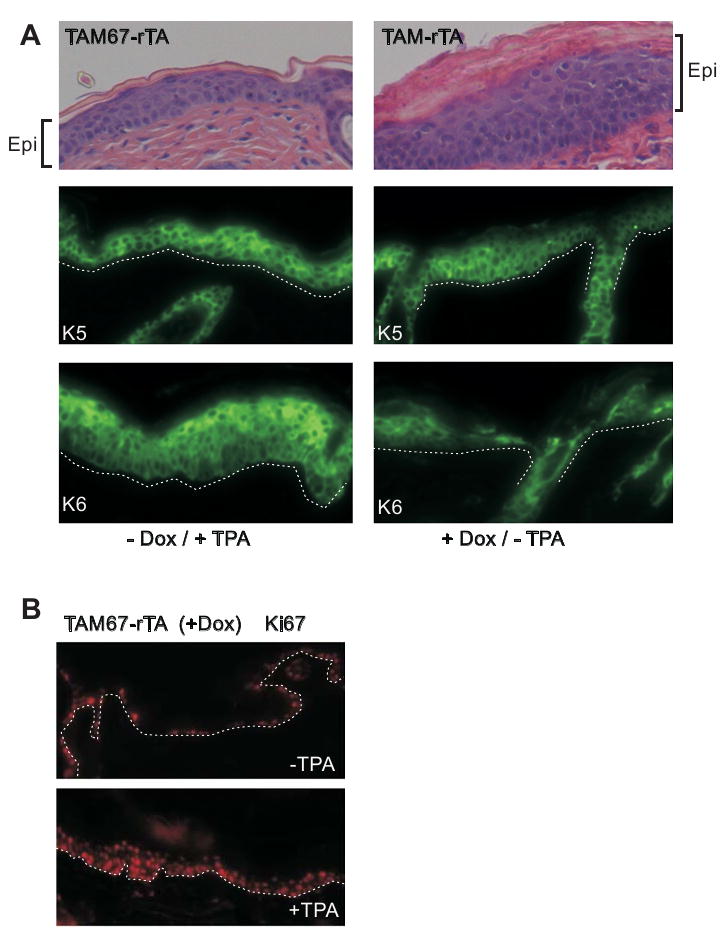
Response to acute TPA treatment. A Suprabasal TAM67 positive epidermis resembles TPA-treated skin. TAM67-rTA mice were maintained on drinking water with or without doxycycline for 12 d followed by topical application of TPA as indicated. After 24 h of TPA treatment the tissue was harvested and sectioned for H&E staining or incubation with the indicated antibody. The fluorescent pictures are confocal 1 μm optical sections. B Acute TPA treatment increases proliferation in TAM67-positive epidermis. TAM67-rTA mice were maintained on dietary doxycycline for 12 d. The epidermis was then challenged with 10 μg TPA/100 μl acetone and tissue was harvested 24 h later for staining to Ki67.
We next examined whether the hyperproliferation observed in epidermis is also detected in cornea. As shown in Fig. 5A, TAM67-FLAG is expressed at comparable levels in both the epidermis and corneal epithelium. However, in contrast to the epidermis, TAM67 expression in the corneal epithelium does not provoke hyperproliferation (Fig. 5B)., suggesting that TAM67-dependent hyperproliferation is only observed in selected epithelia.
Fig. 5.
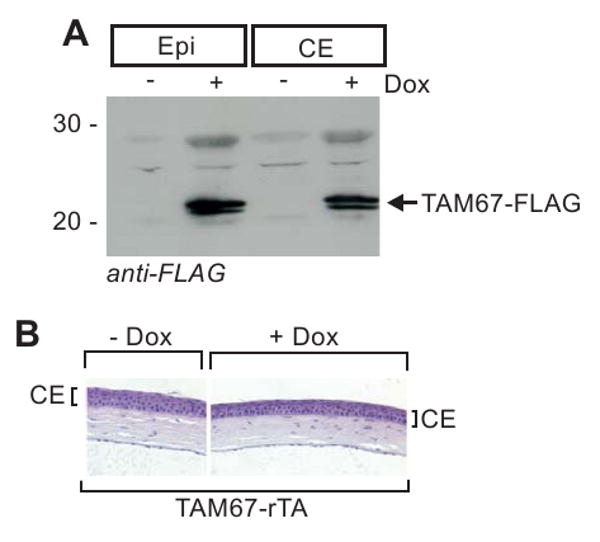
Lack of response in corneal epithelium. A/B High level expression of TAM67-FLAG in mouse corneal epithelium. TAM67-rTA mice were treated with or without doxycycline for 12 d and the epidermal and corneal epithelium were harvested for assay of TAM67-FLAG level and to prepare histological sections. The immunoblot results are normalized based on detection of β-actin (not shown).
Discussion
AP1 factors - a key role in normal epidermis
The epidermal barrier protects against infection, abrasion and loss of critical body fluids by balancing cell proliferation, differentiation and apoptosis. This is achieved by assembly of a multilayered tissue that includes proliferating, viable/non-proliferating and cornified layers. Proliferative basal undifferentiated keratinocytes (Dunnwald et al., 2001; Bickenbach and Dunnwald 2000) give rise to suprabasal viable differentiating cells that cannot proliferate. This process is tightly controlled and abnormal rates of keratinocyte proliferation contribute to impaired barrier function and disease (Nickoloff et al., 2002; Nickoloff 1999). The suprabasal cells produce a host of proteins that are involved in barrier assembly including involucrin, cornifin, keratin K1, loricrin and filaggrin (Eckert 1989). The transition zone is a region of extensive remodeling in the upper epidermis and also site of type I transglutaminase-mediated cornified envelope assembly (Simon and Green 1985). The epidermis also has apoptotic machinery designed to respond to stress. For example, keratinocytes undergo apoptosis when challenged with high level UVB exposure (Gandarillas et al., 1999; Gandarillas 2000; Grossman et al., 2001; Nickoloff and Denning 2001; Young 1987).
The AP1 transcription factor network includes jun (c-jun, junB, junD) and fos (c-fos, FosB, Fra-1, Fra-2) members. They form jun-jun and jun-fos homodimers and heterodimers that interact with consensus DNA binding sites on target genes to regulate gene expression. AP1 factors are key signal transduction integrators in epidermis that mediate input from cytokines, growth factors, differentiating agents and growth promoters. These signals flow through the MAPK cascade leading to altered AP1 factor function. AP1 factors control keratinocyte proliferation (Mizuno et al., 2006; Shi and Isseroff 2005; Takahashi et al., 2002), differentiation (Adhikary et al., 2004; Eckert et al., 2003; Eckert et al., 2004) and apoptosis (Efimova et al., 2004; Raj et al., 2006), and are important in tumor progression and psoriasis development (Saez et al., 1995; Kahn et al., 1993; She et al., 2002; Rutberg et al., 2000; Iizuka et al., 2004; Eckert et al., 2002; Eckert et al., 2003; Eckert et al., 2004; Efimova et al., 2002; Efimova et al., 2003; Efimova et al., 2004).
Treatment with differentiating agents increase the level of selected AP1 factors (junB, junD, Fra-1) leading to increased involucrin gene expression (Eckert and Welter 1996b; Eckert and Welter 1996a). Moreover, the specific mix of AP1 factors within regulatory complexes influences activity, since AP1 factors can form eighteen different heterodimeric pairs. For example, co-expression of c-fos with c-jun, leading to c-fos:c-jun dimer formation, enhances the transforming capacity of c-jun, whereas pairing c-jun with junB inhibits c-jun transforming capacity (Schutte et al., 1989; Bakiri et al., 2000; Passegue and Wagner 2000). These differences are related to the higher DNA binding and transcriptional activity of c-jun:c-fos heterodimer in comparison to c-jun:junB heterodimer (Deng and Karin 1993). Phosphorylation also modulates activity (Karin 1995; Karin 1998). For example, c-jun expression levels do not significantly change during the cell cycle, but the protein undergoes transient N-terminal phosphorylation as cells proceed from G2 to M that persists until the cells complete mitosis (Bakiri et al., 2000). A crucial conclusion from these studies is that the mix of AP1 factors and their level and phosphorylation state influence biological outcome. The fact that these factors form multiple hetero- and homodimers indicates that manipulating the function of one AP1 factor will modify the function of other members. An additional complication in epidermis is that expression of most AP1 family members changes during differentiation (Welter and Eckert 1995; Mehic et al., 2005). This means that different sets of pairing combinations exist in the basal versus suprabasal layers and that this may drive differences in function.
Impact of suprabasal inactivation of AP1 factor activity
In the present study we examined the impact of suprabasal AP1 factor inactivation on keratinocyte differentiation and proliferation. We chose as a model, targeting dominant-negative c-jun (TAM67) expression to the suprabasal epidermis. This approach has several advantages. First, TAM67 interferes with the function of all AP1 transcription factors (Brown et al., 1994). Second, use of the involucrin promoter permits targeting of TAM67 to the suprabasal epidermis and alleviates problems that are observed with knockout mice where the protein is lost from all layers. Third, a basal layer TAM67-targeted model already exists (Young et al., 1999; Thompson et al., 2002; Young et al., 2002; Cooper et al., 2003; Matthews et al., 2007) which permits a comparison of the impact of basal versus suprabasal AP1 factor inactivation.
Our studies show that targeted expression of TAM67 in the suprabasal epidermis results in hyperplasia and hyperkeratosis. This is associated with a substantial increase in keratinocyte proliferation as measured by increased BrdU incorporation and increased appearance of Ki67-positive cells. This increase in proliferation is most evident in the basal epidermal layer, but some proliferating cells are also detected in the suprabasal epidermis. It is interesting that the major increase in proliferation is localized to basal layer cells. We argue that this is not a direct effect of TAM67 expression on the basal epidermis, since two different staining methods reveal that the TAM67-FLAG expression is confined to the suprabasal layers. This suggests that inactivating suprabasal AP1 factor function may feedback in a manner that stimulates basal layer proliferation. In spite of this increase in proliferation, the epidermal keratinocytes still undergo differentiation, as evidence by the presence of suprabasal cornified cells, and that fact that keratin K1, filaggrin and involucrin display a normal pattern of suprabasal expression. However, differentiation appears to be delayed and may not be complete. This is evidenced by the fact that involucrin and loricrin levels are reduced suggesting that the cornified envelope formation is likely to be abnormal. Moreover, instead of forming an immunoreactive band in the uppermost epidermal layers, loricrin is diffusely present in multiple epidermal layers. The lack of involucrin expression provides supporting evidence that TAM67 is inactivating AP1 factor function the suprabasal epidermis, as an AP1 transcription factor binding site in the involucrin distal promoter is absolutely required for expression in vivo (Crish et al., 1998; Crish et al., 2002; Crish et al., 2006; Crish and Eckert 2008) and AP1 factors bind at this site (Banks et al., 1998; Welter et al., 1995). Moreover, TAM67 expression in normal human keratinocytes inactivates AP1-dependent responses, including phorbol ester-dependent activation of involucrin expression (Efimova et al., 1998). Thus, the reduced involucrin expression provides evidence that TAM67 is interfering with AP1 factor function.
Consistent with delayed differentiation, keratins K5 and K14, which are normally expressed exclusively in the basal layer, are detected in all epidermal layers. This suggests that events required to inactivate K5 and K14 expression are compromised. In addition, K6 is expressed in all epidermal layers. K6 is a keratin that is expressed under conditions of hyperproliferation, but is not expressed in normal epidermis. K6 is expressed in psoriasis, in phorbol ester-treated epidermis and in other conditions associated with keratinocyte hyperproliferation (Sun et al., 1984; Heyden et al., 1994b; Heyden et al., 1994a; Iizuka et al., 2004). Thus, suprabasal TAM67 expression leads to increased basal layer proliferation and delayed differentiation and ultimately results in extensive hyperkeratosis.
Because of the hyperproliferative phenotype, we anticipated that suprabasal TAM67 mice would be susceptible to tumor formation. No tumors were formed in TAM67-positive or negative mice treated with DMBA; however, DMBA-TPA treatment resulted in tumor formation. It was a surprise to discover that DMBA-TPA dependent tumor formation was markedly reduced in suprabasal TAM67 mice. This suggests that although TAM67 leads to increased epidermal proliferation, this does not translate to increased tumor formation. We considered the possibility that TAM67 may interfere with the proliferation promoting activity of TPA in the carcinogenesis protocol. However, our experiments suggest that TAM67-expressing epidermis is fully competent to respond to TPA. TAM67-positive epidermis has five times more Ki67-positive basal cells, and most suprabasal cells are also positive.
These studies indicate that TAM67 has a clear impact on epidermal function. However, it is interesting that TAM67 expression does not impact the corneal epithelium (Fig. 5). This finding suggests that not all tissues may respond to perturbation of AP1 function. It is not clear why the corneal epithelium would be resistant to TAM67 interference with AP1 factor function, but it is interesting that the corneal epithelium is highly resistant to cancer (Cutler 2004).
In an elegant series of experiments, Colburn and colleagues studied the impact of AP1 factor inactivation in the epidermal basal layer by targeting TAM67 to the epidermal basal layer using the keratin 14 promoter (K14-TAM67) and found no overt phenotype under resting conditions (Young et al., 1999). In these mice basal TAM67 produces no obvious changes in keratinocyte proliferation or epidermal morphology, nor does it block 12-O-tetradecanoylphorbol-13-acetate (TPA)-, okadaic acid-, or UVB-dependent epidermal hyperplasia (Young et al., 1999; Thompson et al., 2002; Cooper et al., 2003). These findings suggest that reducing AP1 factor function in the basal compartment has no impact on the epidermis. However, basal layer TAM67 expression suppresses DMBA-TPA dependent tumor formation and this is associated with a reduction in cyclin D1 level (Young et al., 1999; Thompson et al., 2002; Cooper et al., 2003). These mice share several features in common with the mice described in the present paper, but also one major difference. First, TAM67 basal and suprabasal mouse strains both respond to stress agent (okadaic acid, TPA, etc.) with increased basal cell proliferation and that this response is not reduced when compared to normal mice. Second, both strains display a reduced sensitivity to DMBA-TPA induced tumor formation. The details of the mechanism are not known; however, these studies, which show that inactivating AP1 factor function in the basal or suprabasal epidermis reduces tumor formation, clearly suggest that AP1 factors have an essential role in driving tumor formation. The most surprising finding, however, is that suprabasal TAM67 mice display extensive epidermal hyperproliferation and hyperkeratosis under resting conditions, while no phenotype is observed in basal TAM67 mice. Based on these studies, it is possible that inactivating AP1 factor function in the suprabasal compartment may, as a primary mechanism of action, suppress the progress of normal differentiation. We further propose that normal differentiation leads to accumulation of signals that suppress basal layer cell proliferation and that inhibiting differentiation opens this feedback loop leading to increased basal keratinocyte proliferation. We anticipate that this model will prove useful in dissection the role of AP1 factor in normal epidermis versus squamous cell carcinoma.
Materials and Methods
TAM67 transgenic mice
The TAM67 sequence (Brown et al., 1994) was amplified from a plasmid provided by Dr. Peter Steinert using primers designed to attach a FLAG epitope tag to the carboxyl terminus (upstream primer: 5′-GG GAA TCC
 ACT AGC CAG AAC ACG CTG, BamHI underlined, translation start bold-dashed underline; downstream primer: GG AAG CTT
ACT AGC CAG AAC ACG CTG, BamHI underlined, translation start bold-dashed underline; downstream primer: GG AAG CTT

 AAA TGT TTG CAA CTG CTG CGT, HindIII underlined, FLAG epitope double underlined, stop bold-dashed underline). The BamHI/HindIII-restricted PCR product was cloned into pTRE-Tight (Clontech) to create pTRE-Tight-TAM67-FLAG. Digestion of this plasmid with XhoI releases TetO-TAM67-FLAG-SV40 in which the TetO sequence controls TAM67-FLAG expression and the transcript is terminated by an SV40 polyadenylation signal. The TetO-TAM67-FLAG-SV40 sequence was released from the plasmid using XhoI and injected into fertilized mouse embryos as previously described (Crish et al., 1998; Crish et al., 1993). We included the FLAG epitope at the carboxyl terminus of TAM67 so that expression could be easily monitored. Our previous reports, using TAM67-FLAG in cultured keratinocytes, indicate that FLAG at this position does not interfere with TAM67 function (Efimova et al., 1998). Founder mouse tail-DNA was screened for the presence of TAM67-FLAG using polymerase chain reaction (PCR) and primers designed to amplify a unique fragment spanning the TAM67-FLAG coding region. We also performed Southern blot analysis to assure that the integrated transgene was intact (not shown). These B6SJL TetO-TAM67-FLAGTG/- (TAM67) mice were then mated to a FVB hINV-rTATG/- mice (Jaubert et al., 2004) to produce bi-transgenic B6SJL/FVB TAM67-FLAGTG/-, hINV-rTATG/- (TAM67-rTA) mice for experimental use. To confirm that the offspring were bi-transgenic, we perform DNA-dependent PCR using primers that detect each transgene (Diamond et al., 2000). All four of the independently-derived B6SJL TetO-TAM67-FLAGTG/- mouse lines we developed have been mated to FVB hINV-rTATG/- mice to produce bi-transgenic mice, and all (following doxycycline treatment) display the phenotype described in this manuscript. For the experiments outlined in the present study we utilize the TAM67-44 strain. The TetO-TAM67-FLAGTG/- mice were maintained in a B6SJL background and the hINV-rTATG/- mice in a FVB background. Mice were maintained in the University of Maryland School of Medicine animal facility in compliance with NIH regulations with laboratory chow and water accessible ad libitum.
AAA TGT TTG CAA CTG CTG CGT, HindIII underlined, FLAG epitope double underlined, stop bold-dashed underline). The BamHI/HindIII-restricted PCR product was cloned into pTRE-Tight (Clontech) to create pTRE-Tight-TAM67-FLAG. Digestion of this plasmid with XhoI releases TetO-TAM67-FLAG-SV40 in which the TetO sequence controls TAM67-FLAG expression and the transcript is terminated by an SV40 polyadenylation signal. The TetO-TAM67-FLAG-SV40 sequence was released from the plasmid using XhoI and injected into fertilized mouse embryos as previously described (Crish et al., 1998; Crish et al., 1993). We included the FLAG epitope at the carboxyl terminus of TAM67 so that expression could be easily monitored. Our previous reports, using TAM67-FLAG in cultured keratinocytes, indicate that FLAG at this position does not interfere with TAM67 function (Efimova et al., 1998). Founder mouse tail-DNA was screened for the presence of TAM67-FLAG using polymerase chain reaction (PCR) and primers designed to amplify a unique fragment spanning the TAM67-FLAG coding region. We also performed Southern blot analysis to assure that the integrated transgene was intact (not shown). These B6SJL TetO-TAM67-FLAGTG/- (TAM67) mice were then mated to a FVB hINV-rTATG/- mice (Jaubert et al., 2004) to produce bi-transgenic B6SJL/FVB TAM67-FLAGTG/-, hINV-rTATG/- (TAM67-rTA) mice for experimental use. To confirm that the offspring were bi-transgenic, we perform DNA-dependent PCR using primers that detect each transgene (Diamond et al., 2000). All four of the independently-derived B6SJL TetO-TAM67-FLAGTG/- mouse lines we developed have been mated to FVB hINV-rTATG/- mice to produce bi-transgenic mice, and all (following doxycycline treatment) display the phenotype described in this manuscript. For the experiments outlined in the present study we utilize the TAM67-44 strain. The TetO-TAM67-FLAGTG/- mice were maintained in a B6SJL background and the hINV-rTATG/- mice in a FVB background. Mice were maintained in the University of Maryland School of Medicine animal facility in compliance with NIH regulations with laboratory chow and water accessible ad libitum.
Carcinogenesis studies
For skin carcinogenesis studies, the dorsal skin was shaved followed after two days by a single topical application of 100 μg of 7,12-dimethylbenzanthracene (DMBA) delivered in 200 μl of acetone. Beginning 1 wk later, mice were painted twice weekly with 10 μg 12-O-tetradecanoylphorbol-13-acetate (TPA) delivered in 200 μl acetone for 22 wks. At the time of the first TPA treatment and continuing thereafter, half of the mice received doxycycline (2 mg/ml) in the drinking water. The DMBA treatment was administered one week prior to induction of TAM67 expression or treatment with TPA to assure that these treatments did not alter DMBA metabolism. TPA and DMBA were obtained from Sigma (St Louis, MO). Mice were observed weekly for tumor onset, number and size. At 22 wks the tumors were harvested and processed for preparation of protein extracts and sectioned for histology. To monitor the acute epidermal proliferative response following TPA challenge, dorsal skin was shaved and treated with Nair to remove hair. After two days, 5 μg of TPA was painted onto the epidermis in 100 μl acetone. At 24 h post-treatment, the mice were euthanized and the skin was removed for histological analysis and preparation of protein extract. Epidermal and dermal thickness was measured using a stage micrometer, and the number of epitope-positive cells was counted and expressed as positive cells per unit length of epidermal basal layer.
Antibodies and immunological methods
Immunofluorescence was performed using paraffin-embedded formalin-fixed sections as previously reported (Crish et al., 1998; Crish et al., 2002; Crish et al., 2006). K1 (PRB-165P), K5 (PRB-160P), K6 (PRB-169P), K14 (PRB-155P), filaggrin (PRB-417P) and loricrin (PRB-145P) antibodies were purchased form Covance (Emeryville, CA). Ki67 (TEC-3) antibody was from Dako (Carpinteria, CA), and β-actin (A5441) and FLAG (M2) (F4049) specific antibodies were obtained from Sigma (St. Louis, MO). BrdU was purchased from BD Pharmingen (550891) and BrdU was detected using the Vector Laboratories anti-mouse kit (MP-7402). Primary antibody localization was visualized using an appropriate fluorophore-conjugated secondary antibody. For immunoblot, epidermis was separated from dermis, frozen in liquid nitrogen, pulverized and suspended in dye-free Laemmli sample buffer. The suspension was sonicated, particulates were removed by centrifugation, and soluble extract was electrophoresed on a polyacrylamide gel and transferred to nitrocellulose for immunoblot (Crish et al., 1998; Crish et al., 2002; Crish et al., 2006). Unless otherwise indicated in the figure legends, immunohistological and immunoblot results were repeated in three separate experiments and sections and extracts were monitored from epidermis of three mice per treatment group.
Acknowledgments
This work was supported by NIH RO1 AR046494 (R. Eckert)
Abbreviations
- TRE or TetO
tetracycline response element
- TAM67
dominant-negative c-jun
- K1
keratin 1
- K14
keratin 14
- K5
keratin 5
- rTA
tetracycline-responsive activator protein
- TPA
12-O-tetradecanoylphorbol-13-acetate
- DMBA
7,12-dimethylbenzanthracene
Footnotes
Conflict of Interest: The authors have no conflict of interest financial or otherwise.
Reference List
- Adhikary G, Crish J, Lass J, Eckert RL. Regulation of involucrin expression in normal human corneal epithelial cells: a role for activator protein one. Invest Ophthalmol Vis Sci. 2004;45:1080–1087. doi: 10.1167/iovs.03-1180. [DOI] [PubMed] [Google Scholar]
- Angel P, Szabowski A, Schorpp-Kistner M. Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene. 2001;20:2413–2423. doi: 10.1038/sj.onc.1204380. [DOI] [PubMed] [Google Scholar]
- Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 2000;19:2056–2068. doi: 10.1093/emboj/19.9.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks EB, Crish JF, Welter JF, Eckert RL. Characterization of human involucrin promoter distal regulatory region transcriptional activator elements-a role for Sp1 and AP1 binding sites. Biochem J. 1998;331(Pt 1):61–68. doi: 10.1042/bj3310061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickenbach JR, Dunnwald M. Epidermal stem cells: characteristics and use in tissue engineering and gene therapy. Adv Dermatol. 2000;16:159–183. [PubMed] [Google Scholar]
- Brown PH, Chen TK, Birrer MJ. Mechanism of action of a dominant-negative mutant of c-Jun. Oncogene. 1994;9:791–799. [PubMed] [Google Scholar]
- Cooper SJ, MacGowan J, Ranger-Moore J, Young MR, Colburn NH, Bowden GT. Expression of dominant negative c-jun inhibits ultraviolet B-induced squamous cell carcinoma number and size in an SKH-1 hairless mouse model. Mol Cancer Res. 2003;1:848–854. [PubMed] [Google Scholar]
- Crish JF, Bone F, Banks EB, Eckert RL. The human involucrin gene contains spatially distinct regulatory elements that regulate expression during early versus late epidermal differentiation. Oncogene. 2002;21:738–747. doi: 10.1038/sj.onc.1205038. [DOI] [PubMed] [Google Scholar]
- Crish JF, Eckert RL. Synergistic activation of human involucrin gene expression by Fra-1 and p300--evidence for the presence of a multiprotein complex. J Invest Dermatol. 2008;128:530–541. doi: 10.1038/sj.jid.5701049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crish JF, Gopalakrishnan R, Bone F, Gilliam AC, Eckert RL. The distal and proximal regulatory regions of the involucrin gene promoter have distinct functions and are required for in vivo involucrin expression. J Invest Dermatol. 2006;126:305–314. doi: 10.1038/sj.jid.5700019. [DOI] [PubMed] [Google Scholar]
- Crish JF, Howard JM, Zaim TM, Murthy S, Eckert RL. Tissue-specific and differentiation-appropriate expression of the human involucrin gene in transgenic mice: an abnormal epidermal phenotype. Differentiation. 1993;53:191–200. doi: 10.1111/j.1432-0436.1993.tb00708.x. [DOI] [PubMed] [Google Scholar]
- Crish JF, Zaim TM, Eckert RL. The distal regulatory region of the human involucrin promoter is required for expression in epidermis. J Biol Chem. 1998;273:30460–30465. doi: 10.1074/jbc.273.46.30460. [DOI] [PubMed] [Google Scholar]
- Cutler TJ. Corneal epithelial disease. Vet Clin North Am Equine Pract. 2004;20:319–43. doi: 10.1016/j.cveq.2004.04.014. vi. [DOI] [PubMed] [Google Scholar]
- Deng T, Karin M. JunB differs from c-Jun in its DNA-binding and dimerization domains, and represses c-Jun by formation of inactive heterodimers. Genes Dev. 1993;7:479–490. doi: 10.1101/gad.7.3.479. [DOI] [PubMed] [Google Scholar]
- Diamond I, Owolabi T, Marco M, Lam C, Glick A. Conditional gene expression in the epidermis of transgenic mice using the tetracycline-regulated transactivators tTA and rTA linked to the keratin 5 promoter. J Invest Dermatol. 2000;115:788–794. doi: 10.1046/j.1523-1747.2000.00144.x. [DOI] [PubMed] [Google Scholar]
- Dunnwald M, Tomanek-Chalkley A, Alexandrunas D, Fishbaugh J, Bickenbach JR. Isolating a pure population of epidermal stem cells for use in tissue engineering. Exp Dermatol. 2001;10:45–54. doi: 10.1034/j.1600-0625.2001.100106.x. [DOI] [PubMed] [Google Scholar]
- Eckert RL. Structure, function, and differentiation of the keratinocyte. Physiol Rev. 1989;69:1316–1346. doi: 10.1152/physrev.1989.69.4.1316. [DOI] [PubMed] [Google Scholar]
- Eckert RL, Crish JF, Efimova T, Dashti SR, Deucher A, Bone F, et al. Regulation of involucrin gene expression. J Invest Dermatol. 2004;123:13–22. doi: 10.1111/j.0022-202X.2004.22723.x. [DOI] [PubMed] [Google Scholar]
- Eckert RL, Crish JF, Robinson NA. The epidermal keratinocyte as a model for the study of gene regulation and cell differentiation. Physiol Rev. 1997;77:397–424. doi: 10.1152/physrev.1997.77.2.397. [DOI] [PubMed] [Google Scholar]
- Eckert RL, Efimova T, Balasubramanian S, Crish JF, Bone F, Dashti S. p38 Mitogen-Activated Protein Kinases on the Body Surface - A Function for p38delta. J Invest Dermatol. 2003;120:823–828. doi: 10.1046/j.1523-1747.2003.12120.x. [DOI] [PubMed] [Google Scholar]
- Eckert RL, Efimova T, Dashti SR, Balasubramanian S, Deucher A, Crish JF, et al. Keratinocyte survival, differentiation, and death: many roads lead to mitogen-activated protein kinase. J Invest Dermatol Symp Proc. 2002;7:36–40. doi: 10.1046/j.1523-1747.2002.19634.x. [DOI] [PubMed] [Google Scholar]
- Eckert RL, Welter JF. Epidermal keratinoctyes - genes and their regulation. Cell Death Differ. 1996a;3:373–383. [PubMed] [Google Scholar]
- Eckert RL, Welter JF. Transcription factor regulation of epidermal keratinocyte gene expression. Mol Biol Rep. 1996b;23:59–70. doi: 10.1007/BF00357073. [DOI] [PubMed] [Google Scholar]
- Efimova T, Broome AM, Eckert RL. A regulatory role for p38 delta MAPK in keratinocyte differentiation. Evidence for p38 delta-ERK1/2 complex formation. J Biol Chem. 2003;278:34277–34285. doi: 10.1074/jbc.M302759200. [DOI] [PubMed] [Google Scholar]
- Efimova T, Broome AM, Eckert RL. Protein kinase Cdelta regulates keratinocyte death and survival by regulating activity and subcellular localization of a p38delta-extracellular signal-regulated kinase 1/2 complex. Mol Cell Biol. 2004;24:8167–8183. doi: 10.1128/MCB.24.18.8167-8183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimova T, Deucher A, Kuroki T, Ohba M, Eckert RL. Novel protein kinase C isoforms regulate human keratinocyte differentiation by activating a p38 delta mitogen-activated protein kinase cascade that targets CCAAT/enhancer-binding protein alpha. J Biol Chem. 2002;277:31753–31760. doi: 10.1074/jbc.M205098200. [DOI] [PubMed] [Google Scholar]
- Efimova T, LaCelle P, Welter JF, Eckert RL. Regulation of human involucrin promoter activity by a protein kinase C, Ras, MEKK1, MEK3, p38/RK, AP1 signal transduction pathway. J Biol Chem. 1998;273:24387–24395. doi: 10.1074/jbc.273.38.24387. [DOI] [PubMed] [Google Scholar]
- Fleischmann A, Hafezi F, Elliott C, Reme CE, Ruther U, Wagner EF. Fra-1 replaces c-Fos-dependent functions in mice. Genes Dev. 2000;14:2695–2700. doi: 10.1101/gad.187900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin L, Knebel J, Zigrino P, Vonderstrass B, Mauch C, Schorpp-Kistner M, et al. Delayed wound healing and epidermal hyperproliferation in mice lacking JunB in the skin. J Invest Dermatol. 2006;126:902–911. doi: 10.1038/sj.jid.5700123. [DOI] [PubMed] [Google Scholar]
- Gandarillas A. Epidermal differentiation, apoptosis, and senescence: common pathways? Exp Gerontol. 2000;35:53–62. doi: 10.1016/s0531-5565(99)00088-1. [DOI] [PubMed] [Google Scholar]
- Gandarillas A, Goldsmith LA, Gschmeissner S, Leigh IM, Watt FM. Evidence that apoptosis and terminal differentiation of epidermal keratinocytes are distinct processes. Exp Dermatol. 1999;8:71–79. doi: 10.1111/j.1600-0625.1999.tb00350.x. [DOI] [PubMed] [Google Scholar]
- Grossman D, Kim PJ, Blanc-Brude OP, Brash DE, Tognin S, Marchisio PC, et al. Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J Clin Invest. 2001;108:991–999. doi: 10.1172/JCI13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyden A, Lutzow Holm C, Clausen OP, Thrane EV, Brandtzaeg P, Roop DR, et al. Application of cantharidin or 12-O-tetradecanoylphorbol-13- acetate on mouse epidermis induces a cell population shift that causes altered keratin distribution. Differentiation. 1994a;57:187–193. doi: 10.1046/j.1432-0436.1994.5730187.x. [DOI] [PubMed] [Google Scholar]
- Heyden A, Lutzow-Holm C, Clausen OP, Brandtzaeg P, Huitfeldt HS. Expression of keratins K6 and K16 in regenerating mouse epidermis is less restricted by cell replication than the expression of K1 and K10. Epithelial Cell Biol. 1994b;3:96–101. [PubMed] [Google Scholar]
- Iizuka H, Takahashi H, Honma M, Ishida-Yamamoto A. Unique keratinization process in psoriasis: late differentiation markers are abolished because of the premature cell death. J Dermatol. 2004;31:271–276. doi: 10.1111/j.1346-8138.2004.tb00672.x. [DOI] [PubMed] [Google Scholar]
- Jaubert J, Patel S, Cheng J, Segre JA. Tetracycline-regulated transactivators driven by the involucrin promoter to achieve epidermal conditional gene expression. J Invest Dermatol. 2004;123:313–318. doi: 10.1111/j.0022-202X.2004.23203.x. [DOI] [PubMed] [Google Scholar]
- Kahn CR, Young E, Lee IH, Rhim JS. Human corneal epithelial primary cultures and cell lines with extended life span: in vitro model for ocular studies. Invest Ophthalmol Vis Sci. 1993;34:3429–3441. [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Karin M. Mitogen-activated protein kinase cascades as regulators of stress responses. Ann N Y Acad Sci. 1998;851:139–146. doi: 10.1111/j.1749-6632.1998.tb08987.x. [DOI] [PubMed] [Google Scholar]
- Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- Matthews CP, Birkholz AM, Baker AR, Perella CM, Beck GR, Jr, Young MR, et al. Dominant-negative activator protein 1 (TAM67) targets cyclooxygenase-2 and osteopontin under conditions in which it specifically inhibits tumorigenesis. Cancer Res. 2007;67:2430–2438. doi: 10.1158/0008-5472.CAN-06-0522. [DOI] [PubMed] [Google Scholar]
- Mehic D, Bakiri L, Ghannadan M, Wagner EF, Tschachler E. Fos and jun proteins are specifically expressed during differentiation of human keratinocytes. J Invest Dermatol. 2005;124:212–220. doi: 10.1111/j.0022-202X.2004.23558.x. [DOI] [PubMed] [Google Scholar]
- Mizuno H, Cho YY, Ma WY, Bode AM, Dong Z. Effects of MAP kinase inhibitors on epidermal growth factor-induced neoplastic transformation of human keratinocytes. Mol Carcinog. 2006;45:1–9. doi: 10.1002/mc.20160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy CJ, Laskin JD. Specific alterations in keratin biosynthesis in mouse epidermis in vivo and in explant culture following a single exposure to the tumor promoter 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1987;47:4674–4680. [PubMed] [Google Scholar]
- Nickoloff BJ. Animal models of psoriasis. Expert Opin Investig Drugs. 1999;8:393–401. doi: 10.1517/13543784.8.4.393. [DOI] [PubMed] [Google Scholar]
- Nickoloff BJ, Denning M. Life and death signaling in epidermis: following a planned cell death pathway involving a trail that does not lead to skin cancer. J Invest Dermatol. 2001;117:1–2. doi: 10.1046/j.1523-1747.2001.117001001.x. [DOI] [PubMed] [Google Scholar]
- Nickoloff BJ, Qin JZ, Chaturvedi V, Bacon P, Panella J, Denning MF. Life and death signaling pathways contributing to skin cancer. J Investig Dermatol Symp Proc. 2002;7:27–35. doi: 10.1046/j.1523-1747.2002.19633.x. [DOI] [PubMed] [Google Scholar]
- Passegue E, Jochum W, Behrens A, Ricci R, Wagner EF. JunB can substitute for Jun in mouse development and cell proliferation. Nat Genet. 2002;30:158–166. doi: 10.1038/ng790. [DOI] [PubMed] [Google Scholar]
- Passegue E, Wagner EF. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 2000;19:2969–2979. doi: 10.1093/emboj/19.12.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj D, Brash DE, Grossman D. Keratinocyte apoptosis in epidermal development and disease. J Invest Dermatol. 2006;126:243–257. doi: 10.1038/sj.jid.5700008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutberg SE, Adams TL, Glick A, Bonovich MT, Vinson C, Yuspa SH. Activator protein 1 transcription factors are fundamental to v-rasHa-induced changes in gene expression in neoplastic keratinocytes. Cancer Res. 2000;60:6332–6338. [PubMed] [Google Scholar]
- Saez E, Rutberg SE, Mueller E, Oppenheim H, Smoluk J, Yuspa SH, et al. c-fos is required for malignant progression of skin tumors. Cell. 1995;82:721–732. doi: 10.1016/0092-8674(95)90469-7. [DOI] [PubMed] [Google Scholar]
- Schreiber M, Wang ZQ, Jochum W, Fetka I, Elliott C, Wagner EF. Placental vascularisation requires the AP-1 component fra1. Development. 2000;127:4937–4948. doi: 10.1242/dev.127.22.4937. [DOI] [PubMed] [Google Scholar]
- Schutte J, Minna JD, Birrer MJ. Deregulated expression of human c-jun transforms primary rat embryo cells in cooperation with an activated c-Ha-ras gene and transforms rat-1a cells as a single gene. Proc Natl Acad Sci U S A. 1989;86:2257–2261. doi: 10.1073/pnas.86.7.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- She QB, Chen N, Bode AM, Flavell RA, Dong Z. Deficiency of c-Jun-NH(2)-terminal kinase-1 in mice enhances skin tumor development by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 2002;62:1343–1348. [PubMed] [Google Scholar]
- Shi B, Isseroff RR. Epidermal growth factor (EGF)-mediated DNA-binding activity of AP-1 is attenuated in senescent human epidermal keratinocytes. Exp Dermatol. 2005;14:519–527. doi: 10.1111/j.0906-6705.2005.00317.x. [DOI] [PubMed] [Google Scholar]
- Simon M, Green H. Enzymatic cross-linking of involucrin and other proteins by keratinocyte particulates in vitro. Cell. 1985;40:677–683. doi: 10.1016/0092-8674(85)90216-8. [DOI] [PubMed] [Google Scholar]
- Sun TT, Eichner R, Nelson WG, Tseng SC, Weiss RA, Jarvinen M, et al. Keratin classes: molecular markers for different types of epithelial differentiation. J Invest Dermatol. 1983;81:109s–115s. doi: 10.1111/1523-1747.ep12540831. [DOI] [PubMed] [Google Scholar]
- Sun TT, Eichner R, Schermer A, Cooper D, Nelson WG, Weiss RA. Classification, expression and possible mechanisms of evolution of mammalian epithelial keratins: a unifying model. Cancer Cells. 1984;1:169–176. [Google Scholar]
- Takahashi H, Ibe M, Nakamura S, Ishida-Yamamoto A, Hashimoto Y, Iizuka H. Extracellular regulated kinase and c-Jun N-terminal kinase are activated in psoriatic involved epidermis. J Dermatol Sci. 2002;30:94–99. doi: 10.1016/s0923-1811(02)00064-6. [DOI] [PubMed] [Google Scholar]
- Thompson EJ, MacGowan J, Young MR, Colburn N, Bowden GT. A dominant negative c-jun specifically blocks okadaic acid-induced skin tumor promotion. Cancer Res. 2002;62:3044–3047. [PubMed] [Google Scholar]
- Toftgard R, Yuspa SH, Roop DR. Keratin gene expression in mouse skin tumors and in mouse skin treated with 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1985;45:5845–5850. [PubMed] [Google Scholar]
- Welter JF, Crish JF, Agarwal C, Eckert RL. Fos-related antigen (Fra-1), junB, and junD activate human involucrin promoter transcription by binding to proximal and distal AP1 sites to mediate phorbol ester effects on promoter activity. J Biol Chem. 1995;270:12614–12622. doi: 10.1074/jbc.270.21.12614. [DOI] [PubMed] [Google Scholar]
- Welter JF, Eckert RL. Differential expression of fos and jun family members c-fos, fosB, Fra-1, Fra-2, c-jun, junB and junD during human epidermal keratinocyte differentiation. Oncogene. 1995;11:2681–2687. [PubMed] [Google Scholar]
- Young AR. The sunburn cell. Photodermatol. 1987;4:127–134. [PubMed] [Google Scholar]
- Young MR, Farrell L, Lambert P, Awasthi P, Colburn NH. Protection against human papillomavirus type 16-E7 oncogene-induced tumorigenesis by in vivo expression of dominant-negative c-jun. Mol Carcinog. 2002;34:72–77. doi: 10.1002/mc.10050. [DOI] [PubMed] [Google Scholar]
- Young MR, Li JJ, Rincon M, Flavell RA, Sathyanarayana BK, Hunziker R, et al. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc Natl Acad Sci U S A. 1999;96:9827–9832. doi: 10.1073/pnas.96.17.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenz R, Eferl R, Kenner L, Florin L, Hummerich L, Mehic D, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–375. doi: 10.1038/nature03963. [DOI] [PubMed] [Google Scholar]