

Abstract
The gene cluster from Pantoea agglomerans responsible for biosynthesis of the dapdiamide antibiotics encodes an adenylation-thiolation didomain protein, DdaD, and an Fe(II)/α-ketoglutarate-dependent dioxygenase homolog, DdaC. Here we show that DdaD, a nonribosomal peptide synthetase module, activates and sequesters Nβ-fumaramoyl-L-2,3-diaminopropionic acid as a covalently tethered thioester for subsequent oxidative modification of the fumaramoyl group. DdaC catalyzes Fe(II)- and α-ketoglutarate-dependent epoxidation of the covalently bound Nβ-fumaramoyl-L-2,3-diaminopropionyl-S-DdaD species to generate Nβ-epoxysuccinamoyl-DAP in thioester linkage to DdaD. After hydrolytic release, Nβ-epoxysuccinamoyl-DAP can be ligated to L-valine by the ATP-dependent ligase DdaF to form the natural antibiotic Nβ-epoxysuccinamoyl-diaminopropionyl-valine.
Introduction
The dapdiamide antibiotics are a family of five N-acylated dipeptides produced by Pantoea agglomerans (Figure 1A).1 The “dap” prefix refers to the presence of the nonproteinogenic amino acid 2,3-diaminopropionate (DAP, blue in Figure 1) while the “diamide” suffix reflects the two backbone amide bonds. The DAP moiety, which can be acylated either on Nβ or Nα, is the first residue of the dipeptide and is attached via a standard peptide linkage to a terminal valine (Val), isoleucine (Ile), or leucine (Leu) (red in Figure 1). The N-acyl moiety (green in Figure 1) is a fumaramoyl group in dapdiamides A-D and an epoxysuccinamoyl group in dapdiamide E. The fumaramoyl or epoxysuccinamoyl functionality most likely provides the electrophilic moiety that accounts for the antibiotic activity of this class of compounds.2 The dapdiamides are likely cleaved intracellularly to generate acyl-DAP warheads that target glucosamine-6-phosphate synthase via capture of the nucleophilic active site cysteine (Cys).2,3
Figure 1.

(A) The dapdiamide family of antibiotics. (B) The dapdiamide gene cluster. (C) Dapdiamide-related epoxide natural products.
The dapdiamides A-E were isolated by activity-based cloning of dapdiamide biosynthetic genes from P. agglomerans CU0119 into Escherichia coli.1 This allowed for sequencing of the responsible gene cluster which revealed nine genes, annotated as ddaA-I (Figure 1B) that are necessary and sufficient for E. coli to make the dapdiamides. In our initial studies on the Dda enzymes, we determined that DdaG and DdaF are ATP-dependent ligases that build the N-acyl-dipeptide scaffolds.4 DdaG is an AMP-generating ligase that makes the regiospecifically N-acylated Nβ-fumaroyl-DAP (NβFmDAP). Our data indicated that DdaF catalyzes the last step in the pathway and forms the dipeptide linkage of N-acyl-DAP with Val, Ile, or Leu. DdaF cleaves ATP to ADP (not AMP), generating the N-acyl-DAP-phosphate as an activated intermediate for capture by Val, Ile, or Leu. This enzyme accepts only Nβ-fumaramoyl-DAP (NβFmmDAP), not NβFmDAP, as the carboxylate substrate, suggesting DdaH, the putative fumaroyl to fumaramoyl amide synthase, acts after DdaG but before DdaF.
In this study, we have turned our attention to the epoxidation event in the dapdiamide biosynthetic pathway. An epoxysuccinamoyl moiety is present in both dapdiamide E and its Nβ-acyl-DAP isomer Nβ-epoxysuccinamoyl-DAP-Val (NβEpSmDAP-Val),i a natural product produced by Serratia plymuthica and P. agglomerans strains Pa48b, Pa39b, and C9-1 (Figure 1C).1,5,6 NMR evidence suggests that each of these compounds contains a trans-epoxide, but the absolute stereochemistry of the epoxide carbons has not been determined.1,5,6 The most similar epoxysuccin(am)ate-containing natural product for which the epoxide stereochemistry has been determined is Sch37137 (Figure 1C), and in this compound the epoxide carbons have an (R,R) configuration.7,8 However, there are other known natural products that contain both (R,R)- and (S,S)-trans-epoxysuccin(am)ate.9,10
Here we report that DdaF catalyzes ATP-dependent ligation of both of the two Nβ-trans-epoxysuccinamoyl-DAP (Nβ-trans-EpSmDAP) diastereomers and Val, with substrate specificity for the (R,R) over the (S,S) diastereomer. Our studies were based on the hypothesis that DdaC and DdaD are required to form Nβ-trans-EpSmDAP from an olefin-containing acyl-DAP intermediate. DdaD is a third type of ATP-utilizing enzyme in this pathway, a predicted nonribosomal peptide synthetase (NRPS) module,11 while DdaC has homology to mononuclear nonheme iron oxygenases.12 In this study, we demonstrate that DdaC and DdaD are the relevant catalysts and determine the timing of epoxidation during Nβ-acyl-DAP-Val assembly.
Materials and Methods
Materials and General Methods
Oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA). Polymerase chain reaction (PCR) was performed with Phusion High-Fidelity PCR Mastermix (New England Biolabs). Cloning was performed using the Gateway System (Invitrogen). One Shot Chemically Competent TOP10 E. coli (Invitrogen) and NovaBlue(DE3) (Novagen) were used for routine cloning and propagation of DNA vectors. Recombinant plasmid DNA was purified with a Qiaprep kit (Qiagen). DNA sequencing was performed at the Molecular Biology Core Facilities of the Dana Farber Cancer Institute (Boston, MA). Nickel-nitrilotriacetic acid-agarose (Ni-NTA) superflow resin and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels were purchased from Qiagen and Biorad, respectively. Protein concentrations were determined by Bradford assay13 with bovine serum albumin (BSA) as a standard or by Nanodrop 1000 spectrophotometer (Thermo Scientific) based on the absorbance at 280 nm with the predicted molar extinction coefficient.
Anaerobic manipulations were performed under a nitrogen atmosphere using an Mbraun Labmaster glovebox (Stratham, NH) maintained at 2 parts-per-million (ppm) O2 or less. Buffers were sparged with argon for 20–30 min and equilibrated overnight with the nitrogen atmostphere in the glovebox before use.
A pyruvate kinase/lactate dehydrogenase (PK/LDH) enzyme mix from rabbit muscle was purchased from Sigma as a buffered aqueous glycerol solution. Synthetic dapdiamide A and the plasmid containing the dapdiamide gene cluster, pUC19 A10A, were provided by Jessica Dawlaty (Harvard Medical School, Boston, MA).1 BODIPY-CoA14 and Sfp15,16 were prepared according to published procedures. N-His6-tagged DdaF was purified as described previously.4 NβFmDAP, NαFmDAP, NβFmmDAP, NαFmmDAP, and fumaramate (Fmm) were synthesized as described previously.4
DdaF ADP Production Assay
250 μL reaction mixtures were incubated at room temperature and contained 250 nM DdaF, 5 mM Val, 10 mM ATP, 12 mM MgCl2, 200 μM NADH, 500 μM phosphoenolpyruvate, 59 units/mL pyruvate kinase, 41 units/mL lactate dehydrogenase, 5 mM borate·NaOH (pH 9.5) and 100 mM 4-(2-hyhdroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 8). The consumption of NADH was monitored continuously in Plastibrand micro UV-cuvettes in a Varian Cary 50 UV-visible spectrophotometer by measuring the absorbance at 340 nm. Kinetic constants were derived from velocity versus substrate concentration data with GraphPad Prism using a non-linear, least-squares fitting method for NβFmmDAP and NβRREpSmDAP and a linear regression for NβSSEpSmDAP.
DdaD ATP-[32P]PPi Exchange Assays
Reaction mixtures (170–350 μL) contained 5 μM DdaD, 125 μM substrate, 1 mM ATP, 1 mM MgCl2, 40 mM KCl, 5 mM Na[32P]PPi (approximately 2–4 × 107 counts-per-min (cpm)/mL), and 50 mM HEPES (pH 7.5). Reactions were incubated at room temperature for 10 min, and then 50 μL aliquots were removed and quenched with 250 μL of a charcoal suspension (100 mM NaPPi, 350 mM HClO4, and 16 g/L charcoal). The samples were mixed using a vortex and then centrifuged at 16,000 × g for 3 min. The pellets were washed twice with 250 μL of wash solution (100 mM NaPPi, and 350 mM HClO4). Charcoal-bound radioactivity was measured on a Beckman LS 6500 scintillation counter.
DdaC/D Enzymatic Assays with Anaerobically Purified DdaC
Phosphopantetheinylation reactions (12–270 μL) contained 25 μM DdaD, 12 μM Sfp, 400 μM coenzyme A (CoA), 10 mM MgCl2, 1.5 mM dithiothreitol (DTT), and 50 mM HEPES (pH 7.5). Reactions were incubated at room temperature for 1 h. To form aminoacyl-S-DdaD, 120–250 μM substrate and 1 mM ATP were added (final volume 12.5–156 μL) and the reactions incubated an additional 5–60 min at room temperature.
50 μM Fe(NH4)2(SO4)2 (stock solution 2.5 mM in 200 μM HCl), 300 μM α-KG (stock solution 6 mM in 50 mM HEPES (pH 8)), 4 mM ascorbic acid (stock solution 100 mM in 50 mM HEPES (pH 8)), and 10 μM DdaC were added to the NβFmmDAP-S-DdaD preparations (final volume 73–79 μL). Reactions were incubated for 5–60 min at room temperature.
Reactions were quenched by flash freezing in N2(l) for subsequent trypsin digestion, or by the addition of an equal volume of 25% formic acid followed by flash freezing in N2(l) for reverse phase liquid chromatography (RPLC)-Fourier-Transform mass spectrometry (FTMS) analysis of intact DdaD.
H218O incubations were carried out as described above with the exception that the final reaction mixtures contained 63% H218O (Cambridge Isotope Laboratories).
Trypsin Digestion of DdaD-Containing Reactions
The following digestion procedure was used for analysis of 1) conversion of apo-DdaD to holo-DdaD (HS-DdaD), 2) loading of DdaD with NβFmmDAP to generate NβFmmDAP-S-DdaD, 3) conversion of NβFmmDAP-S-DdaD to NβEpSmDAP-S-DdaD by DdaC, 4) dependence of epoxide formation on α-KG, 5) loading of dapdiamide A onto HS-DdaD, and 6) incorporation of 18O into NβEpSmDAP-S-DdaD using H218O.
All reactions were stored at −80 °C until trypsin digestion. Prior to trypsin digestion, samples were thawed at room temperature. Trypsin (Promega Sequencing Grade) was resuspended in the buffer provided by the manufacturer to a final concentration of 1 μg/μL and incubated at 30 °C for 15 min. An aliquot of the reaction containing 50 μg of DdaD was removed and added to an equal volume of 0.1 M NH4HCO3 (pH 7.8–8) and 2 mM Tris(2-carboxyethyl)phosphine (TCEP) (pH 6). This mixture was incubated for 4 min at room temperature, and trypsin was added at a mass ratio of 1:5 trypsin/total protein in the digestion reaction. After addition of the trypsin, the reaction was incubated at 30 °C for 5 min. The reaction was quenched by the addition of one half the reaction volume of 25% formic acid and stored at −80 °C until mass spectrometric analysis.
RPLC-FTMS Analysis of Trypsin Digests
All RPLC-FTMS analyses were conducted using an Agilent 1200 high performance LC (HPLC) system with autosampler coupled directly to a ThermoFisher Scientific LTQ-FT hybrid linear ion trap-FTMS system operating at 11 Tesla. The mass spectrometer was calibrated weekly using the calibration mixture and instructions specified by the manufacturer. All instrument parameters were tuned according to the manufacturer’s instructions (employing bovine ubiquitin (Sigma) for tuning purposes).
For all analyses of trypsin digests of DdaD-containing reactions, a 1 mm × 150 mm Jupiter C18 column (Phenomenex, 300 Å, 5 μm) was connected in-line with the electrospray ionization (ESI) source (operated at ~5 kV with a capillary temperature of 200–250 °C) for the MS system. The 70 min separation gradient used for all RPLC analyses is shown in Table S1, where solvent A was H2O + 0.1% formic acid and solvent B was acetonitrile (MeCN) + 0.1% formic acid. A trypsin-digested reaction mixture was loaded onto the column using the autosampler and separated according to the gradient shown.
All ionized peptide species entering the mass spectrometer were subjected to an MS method with five MS and MS/MS events: 1) full scan measurement of all intact peptides (all ions detected in the FTMS in profile mode; resolution: 100,000; m/z range detected: 400–2000), 2) the phosphopantetheinyl (Ppant) ejection assay using nozzle-skimmer dissociation (NSD) (all ions detected in the FTMS in profile mode; resolution: 50,000; m/z range: 250–500; surface-induced dissociation (SID) = 75 V), 3–5) data-dependent MS/MS on the first, second and third most abundant ions from scan (1) using collision induced dissociation (CID) (all ions detected in the FTMS in profile mode; minimum target signal counts: 5,000; resolution: 50,000; m/z range detected: dependent on target m/z, default charge state: 2, isolation width: 5 m/z, normalized collision energy (NCE): 35; activation q value: 0.40; activation time: 30 ms). During all analyses, dynamic exclusion was enabled with the following settings: repeat count − 2, repeat duration − 30 s, exclusion list size − 300, exclusion duration − 60 s.
All data were analyzed using Qualbrowser (Xcalibur) and ProSightPC, both provided with the LTQ-FT system. ProsightPC was used to search all MS/MS data against a database containing the apo-DdaD protein sequence for peptide identification, with an intact peptide mass tolerance of 750 Da (which allowed for observation of HS-DdaD and HS-DdaD loaded with a variety of substrates) and a fragment ion mass tolerance of 10 ppm (Δm mode was enabled for all searches). A minimum of 5 matching fragment ions was required for peptide identification. In Qualbrowser, ions of interest were searched within a range of 0.01 m/z around the isotopic peak of interest, within a tolerance of 5 ppm.
RPLC-FTMS Analysis of Intact DdaD
RPLC-FTMS analysis of intact DdaD by the Ppant ejection assay was employed for analyses of the loading of Nβ-(R,R)-epoxysuccinamoyl-DAP (NβRREpSmDAP) onto HS-DdaD, the loading of Nβ-(S,S)-epoxysuccinamoyl-DAP (NβSSEpSmDAP) onto HS-DdaD, and the analysis of 18O incorporation into NβEpSmDAP-S-DdaD using 18O2. All reactions were quenched with an equal volume of 25% formic acid and stored at −80 °C prior to analysis.
For all analyses of intact proteins in DdaD-containing reactions, a 1 mm × 150 mm Jupiter C4 column (Phenomenex, 300 Å, 5 μm) was connected in-line with the ESI source (operated at ~5 kV with a capillary temperature of 200–250 °C) for the MS system. A reaction aliquot containing 12–15 μg DdaD was injected for each sample. The 45 min separation gradient used for all RPLC analyses is shown in Table S2, where solvent A was H2O + 0.1% formic acid and solvent B was MeCN + 0.1% formic acid. The reaction mixture was loaded onto the column using the autosampler and separated according to the gradient shown.
All ionized protein species entering the mass spectrometer were subjected to an MS method with two MS and MS/MS events: 1) full scan measurement of all intact peptides (all ions detected in the ion trap MS in profile mode; m/z range detected: 400–2000), 2) the Ppant ejection assay using NSD (all ions detected in the FTMS in profile mode; resolution: 50,000; m/z range: 250–500; SID = 75 V). All data were analyzed using Qualbrowser (Xcalibur), provided for analysis with the LTQ-FT system. Ppant ejection ions of interest were searched within a range of 0.01 m/z around the isotopic peak of interest, within a tolerance of 5 ppm.
Determination of the Source of the Epoxide Oxygen by Use of 18O2(g)
Phosphopantetheinylation reactions (506 μL) contained 25 μM DdaD, 12 μM Sfp, 400 μM CoA, 1 mM MgCl2, 1.5 mM DTT, and 50 mM HEPES (pH 7.5). Reactions were incubated at room temperature for 1 h. To form NβFmmDAP-S-DdaD, 123 μM NβFmmDAP and 5.5 mM ATP were added (final volume 522 μL) and the reactions incubated an additional 5 min at room temperature.
O2-free solutions (prepared as previously described17) (225 μL) containing NβFmmDAP-S-DdaD and α-KG (0.13 mM) in the absence or presence of DdaC (4.2 μM) were placed in septum-sealed flasks. The flasks were stirred on ice and briefly evacuated to ~30 torr. One flask was subsequently refilled with 1.3 atm of a gas mixture containing 80% 18O2(g) (95–98% isotopic enrichment) and 20% N2(g), and the second flask was refilled with 1.1 atm of natural-abundance O2(g). Fe(NH4)2(SO4)2 and ascorbic acid were subsequently added to the flasks (to concentrations of 21 μM and 1.7 mM, respectively) via a gas-tight syringe. The solutions were stirred for 5 min at 0 °C to allow the reactions to reach completion. The flasks were opened to air, the reactions were terminated by addition of each solution to an equal volume of 25% formic acid, and the acidified mixtures were flash frozen in N2(l).
Nucleotide sequence accession number
The nucleotide sequence of the dapdiamide gene cluster from P. agglomerans strain CU0119 has been deposited in the NCBI GenBank database under accession number HQ130277.
Results
DdaF ligates NβEpSmDAP and Val in an ATP-dependent manner
Guided by the hypothesis that the dapdiamide pathway produces an antibiotic containing a trans-epoxysuccinamate moiety, we synthesized both diastereomers of Nβ-trans-EpSmDAP (see Supporting Information for synthetic protocols and characterization) and tested the activity of DdaF in ligating these compounds to Val.
Anaysis of the enzymatic assays by LC-MS (see Supporting Information for method) revealed that DdaF can catalyze the ligation of both Nβ-trans-EpSmDAP diastereomers to Val to produce the NβEpSmDAP-Val dipeptide antibiotics (Figure S1). A coupled spectrophotometric ADP production assay was used to kinetically characterize the activity of DdaF with respect to the two Nβ-trans-EpSmDAP diastereomers. DdaF was found to use NβRREpSmDAP as a saturable substrate, whereas saturation was not achieved with NβSSEpSmDAP at concentrations up to 590 μM (Table 1, Figure S2). These findings suggested that NβRREpSmDAP may be an on-pathway intermediate, with catalytic efficiency approximately 40-fold greater than the corresponding (S,S)-epoxide.
Table 1.
DdaF kinetics with respect to NβFmmDAP and NβEpSmDAPs. Values were determined by coupled spectrophotometric ADP production assay with a fixed Val concentration of 5 mM.ii
| Substrate | kcat (min−1) | Km (μM) | kcat/Km (min−1·mM−1) |
|---|---|---|---|
| NβFmmDAP | 21 ± 3 | 72 ± 34 | 290 |
| NβRREpSmDAP | 181 ± 7 | 53 ± 7 | 3400 |
| NβSSEpSmDAP* | ND | ND | 82 ± 8 |
Saturation not achieved at concentrations up to 590 μM.
ND = not determined
Expression and purification of DdaC and DdaD in E. coli
The ddaC and ddaD genes were amplified from pUC19 A10A, a plasmid containing the dapdiamide gene cluster,1 and cloned into an expression vector encoding an N-terminal His6 tag for DdaC or a C-terminal His6 tag for DdaD. The proteins were overexpressed in E. coli BL21(DE3) and purified by Ni-NTA affinity chromatography (See Supporting Information for methods, Figure S3). Yields ranged from 4 to 6 mg/L for DdaC and 11 to 14 mg/L for DdaD.
Following Ni-NTA chromatography, DdaC was either flash frozen to yield an aerobic preparation or gel filtered into an anaerobic atmosphere. The anaerobic preparations were incubated with Fe(NH4)2(SO4)2, α-KG, and DTT and subsequently desalted, giving a preparation with an iron occupancy of 47 ± 4% (average ± standard deviation, data from two independent experiments) by ferene spectrophotometric assay.18
DdaD activates and covalently tethers NβFmmDAP
The substrate specificity of the DdaD adenylation (A) domain was probed using ATP-[32P]PPi exchange assays,19 which revealed that NβFmmDAP is the preferred substrate (Figure 2). The kinetics of NβFmmDAP adenylation by DdaD were determined by this assay; the Km is 420 ± 80 μM and the kcat is 64 ± 5 min−1 (Figure S4). DdaD was also active with NβSSEpSmDAP but to a much lesser extent, with a kcat/Km of 2.3 min−1mM−1 compared with 150 min−1mM−1 for NβFmmDAP (Figure S4). No exchange was observed with NβRREpSmDAP.
Figure 2.

DdaD ATP-[32P]PPi exchange data. In these experiments, DdaD, [32P]PPi, unlabeled ATP, and substrate were incubated and then ATP separated from PPi by its specific adsorption to charcoal. The graph shows counts per minute arising from scintillography of the charcoal. (A) Adenylation activity with a variety of potential substrates. Reactions were quenched after 10 min. (B) Timecourse with NFmmDAPs, NβEpSmDAPs, and dapdiamide A.
The competence of the DdaD T domain to be phosphopantetheinylated (PPTated) was initially validated by the observation that it could be modified by the promiscuous Bacillus subtilis phosphopantetheinyl transferase (PPTase) Sfp15,16 with BODIPY-CoA14 to produce a fluorescent band during SDS-PAGE analysis (Figure S5).
Next, we turned to FTMS and the Ppant ejection assay20,21 (Scheme S1) to further characterize intermediates tethered to DdaD. The experiments were carried out in one of two fashions. For some analyses, enzymatic incubations were subjected to trypsin digestion, and tryptic peptides were separated by RPLC coupled directly to a hybrid linear ion trap-FTMS system (ThermoFisher Scientific LTQ-FT), allowing determination of the masses of intact peptides, identification of the DdaD active site peptide (containing the site of serine (Ser) phosphopantetheinylation (PPTation)), and observation of Ppant ejection products. In other experiments, undigested reactions were subjected to RPLC-MS and Ppant ejection was observed directly from intact DdaD.
During LC-MS analysis of a tryptic digest of apo-DdaD incubated with Sfp and CoA, we observed a thiolation (T) domain tryptic peptide that underwent the expected 340.09 Da mass shift for PPTation (Figure 3A-B, Table S3). The PPTated peptide was subjected to two different types of MS/MS fragmentation; NSD resulted in the formation of the predicted small molecule Ppant ejection product (a 1+ ion at m/z 261.127) (Figure 3C, Table S3), and CID resulted in the formation of the predicted Ppant ejection peptide marker ions (Figure 3D). These are the species corresponding to apo-DdaD – 18 Da, from the formation of dehydroalanine (Dha) at the active site Ser (brown in Figure 3D; 2+ ion at m/z 1451.6994, with an intact mass of 2901.3828 Da, an error of -7 ppm from the theoretical mass), and to apo-DdaD + 80 Da, from the retention of the phosphate moiety on the active site Ser during loss of the small molecule ejection ion with m/z 261.127 (tan in Figure 3D; 2+ ion at m/z 1500.7010, with an intact mass of 2999.386 Da, an error of 1 ppm from the theoretical mass). Localization of the site of Ppant modification was achieved by CID MS/MS fragmentation of the PPTated T domain active site tryptic peptide. This analysis demonstrated that Ser533, which aligns with known T domain active site Ser residues, is indeed the site of PPTation (Figure S7).
Figure 3.

Apo-DdaD is converted to holo-DdaD (HS-DdaD) in the presence of Sfp and CoA. In the presence of ATP and NβFmmDAP, HS-DdaD is loaded with NβFmmDAP to form NβFmmDAP-S-DdaD. (A) In the presence of Sfp but the absence of CoA, only the active site peptide from apo-DdaD (green) is observed. (B) In the presence of Sfp and CoA, the active site peptide from HS-DdaD (red) is observed. This peptide shows a mass shift of +340.09 Da from the apo-DdaD active site peptide, the exact mass shift expected for PPTation. (C) The small molecule Ppant ejection ion (red) is observed from the HS-DdaD active site peptide when the peptide is subjected to MS/MS by NSD. (D) Both predicted Ppant ejection peptide marker ions are observed when the HS-DdaD active site peptide is subjected to MS/MS using CID. (E) In the presence of ATP but the absence of NβFmmDAP, only the HS-DdaD active site peptide (red) is observed. (F) In the presence of both ATP and NβFmmDAP, both the HS-DdaD (red) and NβFmmDAP-S-DdaD (blue) active site peptides are observed. (G) When the HS-DdaD and NβFmmDAP-S-DdaD active site peptides are subjected to MS/MS using NSD, both expected small molecule Ppant ejection ions are observed. The HS-DdaD Ppant ejection ion is shown in red and the NβFmmDAP-S-DdaD Ppant ejection is shown in blue. For the predicted structures of the Ppant ejection ions, see Figure S6.
When DdaD, ATP, and NβFmmDAP were incubated, we observed loading of the substrate onto the T domain both by the mass shift of the intact peptide (a mass shift of +183.066 Da from the HS-DdaD active site peptide, where the theoretical mass shift is +183.064 Da) and Ppant ejection to produce an ion at m/z 444.2 (Figure 3E-3G, Table S3). Additional confirmation that NβFmmDAP is covalently tethered to the DdaD T domain came from LC-MS analysis of the NβFmmDAP compound following nonenzymatic hydrolysis from the T domain (Figure S8). Using LC-MS and the Ppant ejection assay to analyze intact DdaD, we also found that DdaD is capable of loading both diastereomers of Nβ-trans-EpSmDAP (Figure S9). We did not observe any loading of dapdiamide A (Figure S10).
DdaC is an Fe(II)/a-KG-dependent epoxidase
When DdaC was added to reaction mixtures containing NβFmmDAP-S-DdaD and a-KG, a +16 Da mass shift in both the intact mass of the T domain active site tryptic peptide and small molecule Ppant ejection ion (to yield an ion at m/z 460.2) was observed (Figure 4, Table S3). This mass shift was observed following two types of enzymatic incubations: 1) reaction mixtures containing DdaC that had been incubated anaerobically with Fe(NH4)2(SO4)2 and a-KG, then desalted (Figure 4) and 2) reaction mixtures containing aerobically purified DdaC to which excess Fe(NH4)2(SO4)2 was added (Figure S11). No qualitative differences in the activities of the two enzyme preparations were observed, so the latter method was used for the remaining experiments. No +16 Da mass shift was observed in DdaC incubations that lacked α-KG (Figure S12). In contrast to the activity of DdaC on NβFmmDAP-S-DdaD, we have observed no evidence for DdaC oxidation of free NβFmmDAP by LC-MS or HPLC assay.
Figure 4.
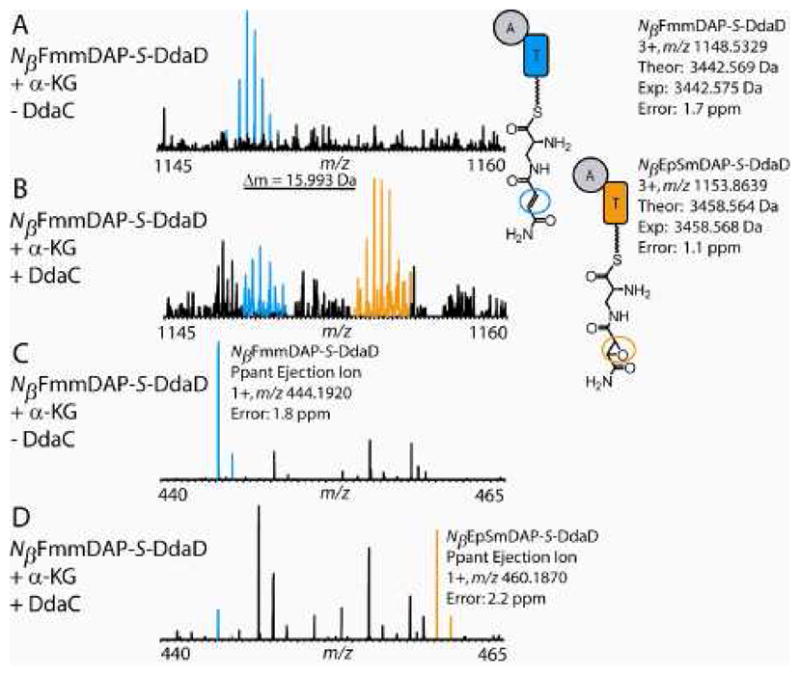
In the presence of both α-KG and DdaC, NβFmmDAP-S-DdaD is converted to NβEpSmDAP-S-DdaD. (A) In the presence of α-KG but the absence of DdaC, only the active site peptide from NβFmmDAP-S-DdaD (blue) is observed. (B) In the presence of both α-KG and DdaC (prepared anaerobically), NβFmmDAP-S-DdaD is converted to NβEpSmDAP-S-DdaD and the active site peptide from NβEpSmDAP-S-DdaD (orange) is observed. The theoretical mass shift of this transformation is 15.995 Da. (C) In the presence of α-KG and the absence of DdaC, only the small molecule Ppant ejection ion from NβFmmDAP-S-DdaD (blue) is observed when the peptides are subjected to MS/MS using NSD. (D) In the presence of both α-KG and DdaC (prepared anaerobically), the Ppant ejection ions from both NβFmmDAP-S-DdaD and NβEpSmDAP-S-DdaD (orange) are observed when the peptides are subjected to MS/MS using NSD. For the predicted structures of the Ppant ejection ions, see Figure S6.
To confirm that the oxidation catalyzed by DdaC was indeed an epoxidation, we compared the MSn fragmentation patterns of the Ppant ejection ion generated from HS-DdaD loaded with synthetic Nβ-trans-EpSmDAP (both diastereomers) and the Ppant ejection ion generated from NβFmmDAP-S-DdaD incubated with DdaC. Fragment ions for MSn analysis were selected that reported a +16 Da shift when compared to similar analyses conducted on the Ppant ejection ion from NβFmmDAP-S-DdaD. In all cases, the fragmentation patterns produced were the same for all epoxide-containing species, confirming the DdaC-catalyzed oxidation as an epoxidation (Figure S13).
To determine the origin of the putative incorporated oxygen, DdaC incubations were carried out either in H218O or under an 18O2 atmosphere. When the reaction was conducted in H218O and then analyzed by trypsin digest followed by LC-MS, a +16 Da mass shift was observed in the DdaD tryptic active site peptide and the corresponding Ppant ejection ion. If the oxygen atom was derived from water, a +18 Da mass shift would be observed (Figure S14). In contrast, incubations under 18O2 followed by LC-MS and Ppant ejection analysis of intact DdaD resulted in a +18 Da (as opposed to a +16 Da mass shift in a 16O2 atmosphere) mass shift in the observed NβEpSmDAP-S-DdaD Ppant ejection ion, indicating that DdaC uses O2 as a co-substrate (Figure 5, Table S3).
Figure 5.
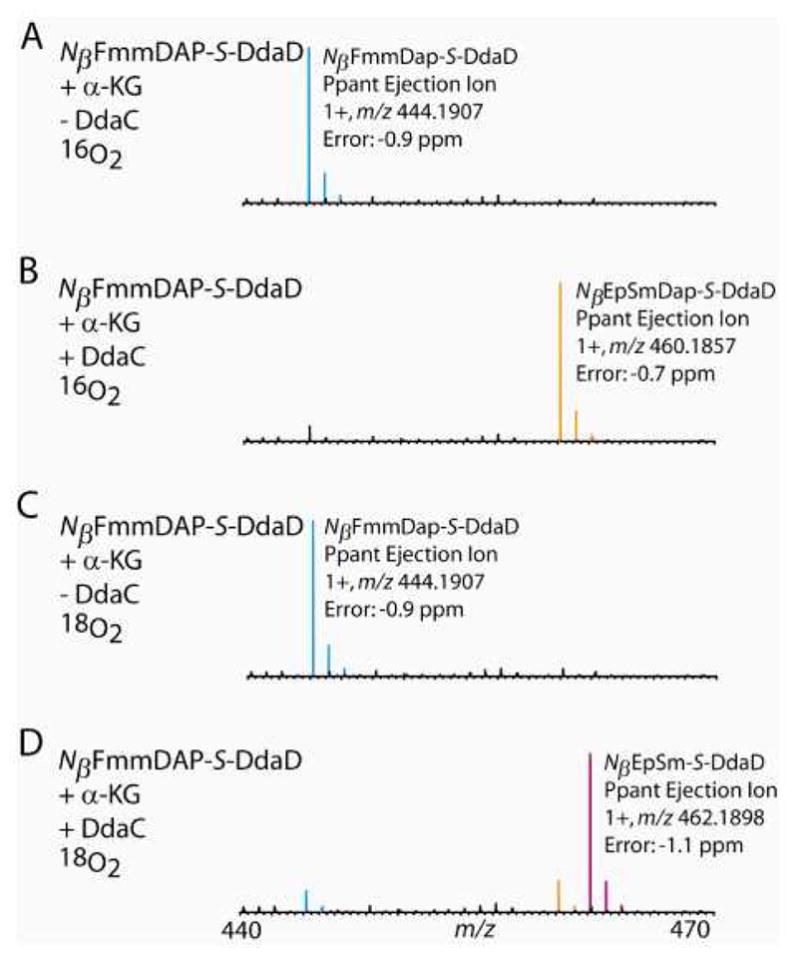
In an atmosphere of 18O2, 18O is incorporated into the substrate loaded onto DdaD and NβFmmDAP is converted to Nβ-trans-EpSmDAP. (A) Under normal reaction conditions (an atmosphere of 16O2), in the presence of α-KG and the absence of DdaC, the only Ppant ejection ion observed (blue) during Ppant ejection analysis (using NSD) of NβFmmDAP-S-DdaD corresponds to the unoxidized starting substrate. (B) Under normal reaction conditions (an atmosphere of 16O2), in the presence of both α-KG and DdaC, Ppant ejection ions corresponding to both NβFmmDAP-S-DdaD and NβEpSmDAP-S-DdaD (orange) are observed. (C) Under an atmosphere of 18O2, in the presence of α-KG but the absence of DdaC, the only Ppant ejection ion observed corresponds to NβFmmDAP-S-DdaD. (D) Under an atmosphere of 18O2, in the presence of both α-KG and DdaC, Ppant ejection ions are observed for both NβFmmDAP-S-DdaD and NβEpSmDAP-S-DdaD. A +2 Da mass shift (from the NβEpSmDAP-S-DdaD Ppant ejection ion in (B)) is observed in the Ppant ejection ion corresponding to NβEpSmDAP-S-DdaD (purple), indicating that the oxygen in the epoxide originates from molecular oxygen. For the predicted structures of the Ppant ejection ions see Figure S6.
Discussion
The dapdiamides comprise a family of acylated dipeptide natural antibiotics that likely inhibit the glucosamine-6-phosphate synthase enzyme of susceptible organisms and consequently disrupt cell wall assembly and integrity.1 The putative target has an active site Cys that can be covalently captured by the warheads of the dapdiamides and related scaffolds.2 Most commonly the electrophilic warhead is the eneamide moiety provided by the fumaramoyl group of this antibiotic class (e.g. dapdiamides A-D) which can be a Michael acceptor for the Cys thiolate. An alternative electrophile is found in the α-epoxysuccinamoyl-containing dapdiamide E and in the corresponding epoxysuccinamoyl β regioisomer NβEpSmDAP-Val (Figure 1C), in which the olefin of the fumaramoyl moiety is replaced with an epoxide that is proposed to be opened by the active site Cys residue. The presumption has been that the fumar(am)oyl group gets epoxidized to the epoxysuccin(am)oyl group to create this second potential electrophile; the timing and the identity of the catalysts responsible for this conversion are the subject of this study.
Regiochemical variation is observed among the dapdiamides with regard to which amino group of 2 (α), 3 (β)-DAP the fumaramoyl/epoxysuccinamoyl moities are attached in amide linkage. Dapdiamides A-C have an NβFmmDAP attachment while dapdiamide D has an NαFmmDAP linkage. Dapdiamide E, with an epoxysuccinamoyl group in α amide linkage, could in principle arise from epoxidation of dapdiamide D or its likely precursor amino acids NαFmDAP or NαFmmDAP. The natural product NβEpSmDAP-Val could arise from comparable epoxidation of dapdiamide A or NβFm(m)DAP. In this work we demonstrate that it is NβFmmDAP that is the substrate for T-domain loading and then epoxidation.
In our prior study of the biosynthetic pathway for this antibiotic family, we demonstrated that DdaG and DdaF are the two ATP-cleaving amide-forming ligases that are responsible for assembling the Fmm-dipeptide scaffold. DdaG uses ATP to activate fumarate to fumaroyl-AMP on the way to making NβFmDAP. DdaF then makes the second amide bond, but only after the acid of FmDAP has been converted enzymatically to the amide in FmmDAP. DdaF is a member of the ATP grasp family,22 and as such cleaves ATP to ADP and Pi, presumably making the FmmDAP-phosphate mixed anhydride as an activated intermediate. To date only dapdiamide enzymes that form and utilize Nβ-acyl-DAP species have been characterized; the enzyme creating the NαFmmDAP regiochemistry has not yet been identified.
In this study, we assayed both synthetic Nβ-trans-EpSmDAP diastereomers and validated that they are substrates for DdaF in the presence of Val and ATP to produce the NβEpSmDAP-Val dipeptide. NβEpSmDAP-Val is the natural product (of as yet unassigned epoxide stereochemistry) recently identified in P. agglomerans strains (48b/90, C9-1, and 39b/90) as well as a strain of Serratia plymuthica.5,6 We observed selectivity of DdaF for NβRREpSmDAP over the (S,S) diastereomer, suggesting that the dapdiamide pathway produces a natural product with (R,R) epoxide stereochemistry. The precedence of (R,R) epoxide stereochemistry in the related natural product Sch37137 is in accord with this hypothesis.8
Sequence analysis of the Dda gene cluster suggested that in addition to DdaG and DdaF, one more ORF should be capable of using ATP to activate an acid cosubstrate, namely DdaD. This protein is predicted to be a member of a third family of ATP-cleaving enzymes, and contains two domains which comprise a minimal NRPS module. The first domain of approximately 50 kDa is predicted to be an A domain while the second of 10 kDa should be a T domain that can be posttranslationally primed with a Ppant group. The adjacent ORF DdaE, encoding a predicted thioesterase, is the only other NRPS-related ORF in the dapdiamide biosynthetic gene cluster, suggesting the adenylation-thiolation (A-T) didomain DdaD acts as a stand-alone module and not as part of a classical NRPS assembly line.
We have shown in several other contexts that stand-alone A-T didomains (or isolated T domains) are used in bacterial metabolism to sequester some fraction of an amino acid pool, tethered as the aminoacyl-S-pantetheinyl T domain thioester. The tethered aminoacyl-S-T domain is then subjected to covalent modification, which is often oxidative. Thus, in the biosynthesis of the vancomycin class of glycopeptides23,24 and the nikkomycin25 and aminocoumarin26 classes of antibiotics, Cβ hydroxylation of the sequestered aminoacyl thioester moiety is effected by either heme iron or nonheme iron oxygenases. In the biogenesis of syringomycin, chlorination occurs at C4 of a tethered threonyl moiety,27 while in the assembly of the jasmonate phytohormone mimic coronatine, cryptic chlorination occurs at Cγ of the allo-Ile moiety prior to subsequent ring closure to the cyclopropane.28,29
With such precedents, we sought an analogous role for modification of an aminoacyl thioester covalently attached to DdaD. The adjacent ORF DdaC has homology to the Fe(II)/α-KG-dependent dioxygenase family of enzymes, which typically catalyze O2-dependent substrate hydroxylations.12 Examples of members of this family include the syringomycin biosynthetic enzyme SyrP30 and the kutzneride pathway enzymes KtzO and P,31 which carry out β-hydroxylations of T-domain tethered aspartate and glutamate, respectively. In addition to hydroxylations, members of this family have been shown to carry out a range of other oxidative transformations.12 Evidence from bioconversion and cell extract studies has implicated Fe(II)/α-KG enzymes in epoxidation reactions,32,33 but to our knowledge no in vitro characterization of a purified epoxidase in this class has been reported previously.
In the context of the known dapdiamide family members (Figure 1A) it seemed likely that DdaC could be an epoxidase that acts on the fumaroyl/fumaramoyl moiety of an intermediate tethered in thioester linkage to the T domain of DdaD. In addition, DdaE is a predicted thioesterase, thus the tandem action of DdaD, C, and E could be a branch pathway for selection and activation of an olefin-containing pathway intermediate, epoxidation, and then hydrolysis to produce an epoxysuccin(am)oyl building block for condensation with another monomer via DdaG and/or DdaF. (We have not been able to heterologously express DdaE in a soluble form in E. coli to establish such a thioesterase role.)
Validation of the proposed roles for DdaD and DdaC started with determination of the selectivity of the A domain of DdaD. Using the classical ATP-[32P]PPi exchange assay, diagnostic for reversible formation of tightly held (amino)acyl-AMPs in enzyme active sites, DdaD showed clear preference for NβFmmDAP. The Km value for DdaD with respect to NβFmmDAP was found to be 420 μM, comparable to Km values reported for other NRPS A domains.28,34,35 These results provided a key early insight: DdaD is indeed selecting an olefin-containing pathway intermediate for activation as the AMP mixed anhydride. This was strongly suggestive that the fumaramoyl moiety of thioester-tethered FmmDAP would be the species epoxidized.
To validate the second step of A-T didomain function, the predicted covalent loading of NβFmmDAP-AMP onto the Ppant arm of the T domain of DdaD, we turned to mass spectrometry. We found that apo-DdaD could be posttranslationally converted to the Ppant-containing holo-DdaD by action of purified Sfp. Incubation of holo-DdaD with ATP and NβFmmDAP allowed detection of the NβFmmDAP-S-Ppant adduct in the T domain by peptide mass analysis and by the release of the NβFmmDAP-S-Ppant thioester fragment ion. Thus, the second step of A-T didomain function, the covalent tethering of the substrate activated by the A domain, was operant.
When DdaC was incubated with the covalent NβFmmDAP-S-DdaD enzyme intermediate, a mass increase of +16 Da was observed for both the T domain active site tryptic peptide containing the tethered acyl-DAP thioester and in the ejected Ppant ion. We found that, as anticipated for a member of the Fe(II)/α-KG family, the activity of DdaC is dependent on α-KG. Additionally, incubation under 18O2(g) resulted in a +18 Da mass shift, demonstrating that DdaC uses molecular oxygen as a cosubstrate.
The ejected pantetheinyl fragment from DdaCD experiments had the M+16 Da mass increase anticipated for the epoxide product. However, it was formally possible that the introduction of one oxygen atom into the FmmDAP moiety arose not by epoxidation of the double bond but by C- or N-hydroxylation of the DAP residue. MSn fragmentation of Ppant ejection ions from both HS-DdaD loaded with authentic Nβ-trans-EpSmDAP and from NβFmmDAP-S-DdaD incubated with DdaC resulted in the same fragmentation pattern, suggesting that DdaC indeed acts as an epoxidase.
Our studies of DdaC have generated a number of questions to be answered in future investigations. We have not attempted to determine single turnover kinetics of the enzyme because of the difficulty of quantifying its substrate, the covalent N-acyl-aminoacyl thioester adduct of DdaD. We have also been unable to obtain sufficient NβEpSmDAP from DdaCD incubations to directly determine the stereochemistry of the epoxide carbons in the product, nor have we yet evaluated the epoxidation mechanism. In analogy to proposed mechanisms for Fe(II)/α-KG hydroxylases, an Fe(IV)-oxo intermediate is the likely oxygen transfer species. But whether C-O bond formations are stepwise and ionic or radical, as suggested in Scheme S2, is yet to be probed.
Additionally, the question arises why P. agglomerans makes both the enamide electrophile (fumaramoyl) and the epoxide electrophile (epoxysuccinamoyl) as parallel N-acyl warheads in this antibiotic family. Two future studies will compare the epoxysuccinamoyl versus the fumaramoyl groups. First, minimum inhibitory concentration (MIC) determinations of the Nβ molecules dapdiamide A and NβEpSmDAP-Val will test for any differences in uptake by susceptible bacteria and fungi. Once taken up by the oligopeptide permease systems, intracellular proteases are thought to liberate the Nβ-acyl-DAPs as the proximal inactivators for glucosamine synthase. Thus, it will be useful to compare FmmDAP and EpSmDAP side by side against the target enzyme to determine inactivation efficiencies. It is possible that the epoxide warhead is more selective than the enamide: the epoxide may require acid catalysis in the enzyme active site for covalent capture whereas Michael addition to the fumaramoyl moiety may not. In that context a proteomics36 study to evaluate how many proteins in a susceptible cell are targeted covalently would offer a global comparison of “off-target” labeling by the two types of electrophilic N-acyl warheads.
Supplementary Material
Scheme 1.

Proposed role of DdaC-F in the formation of NβEpSmDAP-Val.iii
Acknowledgments
We thank Emily Balskus, Christopher Neumann, Elizabeth Sattely, and Albert Bowers for helpful discussions. We thank John Heemstra for providing synthetic BODIPY-CoA, Elizabeth Sattely for providing Sfp, and Jessica Dawlaty for providing synthetic dapdiamide A and the pUC19 A10A plasmid. This work was supported in part by NIH Grant GM 20011 (C.T.W.), NIH Medical Scientist Training Program GM 07753 (M.A.H.), NIH Grant GM 067725-08 (N.L.K), NSF Grant MCB-642058 (J.M.B.) and a fellowship from the American Chemical Society Division of Analytical Chemistry (S.B.B).
Footnotes
This compound has been referred to in the literature variously as CB-25-I, 2-amino-3-(oxirane-2,3-dicarboxamido)-propanoyl-valine, and herbicolin I.
Data are presented as value ± standard deviation. ND = not determined.
The grayed-out step has not been biochemically validated.
Supporting Information Available: Supplemental materials and methods, Tables S1-S5, Figures S1-S14, and Schemes S1-S2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dawlaty J, Zhang X, Fischbach MA, Clardy J. J Nat Prod. 2010;73:441–446. doi: 10.1021/np900685z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kucharczyk N, Denisot MA, Le Goffic F, Badet B. Biochemistry. 1990;29:3668–3676. doi: 10.1021/bi00467a012. [DOI] [PubMed] [Google Scholar]
- 3.Milewski S, Andruszkiewicz R, Kasprzak L, Mazerski J, Mignini F, Borowski E. Antimicrob Agents Chemother. 1991;35:36–43. doi: 10.1128/aac.35.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollenhorst MA, Clardy J, Walsh CT. Biochemistry. 2009;48:10467–10472. doi: 10.1021/bi9013165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shoji J, Hinoo H, Sakazaki R, Kato T, Hattori T, Matsumoto K, Tawara K, Kikuchi J, Terui Y. J Antibiot. 1989;42:869–874. doi: 10.7164/antibiotics.42.869. [DOI] [PubMed] [Google Scholar]
- 6.Sammer UF, Volksch B, Mollmann U, Schmidtke M, Spiteller P, Spiteller M, Spiteller D. Appl Environ Microbiol. 2009;75:7710–7717. doi: 10.1128/AEM.01244-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper R, Horan AC, Gentile F, Gullo V, Loebenberg D, Marquez J, Patel M, Puar MS, Truumees I. Journal of Antibiotics. 1988;41:13–19. doi: 10.7164/antibiotics.41.13. [DOI] [PubMed] [Google Scholar]
- 8.Rane DF, Girijavallabhan VM, Ganguly AK, Pike RE, Saksena AK, McPhail AT. Tetrahedron Lett. 1993;34:3201–3204. [Google Scholar]
- 9.Martin WR, Foster JW. J Bacteriol. 1955;70:405–414. doi: 10.1128/jb.70.4.405-414.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanada K, Tamai M, Ohmura S, Sawada J, Seki T, Tanaka I. Agric Biol Chem. 1978;42:529–536. [Google Scholar]
- 11.Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 12.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21 – 68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 13.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 14.La Clair JJ, Foley TL, Schegg TR, Regan CM, Burkart MD. Chem Biol. 2004;11:195–201. doi: 10.1016/j.chembiol.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 15.Quadri LEN, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 16.Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, Reid R, Khosla C, Walsh CT. Chem Biol. 1996;3:923–936. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 17.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 18.Hennessy DJ, Reid GR, Smith FE, Thompson SL. Can J Chem. 1984;62:721–724. [Google Scholar]
- 19.Lee SG, Lipmann F. Methods Enzymol. 1975;43:585–602. doi: 10.1016/0076-6879(75)43121-4. [DOI] [PubMed] [Google Scholar]
- 20.Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Biochemistry. 2006;45:12756–12766. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bumpus SB, Kelleher NL. Curr Opin Chem Biol. 2008;12:475–482. doi: 10.1016/j.cbpa.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galperin MY, Koonin EV. Protein Sci. 1997;6:2639–2643. doi: 10.1002/pro.5560061218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H, Thomas MG, O’Connor SE, Hubbard BK, Burkart MD, Walsh CT. Biochemistry. 2001;40:11651–11659. doi: 10.1021/bi0115434. [DOI] [PubMed] [Google Scholar]
- 24.Hubbard BK, Walsh CT. Angew Chem, Int Ed. 2003;42:730–765. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- 25.Chen H, Hubbard BK, O’Connor SE, Walsh CT. Chem Biol. 2002;9:103–112. doi: 10.1016/s1074-5521(02)00090-x. [DOI] [PubMed] [Google Scholar]
- 26.Chen H, Walsh CT. Chem Biol. 2001;8:301–312. doi: 10.1016/s1074-5521(01)00009-6. [DOI] [PubMed] [Google Scholar]
- 27.Vaillancourt FH, Yin J, Walsh CT. Proc Natl Acad Sci U S A. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Couch R, O’Connor SE, Seidle H, Walsh CT, Parry R. J Bacteriol. 2004;186:35–42. doi: 10.1128/JB.186.1.35-42.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 30.Singh GM, Fortin P, Koglin A, Walsh CT. Biochemistry. 2008;47:11310–11320. doi: 10.1021/bi801322z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strieker M, Nolan EM, Walsh CT, Marahiel MA. J Am Chem Soc. 2009;131:13523–13530. doi: 10.1021/ja9054417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe M, Sumida N, Murakami S, Anzai H, Thompson CJ, Tateno Y, Murakami T. Appl Environ Microbiol. 1999;65:1036–1044. doi: 10.1128/aem.65.3.1036-1044.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hashimoto T, Matsuda J, Yamada Y. FEBS Lett. 1993;329:35–39. doi: 10.1016/0014-5793(93)80187-y. [DOI] [PubMed] [Google Scholar]
- 34.Mootz HD, Marahiel MA. J Bacteriol. 1997;179:6843–6850. doi: 10.1128/jb.179.21.6843-6850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehmann DE, Shaw-Reid CA, Losey HC, Walsh CT. Proc Natl Acad Sci U S A. 2000;97:2509–2514. doi: 10.1073/pnas.040572897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han X, Aslanian A, Yates JR., III Curr Opin Chem Biol. 2008;12:483–490. doi: 10.1016/j.cbpa.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.