

Abstract
Five studies show that disabling p53, an essential tumour-suppressor protein, improves the efficiency of stem-cell production. Are these results a ‘heads up’ that cancer cells and stem cells are disturbingly similar?
Mutations that inactivate the p53 tumour-suppressor-protein network occur in most human cancers and, consequently, the roles and regulation of p53 activity in tumour formation are the topic of intense research. Inherited germline mutations in the p53 gene promote cancer in mice and humans, and p53 loss interacts with various mutant genes to transform normal cells into tumour cells. p53 is a stress-response protein, which suppresses tumour formation by triggering programmed cell death (apoptosis); by activating cell-cycle checkpoints that prevent damaged cells from proliferating; or by promoting senescence (permanent cell-cycle arrest). Thus, inactivation of p53 facilitates the expansion of aberrant cells and leads to rampant genome instability. Five papers1-5 published online in Nature describe how disruption of the p53 network also enhances the production of induced pluripotent stem (iPS) cells. Although these observations catapult p53 into the centre of stem-cell research, time will tell whether they represent a promise or a warning.
iPS cells were first produced three years ago6 by the enforced expression of genes encoding four transcription factors (c-Myc, Klf4, Sox2 and Oct4) in mouse fibroblast cells. These cells have the same capabilities as embryonic stem (ES) cells isolated from early mammalian embryos — they can self-renew, and are able to give rise to all tissue types of the body. ES cells hold promise as a potential treatment for various diseases; and because iPS cells can be produced from adult cells from any individual, the availability of these cells can, in principle, solve the ethical issues associated with ES-cell use and may also circumvent the need for immunologically matched cell donors. Still, despite much enthusiasm, it is not known whether iPS cells will be an effective treatment for human diseases. In fact, even embryo-derived ES cells have been tested in only a few settings, and their effectiveness and safety are not well established.
Methods for producing iPS cells have so far been inefficient. They initially involved expression of the tumour-promoting oncogene c-myc, and random insertion of foreign DNA, including DNA from viruses used as vectors, into the recipient genome — techniques that could lead to cancer. Subsequently, in a flurry of activity, researchers identified ways to produce iPS cells without c-myc7, devoid of viral integration8, or using only proteins for reprogramming9. Indeed, the ‘factorology’ of iPS-cell production is all the rage, reminiscent of the early days of cancer-gene discovery, when researchers raced to identify the first genes and gene combinations that could convert normal cells to an immortal, cancerous state.
The new studies1-5 add to the frenzy by decisively showing that inactivation of p53 markedly increases the efficiency of iPS-cell production. Furthermore, Kawamura et al.3 show that p53 deficiency simplifies iPS generation by enabling production of iPS cells with only two factors, Oct4 and Sox2. Also, three groups1-3 show that p53-deficient iPS cells can give rise to adult tissues when implanted into mouse embryos.
As p53 inactivation promotes genome instability and cancer, the risks of producing iPS cells that lack this essential tumour suppressor may outweigh its benefits. In line with this, Marion et al.5 reveal that p53-deficient iPS cells are genomically unstable and are not ‘fit’ enough to efficiently produce mice. And even when mice can be generated partly from iPS cells as in the work by Hong and colleagues1, they eventually develop tumours. In addressing such safety concerns, Utikal et al.4 demonstrate that transient, rather than permanent, inhibition of p53 also enhances reprogramming efficiency. Still, for p53-suppressed iPS cells to be used therapeutically, it must be shown that the reconstituted tissues function normally and remain tumour free. Moreover, next-generation sequencing and other genomic technologies must be used to demonstrate that these cells do not acquire deleterious mutations.
Beyond its therapeutic implications, this body of work1-5 presents overlapping but contrasting views of the mechanism by which p53 limits reprogramming and, in particular, is inconclusive as to whether interaction between the p53 network and reprogramming pathways is direct or indirect. Li et al.2 show that the Ink4a/Arf locus, which encodes a cell-cycle inhibitor (p16Ink4a) and an indirect p53 activator (p19Arf), is silenced during iPS reprogramming. They argue that this silencing occurs early in reprogramming, implying a direct effect of the reprogramming factors on this locus. There is, however, no consensus about the timing of downregulation of Ink4a/Arf expression during reprogramming2,4.
Other observations3,4 point to an indirect interaction between reprogramming factors and p53 through activation of a p53-mediated stress response (for instance, apoptosis or senescence). Accordingly, three groups1-3 provide evidence that one key p53 effector in the process is the cell-cycle inhibitor p21. Indeed, Gil and colleagues10 suggest that senescence represents the primary barrier to reprogramming. It is well established that cells with an intact p53 network are prone to senescence in culture11, and perhaps this alone explains why normal cells are more difficult to reprogram. Accordingly, Utikal et al.4 show that spontaneously immortalized cells exhibiting unrestricted growth in culture, with or without obvious p53 impairment, are readily reprogrammed into iPS cells.
At face value, the results of these studies are reminiscent of work12, published 25 years ago, showing that loss of p53 facilitates cellular immortalization — a state of end-less self-renewal that is one of the first steps towards cancer. And more recently, p53 has been implicated as a factor that limits the self-renewal capacity of certain stem cells13,14. Even in the iPS field, previous work had shown that the SV40 T antigen — an immortalizing oncogenic protein that disables p53 — or transient inhibition of p53 using small interfering RNAs, enhance reprogramming efficiency15,16. The current studies substantially extend and expand on these findings, and provide new platforms for more effectively studying reprogramming.
Just as the race to find new reprogramming factors is reminiscent of the hunt for cooperating oncogenes, the remarkable similarities between the reprogramming processes and oncogenic transformation may provide insights into cancer (Fig. 1). Indeed, both processes require specific combinations of collaborating genes that can produce a less differentiated cell able to proliferate and self-renew indefinitely. All four factors initially shown to reprogram cells are overexpressed in at least some types of tumour, and at least two of them — c-myc and Klf4 — are established oncogenes. Now we find that p53 — a tumour suppressor whose loss greatly increases the efficiency of oncogene cooperation in transforming normal cells to tumour cells17 — affects reprogramming similarly. Notably, a gold-standard assay for the formation of iPS cells is in fact a tumorigenesis assay that measures their ability to form germ-cell tumours following transplantation into mice.
Figure 1. Overlapping mechanisms control iPS-cell and cancer-cell production.
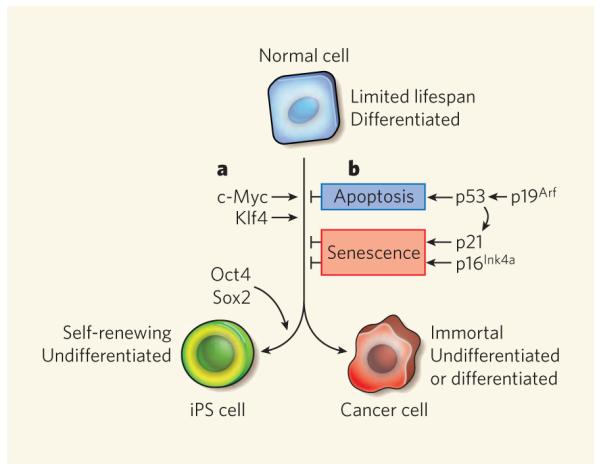
Normal fibroblasts, which are mature, differentiated cells, can be reprogrammed into induced pluripotent stem (iPS) cells or tumour cells by a combination of defined factors. a, The transcription factors c-Myc and Klf4 promote reprogramming of fibroblasts into iPS cells in a manner that conceptually parallels their roles in transformng normal cells into tumour cells. Oct4 and Sox2, although overexpressed in cancers, are currently thought to function specifically to promote iPS-cell formation. b, Conversely, the p53 tumour-suppressor protein, which can be induced by p19Arf, directly or indirectly limits the reprogramming of fibroblasts into iPS cells1-5 or into transformed cancer cells by inducing apoptosis, or cellular senescence through its target protein, the cell-cycle inhibitor p21. Another cell-cycle inhibitor, p16Ink4a, also promotes cellular senescence directly to limit both processes. The Ink4a/Arf locus (not shown), which encodes p19Arf and p16Ink4a, is silenced during iPS reprogramming2,4.
If the processes that lead to the production of iPS cells and tumours overlap, one wonders whether so-called cancer stem cells — cells capable of self-renewal that are considered essential for the propagation of some tumour types — might initially arise through a reprogramming-like mechanism. Moreover, not all of the factors required to trigger reprogramming of cells to iPS cells are necessary for their maintenance8,9. If cancer arises through reprogramming-like processes, then perhaps many of the oncogenes that initiate tumour formation might be dispensable for tumour progression and, hence, be poor targets for new cancer therapies. If this proves to be the case, further studies into reprogramming might eventually point towards new treatments for cancers as well.
References
- 1.Hong H, et al. Nature. 2009 doi:10.1038/nature08235. [Google Scholar]
- 2.Li H, et al. Nature. 2009 doi:10.1038/nature08290. [Google Scholar]
- 3.Kawamura T, et al. Nature. 2009 doi:10.1038/nature08311. [Google Scholar]
- 4.Utikal J, et al. Nature. 2009 doi:10.1038/nature08285. [Google Scholar]
- 5.Marion R, et al. Nature. 2009 doi:10.1038/nature08287. [Google Scholar]
- 6.Takahashi K, Yamanaka S. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 7.Nakagawa M, et al. Nature Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 8.Okita K, et al. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- 9.Zhou H, et al. Cell Stem Cell. 2009;4:381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banito A, et al. Genes Dev. in the press. [Google Scholar]
- 11.Livingstone LR, et al. Cell. 1992;70:923–935. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- 12.Eliyahu D, Raz A, Gruss P, Givol D, Oren M. Nature. 1984;312:646–649. doi: 10.1038/312646a0. [DOI] [PubMed] [Google Scholar]
- 13.Krizhanovsky V, et al. Cold Spring Harb. Symp. Quant. Biol. 2008;73:513–522. doi: 10.1101/sqb.2008.73.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin T, et al. Nature Cell Biol. 2005;7:165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 15.Mali P, et al. Stem Cells. 2008;26:1998–2005. doi: 10.1634/stemcells.2008-0346. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, et al. Cell Stem Cell. 2008;3:475–479. doi: 10.1016/j.stem.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Lowe SW, et al. Science. 1994;266:807–810. doi: 10.1126/science.7973635. [DOI] [PubMed] [Google Scholar]