


Abstract
Missense mutations within the central DNA binding region of p53 are the most prevalent mutations found in human cancer. Numerous studies indicate that ‘hot-spot’ p53 mutants (which comprise ∼30% of human p53 gene mutations) are largely devoid of transcriptional activity. However, a growing body of evidence indicates that some non-hot-spot p53 mutants retain some degree of transcriptional activity in vivo, particularly against strong p53 binding sites. We have modified a previously described yeast-based p53 functional assay to readily identify such partial loss of function p53 mutants. We demonstrate the utility of this modified p53 functional assay using a diverse panel of p53 mutants.
INTRODUCTION
p53 is a transcription factor that regulates the expression of a diverse array of genes involved in growth control. The importance of p53 in controlling cell growth is perhaps best highlighted by the high prevalence of p53 gene mutations in human cancer. The vast majority of p53 gene mutations are missense mutations that occur within the central DNA binding region of the protein (codons 110–307) and disrupt site-specific DNA binding activity (a requisite for p53 transcriptional activity) (1,2). Following a mutational event on one p53 allele the remaining wild-type p53 allele is commonly lost, resulting in loss of heterozygosity. p53 mutations can occur either early or late during tumor progression depending on the tumor type (3).
The six most common p53 missense mutations are commonly referred to as ‘hot-spot’ mutants (1). These mutations occur at codons within the DNA binding region critical for maintaining a correct tertiary conformation, or at direct contact points with DNA (4). Hot-spot mutations severely affect p53 activity in a number of biological assays. However, mutations at the six hot-spot codons together account for <30% of the mutations present in a large database (5). Therefore, most p53 mutations in human cancer occur at non-hot-spot codons and the biological activities of the resulting mutant p53 proteins have not been well characterized. There is evidence to suggest that at least some of these mutations may result in only partial loss of p53 function. For example, p53 with a proline mutation at codon 175 is defective for transactivating the p53-responsive element from the bax promoter but not p21 promoter in transfection studies (6). This is probably due to the relatively weak p53 binding site within the bax promoter (7). A methionine mutation at codon 143 similarly eliminates transcriptional activity against weak, but not strong, p53 binding sites in vivo (8). These studies indicate that not all p53 mutations found in human cancer inhibit p53 activity to the same extent.
Currently, p53 mutations are classified based upon frequency of mutation in human cancer (i.e. hot-spot mutations). A more biological classification based upon degree of loss of transcriptional activity may potentially be more useful but is difficult to employ given the need to individually test each p53 mutant for transcriptional activity in vivo. Iggo and colleagues (9,10) have previously devised a yeast-based functional assay for identifying tumor-derived p53 gene mutations. p53-encoding DNA sequences encompassing the central DNA binding region are amplified through RT–PCR and transferred into a yeast expression plasmid via homologous recombination (gap repair). Yeast containing the repaired p53 expression plasmid are scored for p53 transcriptional activity using a nutritional marker gene under p53 transcriptional control. This assay correctly identifies p53 gene mutations present in tumor samples or tumor cell lines even when p53 mutations are present in only a subset of cells (10,11).
We have modified this assay by employing two marker genes under the transcriptional control of DNA elements with different binding affinities for p53. Utilizing both strong and weak p53 binding elements in the same assay allows for direct selection of yeast expressing mutant p53 as well as detection of residual transcriptional activity in some p53 mutants. Identification and classification of tumor-derived p53 mutants based upon their degree of loss of function may be of clinical value in the future.
MATERIALS AND METHODS
Strains and plasmids
Yeast strain yIG397 with an ADE2 gene open reading frame (ORF) downstream from three copies of a p53-responsive RGC sequence was kindly provided by R.Iggo (ISREC, Epalinges, Switzerland). Yeast strain yCE1128-12C (MATα ade2 his3 leu2 trp1 ura3) was transformed with a HIS3-marked p53-responsive URA3 gene ORF expression plasmid (pRS313-PG-URA) obtained from S.Thiagalingam (Johns Hopkins) yielding strain yGK7. Derivation of yeast strain yCE1116-4C (MATa ade2 his3 leu2 trp1 ura3::URA3::WAF1 promoter element::lacZ) containing an integrated lacZ gene ORF under the transcriptional control of p53-responsive sequences from the human p21 promoter element has been described previously (8). This strain was transformed with shuttle vector pYcDE8 (12) to yield strain yGK13 which grows on tryptophan-deficient media. Strains yCE11128-12C and yCE1116-4C are isogenic with the S288C strain background.
A yeast expression plasmid containing a human wild-type p53 cDNA (pLS76) and a version of this plasmid with an out-of-frame internal deletion in the p53 cDNA (pRDI-22) were obtained from R.Iggo. Plasmids containing mutant p53 cDNA to be used as templates for PCR were derived previously (8).
p53 functional assay
PCR and gap repair were utilized to recombine the various mutant p53 cDNAs into pRDI-22 as described previously (10). Strains yIG397 and yGK7 were transformed using a PEG-based yeast transformation kit (Zymo Reseach) and transformants selected on leucine-deficient media. Several colonies were assayed to ensure the fidelity of the gap repair.
Strain yGK7 transformants were assayed for p53 transcriptional activity by growing yeast to mid-log phase in liquid media and serially diluting onto solid synthetic media lacking the appropriate nutrient and/or supplemented with 5-fluoroorotic acid (FOA; 1 mg/ml). Colony growth was assessed after 3 days at 37°C. Strain yGK7 transformants were also mated with strain yGK13 and diploids (LEU+HIS+TRP+) isolated. Diploids were patched onto sterile filter papers overlying selective solid media and grown for 3 days at 37°C. The filter paper was then freeze–thawed in liquid nitrogen and assayed for β-galactosidase activity using a standard protocol (13).
Strain yIG397 transformants were assayed for p53 transcriptional activity by streaking onto synthetic selective solid media containing limiting amounts of adenine (5 µg/ml). Colony color was assessed after 3 days growth at 37°C.
RESULTS
Previous studies have demonstrated that wild-type p53 is transcriptionally active when expressed in yeast (8–11). We therefore derived a yeast strain in which a URA3 gene ORF is under the transcriptional control of a p53-responsive promoter (PG) element multimer from the human ribosomal gene cluster (RGC) (2; Fig. 1). The PG element is a weak p53 binding sequence both in vitro and in vivo (14,15). As demonstrated in Figure 2, the clonogenic survival of this yeast strain (yGK7) transformed with pLS76 (a yeast single-copy plasmid with a human wild-type p53 cDNA under the transcriptional control of an ADH promoter) is <10% on uracil-deficient media, consistent with weak URA3 expression. However this degree of URA3 expression is enough to sensitize the yeast to the drug FOA which is selectively toxic to URA+ yeast (16). The plating efficiency of strain yGK7 expressing wild-type p53 is decreased by at least four logs in the presence of FOA while the same strain lacking p53 expression is unaffected in its growth (Fig. 2). Use of FOA in this functional assay allows for direct selection of yeast totally lacking p53 activity.
Figure 1.

Comparison of p53-responsive promoter elements. Sequences of the 10 bp p53 binding sites comprising the RGC, PG and p21 promoter elements are shown. The numbers of multimers utilized in the yeast functional assay are indicated.
Figure 2.
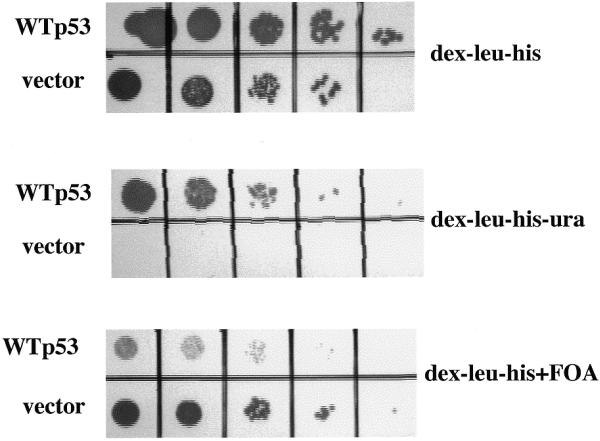
Direct selection of yeast lacking functional p53. Yeast strain yGK7 transformed with pLS76 (WTp53) or pRDI-22 (vector) were grown to mid-log phase in liquid culture and 10-fold serial dilutions were plated on the media indicated.
To determine if this selection strategy correctly identifies yeast expressing mutant p53, different p53 mutants were expressed in the yGK7 strain and analyzed for growth on FOA-containing media. This panel of mutants included one hot-spot p53 mutant (Arg175 changed to His) and five non-hot-spot p53 mutants known to occur in human cancer (8). Strain yGK7 transformed with the plasmid used for gap repair (which contains a p53 cDNA with an out-of-frame central deletion) lacks p53 expression and serves as a positive control for growth on FOA-containing media. As depicted in Figure 3, similar growth was observed in yeast expressing either no p53, or any of the p53 mutants in the presence of FOA. Clonogenicity on FOA was virtually 100% for all mutants tested, allowing each of these mutants to be directly identified under these growth conditions.
Figure 3.
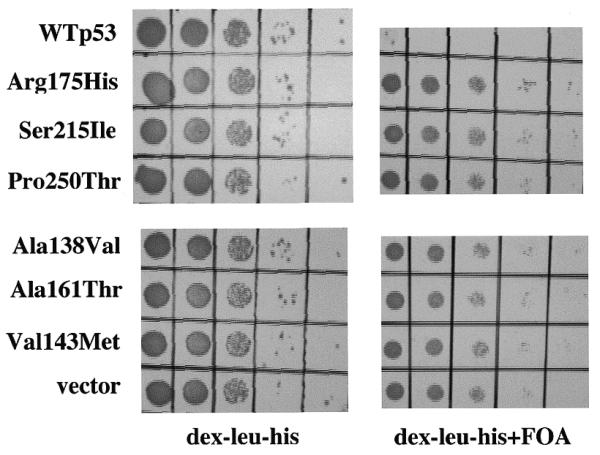
Direct selection of yeast expressing mutant p53. Yeast strain yGK7 transformed with pRDI-22 (vector), pLS76 (WTp53), or plasmids expressing one of the p53 mutants indicated, generated via gap repair, were grown to mid-log phase in liquid culture and 10-fold serial dilutions were plated on the media indicated.
Several non-hot-spot p53 mutants which are inactive against weak p53 binding promoter elements retain residual activity against strong p53 binding promoter elements such as the promoter element from the p21 gene (8,15). We therefore derived a second yeast strain of opposite mating type (yGK13) containing an integrated lacZ gene ORF under the transcriptional control of the p53 binding element from the human p21 promoter (Fig. 1). Mating of this strain to yGK7 transformants identified as expressing mutant p53 (Fig. 3) allows residual transcriptional activity against a strong p53 binding promoter element to be determined. In Figure 4, these diploids were assayed for β-galactosidase activity using a filter lift assay. Several non-hot-spot p53 mutants had significant transcriptional activity in this yeast-based assay. This residual transcriptional activity does not appear to be limited to their expression in yeast since the Val143→Met mutant has previously been shown to be transcriptionally active against the p21 promoter element in mammalian cells as well (8).
Figure 4.
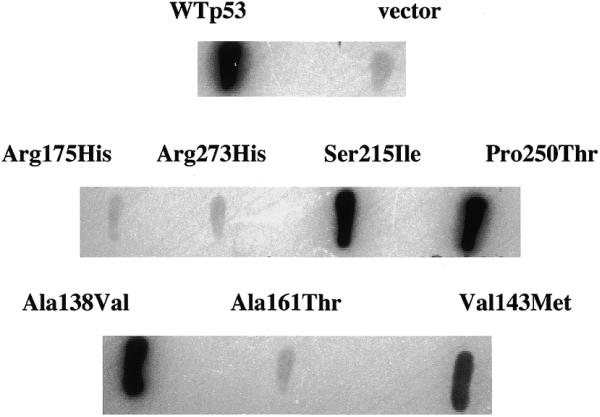
Analysis of p53 activity in yeast utilizing a strong p53 binding promoter element. Yeast strains depicted in Figure 3 were crossed with strain yGK13 and diploids were assayed for β-galactosidase activity using a filter lift assay.
The same panel of non-hot-spot p53 mutants were tested for transcriptional activity in the yeast-based functional assay designed by Flaman et al. (10). This assay utilizes a yeast strain containing the ADE2 gene under transcriptional control of an RGC element multimer (Fig. 1). Yeast expressing wild-type p53 are white in this assay while those devoid of p53 transcriptional activity are red (Fig. 5). Among the non-hot-spot p53 mutants tested, the Pro250→Thr mutant gave rise to intermediate pink yeast in this assay indicating residual transcriptional activity. However, the remaining non-hot-spot mutants gave rise to red yeast even though several express p53 mutants with residual transcriptional activity against the strong p53 binding p21 promoter element (Fig. 4).
Figure 5.
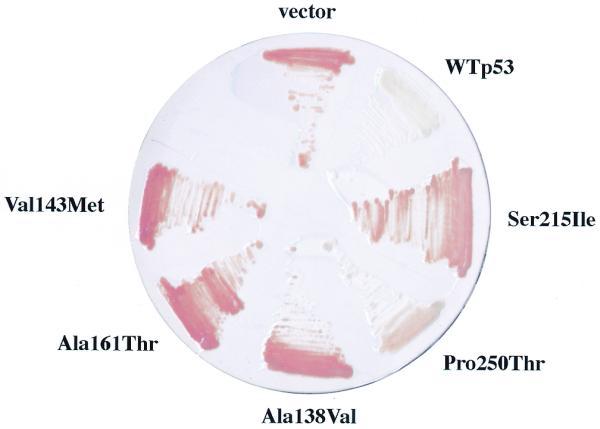
Analysis of p53 activity in yeast utilizing a weak p53 binding promoter element. Yeast strain yIG397 transformed with pRDI-22 (vector), pLS76 (WTp53), or plasmids expressing one of the p53 mutants indicated, generated via gap repair, were pregrown on high-adenine-containing solid media and then streaked on low-adenine-containing solid media to score for adenine auxotrophy.
DISCUSSION
We have modified the yeast-based p53 functional assay designed by Flaman et al. (10) to assess p53 transcriptional activity against promoter elements with strong and weak affinities for p53. This allows p53 mutants with residual transcriptional activity to be identified (Figs 3 and 4). The original published assay makes use of a weak-affinity RGC upstream of an ADE2 gene ORF (10). The weak binding of p53 to this promoter element is perturbed by mutations at both hot-spot and non-hot-spot codons to allow detection of p53 gene mutations based upon adenine auxotrophy. We have similarly utilized a weak p53 binding site (PG) in our modified assay to ensure that all p53 mutations tested, including several partial loss-of-function p53 mutations, are identified. As demonstrated in Figure 3, use of the URA3 gene together with the drug FOA allows for direct selection of yeast expressing mutant p53. In our hands it is easier to screen yeast for growth on FOA-containing media than to score adenine auxotrophy since the latter assay often gives rise to a variable number of ADE+ colonies with an intermediate pink color due to local depletion of adenine in the media (data not shown). These colonies can be confused with colonies expressing a partial loss-of-function mutant p53 (Fig. 5).
We have combined the FOA selection strategy for isolating p53 mutants with a second assay to detect residual p53 transcriptional activity. This is achieved by crossing yeast expressing mutant p53 with a second yeast strain containing a LacZ gene ORF under the transcriptional control of the strong p53 binding element from the p21 promoter. Several non-hot-spot p53 mutants had significant activity against this p53 binding site (Fig. 4) which was not apparent utilizing the original yeast functional assay (Fig. 5). Therefore, the modified yeast functional assay provides a more sensitive readout of residual p53 transcriptional activity.
Implicit in these discussions is that assays for p53 transcriptional activity in yeast mirror p53 activity in mammalian cells. This certainly is the case for hot-spot p53 mutants which are transcriptionally inactive in both yeast and mammalian cells. Several non-hot-spot p53 mutants that retain some degree of residual transcriptional activity in yeast are similarly active when over-expressed in mammalian cells. For example, Arg175 changed to Pro (15) and Val143 changed to Met (8). p53 mutants activate the p21 promoter when expressed in yeast and mammalian cells. Both of these mutants are selected for during human tumorigenesis, indicating that their degree of loss of function is biologically significant. It has been hypothesized that this is due to loss of transactivation of pro-apoptotic genes such as bax which contains a weak p53 binding site (6).
The clinical importance of identifying p53 mutants with residual p53 transcriptional activity using this yeast functional assay remains to be determined. There have been many conflicting studies as to whether tumors with p53 gene mutations have a worse overall prognosis and/or response to treatment. Conceivably, some of this confusion stems from the assumption that all p53 mutations result in total loss of function. In addition, some p53 mutants with residual transcriptional activity may have a gain-of-function phenotype if they selectively transactivate only a partial array of p53-responsive promoter elements as has been suggested (6,7). Identification of p53 mutants with residual transcriptional activity may also be important in the future as these mutants may be more amenable to pharmacological rescue (17) due to the less severe nature of their mutations.
Acknowledgments
ACKNOWLEDGEMENTS
S.G.L. is a Pfizer Scholar and part of this work was supported by an award from the Jattrude Fogarty Trust.
References
- 1.Hollstein M., Sidransky,D., Vogelstein,B. and Harris,C.C. (1991) p53 mutations in human cancers. Science, 253, 49–53. [DOI] [PubMed] [Google Scholar]
- 2.Kern S.E., Kinzler,K.W., Bruskin,A., Jarosz,D., Friedman,P., Prives,C. and Vogelstein,B. (1991) Identification of p53 as a sequence-specific DNA-binding protein. Science, 252, 1708–1711. [DOI] [PubMed] [Google Scholar]
- 3.Kirsch D.G. and Kastan,M.B. (1998) Tumor-suppressor p53: implications for tumor development and prognosis. J. Clin. Oncol., 16, 3158–3168. [DOI] [PubMed] [Google Scholar]
- 4.Cho Y., Gorina,S., Jeffrey,P.D. and Pavletich,N.P. (1994) Crystal structure of a p53 tumor suppressor–DNA complex: understanding tumorigenic mutations. Science, 265, 346–355. [DOI] [PubMed] [Google Scholar]
- 5.Hernandez-Boussard T., Rodriguez-Tome,P., Montesano,R. and Hainaut,P. (1999) IARC p53 mutation database: a relational database to compile and analyze p53 mutations in human tumors and cell lines. International Agency for Research on Cancer. Hum. Mutat., 14, 1–8. [DOI] [PubMed] [Google Scholar]
- 6.Ludwig R.L., Bates,S. and Vousden,K.H. (1996) Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol. Cell. Biol., 16, 4952–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedlander P., Haupt,Y., Prives,C. and Oren,M. (1996) A mutant p53 that discriminates between p53-responsive genes cannot induce apoptosis. Mol. Cell. Biol., 16, 4961–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epstein C., Attiyeh,E., Hobson,D., Silver,A., Broach,J. and Levine,A.J. (1998) p53 mutations isolated in yeast based on loss of transcription factor activity: similarities and differences from p53 mutations detected in human tumors. Oncogene, 16, 2115–2122. [DOI] [PubMed] [Google Scholar]
- 9.Ishioka C., Frebourg,T., Yan,Y.-X., Vidal,M., Friend,S., Schmidt,S. and Iggo,R. (1993) Screening for heterozygous p53 mutations using a functional assay in yeast. Nature Genet., 5, 124–129. [DOI] [PubMed] [Google Scholar]
- 10.Flaman J.M., Frebourg,T., Moreau,V., Charbonnier,F., Martin,C., Chappuis,P., Sappino,A.P., Limacher,J.M., Bron,L., Benhatter,J. et al. (1995) A simple p53 functional assay for screening cell lines, blood and tumors. Proc. Natl Acad. Sci. USA, 92, 3963–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waridel F. and Iggo,R. (1996) Identification of clonal mutations in the morphologically normal mucosa of the aerodigestive tract. Eur. J. Cancer Prev., 5, 67–73. [DOI] [PubMed] [Google Scholar]
- 12.McKnight G.L., Kato,H., Upshall,A., Parker,M.D., Saari,G. and O’Hara,P.J. (1985) Identification and molecular analysis of a third Aspergillus nidulans alcohol dehydrogenase gene. EMBO J., 4, 2093–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guarente L. (1983) Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol., 101, 181–191. [DOI] [PubMed] [Google Scholar]
- 14.Funk W.D., Pak,D.J., Karas,R.H., Wright,W.E. and Shay,J.W. (1992) A transcriptionally active DNA binding site for human p53 protein complexes. Mol. Cell. Biol., 12, 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Como C.J. and Prives,C. (1998) Human tumor-derived p53 proteins exhibit binding site selectivity and temperature sensitivity for transactivation in a yeast-based assay. Oncogene, 16, 2527–2539. [DOI] [PubMed] [Google Scholar]
- 16.Boeke J.D., LaCroute,F. and Fink,G.R. (1984) A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet., 197, 345–346. [DOI] [PubMed] [Google Scholar]
- 17.Foster B.A., Coffey,H.A., Morin,M.J. and Rastinejad,F. (1999) Pharmacological rescue of mutant p53 conformation and function. Science, 286, 2507–2510. [DOI] [PubMed] [Google Scholar]