

Abstract
The aim of this study was to formulate and characterize Eudragit® L100 and Eudragit® L100-poly(lactic-co-glycolic acid) (PLGA) nanoparticles containing diclofenac sodium. Diclofenac generates severe adverse effects with risks of toxicity. Thus, nanoparticles were prepared to reduce these drawbacks in the present study. These nanoparticles were evaluated for surface morphology, particle size and size distribution, percentage drug entrapment, and in vitro drug release in pH 6.8. The prepared nanoparticles were almost spherical in shape, as determined by atomic force microscopy. The nanoparticles with varied size (241–274 nm) and 25.8–62% of entrapment efficiency were obtained. The nanoparticles formulations produced the release profiles with an initial burst effect in which diclofenac sodium release ranged between 38% and 47% within 4 h. The extent of drug release from Eudragit® L100 nanoparticles was up to 92% at 12 h. However, Eudragit®/PLGA nanoparticles showed an initial burst release followed by a slower sustained release. The cumulative release at 72 h was 56%, 69%, and 81% for Eudragit®/PLGA (20:80), Eudragit®/PLGA (30:70) and Eudragit®/PLGA (50:50) nanoparticles, respectively. The release profiles and encapsulation efficiencies depended on the amount of Eudragit in the blend. These data demonstrated the efficacy of these nanoparticles in sustaining the diclofenac sodium release profile.
KEY WORDS: diclofenac sodium, Eudragit® L100, nanoparticles, PLGA, polymer blend
INTRODUCTION
Controlled release systems (microparticles, nanoparticles, and liposome etc.) are prepared to obtain prolonged or controlled drug delivery, to improve bioavailability and to target drug to specific sites. These systems can also to protect drugs from degradation and reduce the toxicity or side effects (1). In particular, nanotechnology pertains to synthetic, engineerable objects which are nanoscale in dimensions or have critical functioning nanoscale components, leading to novel, unique properties. These emergent characteristics arise from the material's large surface area and nanoscopic size (2). Nanoparticle–drug formulation reduces the patient expenses and risks of toxicity. Nanoencapsulation of medicinal drugs (nanomedicines) increases drug efficacy, specificity, tolerability, and therapeutic index of corresponding drugs (3). An increasing number of nanoparticle-based drug delivery systems have been approved for human use or are currently being evaluated in clinical trials. Nanoparticle systems can be engineered to possess a number of desirable features for therapy, including: (1) sustained and controlled release of drugs locally, (2) deep tissue penetration due to their nano-metric size, (3) cellular uptake and sub-cellular trafficking, and (4) protection of cargo therapeutics at both the extracellular and intracellular levels (4). Nanoparticles often have been investigated for the oral delivery of a wide number of drugs (5). Oral delivery is the most preferred route of drug administration due to convenience, patient compliance, and cost-effectiveness (6).
Non-steroidal anti-inflammatory drugs (NSAIDs) are usually good candidates for the development of controlled release preparations, particularly through the oral route. NSAIDs are the world’s most widely used class of therapeutic drugs (7). Members of this class of drugs owe their anti-inflammatory and analgesic properties to inhibition of the enzyme cyclooxygenase (COX). However, the use of NSAIDs is also associated with gastrointestinal, and more recently, cardiovascular side effects (7). The gastrointestinal side effects of NSAIDs appear to be associated with the relative potency of each NSAID for COX-1 over COX-2 such that they are lessened in compounds that are more highly COX-2-selective (7,8). Diclofenac, derived from benzeneacetic acid, is more usually found as sodium or potassium salt with potent anti-inflammatory, analgesic, and antipyretic properties (9).
Several polymers have the characteristics of protecting the drug against the action of the enzymes and gastric fluids, which are in fact very acidic (pH = 1–2), and the reduction of gastrointestinal irritation caused by drugs NSAIDs (10,11). Therefore, enteric coatings using cellulosic polymers and methacrylic acid co-polymers have been extensively studied, such as Eudragit® L 100, as a pH-dependent polymer, that is soluble in intestinal fluid from pH 6 is widely used for the formulation of oral dosage forms (i.e., coating of tablets, matrix tablet, microspheres, nanoparticles; 12–16). Eudragit® L 100 is anionic copolymerization product of methacrylic acid and methyl methacrylate. The ratio of free carboxyl groups to the ester is approximately 1:1 in Eudragit® L 100 having a mean relative molecular mass of about 135,000 Da and apparent viscosity of 50–200 mPas (17). Furthermore, poly(lactic-co-glycolic acid) (PLGA) because of its biocompatibility and biodegradability remain the focus of intensive research effort directed to the controlled release and in vivo localization of drugs (18). PLGA (Resomer® RG 503 H) with the molecular weight of 34,000 Da and inherent viscosity of 0.32–0.44 dl/g has uncapped (free) carboxyl termini (17). In recent years, engineering approaches have been devised to create novel micro- and nano-particles which provide greater control over the drug release profile and present opportunities for drug targeting at the tissue and cellular levels (18,19).
The aim of this study was to develop and characterize diclofenac sodium-loaded Eudragit® L100 and Eudragit®L100/PLGA nanoparticles in order to obtain a controlled-release system. Nanoparticles were prepared by the nanoprecipitation method and characterized the formulation in terms of morphology, size, drug loading, and release. We also investigated the effect of the ratio of Eudragit®L100/PLGA on particle size, encapsulation efficiency, and drug release.
MATERIALS
Acetone, anhydrous ethanol, polyvinyl alcohol (Mw 30,000–70,000) were purchased from Sigma (USA), PLGA ((50:50) Resomer RG 503H, Mw 34000) from Boehringer Ingelheim (Germany). Diclofenac sodium was received as a gift sample from Deva Holding Inc. (Istanbul, Turkey) and Eudragit® L100 (the average molecular weight is approximately 135,000) was supplied by Rohm GmbH&Co. KG (Darmstadt, Germany). All other chemicals and reagents were of analytical grade.
Preparation of Eudragit® L100 and Eudragit®/PLGA-Diclofenac Sodium Nanoparticles
For encapsulating diclofenac sodium into Eudragit nanoparticles, 100 mg of Eudragit® L 100 was dissolved in anhydrous ethanol. This solution was added into 8 mL of PVA solutions (3% w/v) containing diclofenac sodium in the amber glass vial. The mixture was emulsified using 55% power of ultrasonic probe (Sonoplus, HD 2070; Bandelin, Electronics, Berlin, Germany) for 2 min. Finally, organic phases were evaporated under reduced pressure in a rotary evaporator at 40°C. After evaporation of the solvent, nanoparticles were recovered by centrifugation at 15,000 rpm for 40 min and washed with distilled water. The washing step was repeated once before nanoparticles were re-suspended in distilled water and lyophilized overnight. All batches of nanoparticles were produced at least in triplicate (20,21).
For preparation of diclofenac sodium-loaded Eudragit®/PLGA nanoparticles, the mixture of Eudragit®L100-PLGA polymers (50:50, 30:70, 20:80, w/w%) was used for the preparation of nanoparticles. Eudragit® L100 and PLGA were dissolved in anhydrous ethanol and acetone, respectively. Then, the Eudragit solution in ethanol was added slowly to the PLGA solution in acetone (1:3 v/v) with a constant stirring rate of 500 rpm on magnetic stirrer at ambient temperature (13,20,21). Then, the same procedure was used as explained in Eudragit nanoparticles.
Characterization of Nanoparticles
Surface Morphology
The morphological examination of the nanoparticles was performed using atomic force microscopy (AFM; NanoMagnetics Instruments Ltd., UK). The freshly prepared nanoparticles were centrifuged and washed three times with deionized water and later resuspended. A drop of nanoparticle suspension was placed on a glass slide, dried in air, and images were taken (22).
Particle Size and Zeta Potential
The size (Z-average mean) and zeta potential of the nanoparticles were analyzed by photon correlation spectroscopy and laser doppler anemometry, respectively, in triplicate using a Zetasizer 3000HS (Malvern Instruments, UK). Size and zeta potential measurements were performed in triplicate following a dilution of the nanoparticles suspension in distilled water at 25°C. Each measurement was done in triplicate.
HPLC Analysis
The high-performance liquid chromatography (HPLC)–diode array detection (DAD) system consisted of a Thermoquest Spectra System P 1500 isocratic pump coupled with a Spectra System UV 6000 LP photodiode array detection system, a Spectra System AS 3000 autosampler, a SCM 1000 vacuum membrane degasser, a SN 4000 system controller. An ACE reversed-phase C18 column (250 × 4.6 mm, pore size: 5 μm) was used. The mobile phase consisted of a mixture of acetonitrile/water (60:40) and the flow rate was 1 mL/min and diclofenac sodium was monitored spectrophotometrically at 280 nm. An injection volume of 10 μl and diclofenac sodium could be detected at a retention time of 2.4 min. Assay performance was evaluated through determination of specificity, recovery, linearity, the limit of quantification (LOQ), the limit of detection (LOD), precision, accuracy as reported in the International Conference on Harmonization guidelines (23).
Determination of Entrapment Efficiency and Drug Loading of Diclofenac Sodium
To determine the diclofenac sodium content, nanoparticles were dissolved in acetone-ethanol and diclofenac sodium was extracted with phosphate-buffered saline solution (PBS, pH 7.4) and determined by high performance liquid chromatography by a modification of the process previously described (24,25). Briefly, 10 mg of each batch of diclofenac sodium-loaded nanoparticles were vortex mixed for 1 min with acetone-ethanol (1:1 v/v). After vortexing, this mixture in the amber glass vial was sonicated in ultrasonic bath for 20 min. Then, 5 mL of PBS was added into this mixture and mixed by vortexing for 10 min to extract diclofenac sodium. The organic solvents were removed by evaporation under vacuum. The aqueous dispersion was centrifuged at 15,000 rpm for 40 min to remove the polymeric residue and the content of diclofenac sodium in the supernatant was determined by HPLC–DAD under conditions similar to that described above. Experiments were performed in triplicate. The percent drug loading (DL) and percent encapsulation efficiency (EE) of the blend nanoparticles were calculated as (26) :
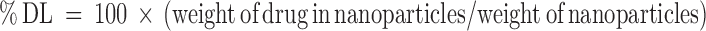 |
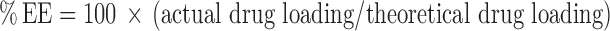 |
In vitro Drug Release Studies
The most commonly used methods for micro/nano particulate systems can be grouped into three broad categories, viz., sample and separate methods (SS), continuous flow, and dialysis. The SS is the most widely used technique. Briefly, drug-loaded micro/nanoparticles are introduced into a vessel containing media and release is assessed over time. Media selection is based on drug solubility and stability over the duration of the release study. Modifications of the basic technique to study drug release include size of container, use of agitation, and sampling methods (27). In the present study, in vitro drug release studies were carried out using the sample and separate methods.
Since diclofenac is a weak acid (pKa = 4), inherently has a negligible solubility in acid and detection of the released drug in acidic solution is not possible (28). Hence, in vitro release of diclofenac sodium from the formulations was carried out by using pH 6.8 phosphate buffer (PB; 28,29). Ten milligrams of nanoparticles were placed in 15 mL of PB at 37°C and 100 rpm in the amber glass vial (30). At different time intervals, the suspension was centrifuged and 15 mL of the release medium was removed and replaced with the same volume of fresh medium. Drug content in supernatant was analyzed using HPLC-DAD as described above. The experiment was carried out six times.
Statistical Analysis
Results are presented as mean ± standard deviation (SD). Wherever appropriate, differences between groups were evaluated with a Student t test (two groups) at an alpha level of 0.05.
RESULTS AND DISCUSSION
In this study, an RP-HPLC method for the determination of diclofenac sodium was developed and validated. A simple sample preparation, short separation time, and a low LOQ were considered when the study started. The calibration curve for diclofenac sodium was constructed under optimum conditions and the linearity of the method was determined by performing injections at six different concentration levels in the linear range over three different days. The method was linear over the range of 1–250 μg/mL and the calibration curve could be described by the equation (Table I). The sensitivity of the analytical method was evaluated by determining the LOD and LOQ. The signal-to noise ratios of 3:1 and 10:1 were taken as LOD and LOQ, respectively. The values of LOQ and LOD were 0.75 μg/mL and 0.1 μg/mL, respectively (Table I). Three different concentrations of standard diclofenac sodium solutions (within the linear range) were analyzed three consecutive days (inter-day precision) and three times within the same day (intra-day precision). The values of intra- and inter-day accuracy and precision were given in Table II.
Table I.
Linearity of Diclofenac Sodium (n = 6)
| Method | Range (μg/ml) | LR | R | LOD (μg/ml) | LOQ (μg/ml) |
|---|---|---|---|---|---|
| HPLC | 1–250 | y = 26,851x − 24,185 | 0.995 | 0.1 | 0.75 |
Based on six calibration curves
LR linear regression, R coefficient of correlation, y peak–area of diclofenac sodium, x diclofenac sodium concentration (μg ml−1).
Table II.
Precision and Accuracy of the Method for Determination of Diclofenac Sodium (mean ± SD, n = 6)
| Intra-day | Inter-day | ||||||
|---|---|---|---|---|---|---|---|
| Method | Added (μg/ml) | Found ± SD | Precision% RSD | Accuracy | Found ± SD | Precision% RSD | Accuracy |
| HPLC | 5 | 5.47 ± 0.43 | 7.86 | 9.4 | 4.85 ± 0.16 | 3.29 | −3.00 |
| 90 | 87.50 ± 4.36 | 4.98 | −2.77 | 92.51 ± 3.31 | 3.57 | 2.78 | |
| 180 | 171.40 ± 3.97 | 2.31 | −4.77 | 174.62 ± 3.77 | 2.15 | −2.98 | |
SD standard deviation of six replicate determinations, RSD relative standard deviation
Average of six replicate determinations, accuracy: (% relative error) (found − added)/added × 100
The nanoprecipitation method was successful in producing diclofenac sodium-loaded Eudragit® L100 and Eudragit®/PLGA nanoparticles employing PVA as stabilizer. The particles were imaged by using AFM. AFM also revealed that all nanoparticles were almost spherical in shape (Fig. 1). The zeta potential values of the nanoparticles of each composition can be observed in Table III. For all the formulations, the nanoparticles exhibited similar low zeta potentials ranging from −1.53 ± 3.1 to 3.47 ± 5.1 mV. The slightly negative charges are attributed to the presence of uncapped end carboxyl groups of the polymer chains at the particle surface. Generally, high negative zeta potential values are expected for pure anionic polymer (PLGA, Eudragit L100, etc.) nanoparticles due to the presence of carboxyl groups on the polymeric chain extremities. However, in this investigation, the zeta potential values are close to zero. The factor which might be responsible for such an effect can be the presence of residual PVA on the nanoparticles surface (31). This subject has been reported by other authors (32–34). The type and concentration of stabilizer affect both particle size and zeta potential of the nanoparticles. Moreover, zeta potential becomes relatively less negative when nanoparticles are formulated using PVA as an emulsifier. This occurs because of coating of emulsifier, thus masking the possible charged groups existing on the particle (32–34).
Fig. 1.

AFM images of drug-loaded Eud/PLGA nanoparticles a 20:80, b 30:70, c 50:50, and d Eudragit®L100 stabilized using polyvinyl alcohol showing the spherical shape of particles
Table III.
Mean Particle Size, Zeta Potential, Drug Loading, and Encapsulation Efficiency of Nanoparticles (mean ± SD, n = 3)
| Formulation | Mean particle size* (nm ± SD) | Zeta potential (mV ± SD) | Entrapment Efficiency* (%) | Drug Loading (%) |
|---|---|---|---|---|
| DS-loaded Eudragit NP | 274 ± 0.19 | −1.53 ± 3.10 | 62.0 ± 2.41 | 14.26 ± 1.60 |
| DS-loaded Eudragit:PLGA (50:50) NP | 263 ± 0.12 | −1.27 ± 2.81 | 53.1 ± 2.73 | 12.21 ± 3.00 |
| DS-loaded Eudragit:PLGA (30:70) NP | 247.4 ± 0.13 | −0.46 ± 4.14 | 45.3 ± 3.00 | 10.51 ± 2.10 |
| DS-loaded Eudragit:PLGA (20:80) NP | 241 ± 0.18 | 3.47 ± 5.10 | 25.82 ± 2.60 | 5.96 ± 1.80* |
DS diclofenac sodium, NP nanoparticles
* p < 0.05
The average mean diameter and polydispersity index of these nanoparticles were determined and listed in Table III. The mean particle size of the nanoparticles prepared using different polymer ratio slightly changed (p < 0.05). Figure 2 shows the particle size distribution of the diclofenac sodium-loaded Eudragit® L100 and Eudragit®/PLGA nanoparticles. The particle size distribution range was relatively narrow, with the drug-loaded nanoparticles having particle size range between 241 and 274 nm. Increasing the concentration of the dissolved Eudragit® L100 polymer increased the viscosity of organic phase and reduced the stirring efficiency resulted in the formation of the bigger emulsion droplets (35). The molecular weight of the polymers is also a critical parameter for drug loading. Higher drug loading was obtained using high molecular weight of polymers (29). In addition, a higher viscosity of the organic phase causes a better distribution of the drug in the matrix. On the contrary, lowering the viscosity of the organic phase allows drugs to come close to the surface during particles formation and to dissolve in the surrounding aqueous medium, resulting in lower drug content (35). The amount of drug in the nanoparticles was determined using HPLC-DAD and the entrapment efficiency percentages, shown in Table III, varied between 25.82% and 62% for all formulations prepared. The entrapment efficiency was affected by the amount and molecular weight of Eudragit® L100 in the blend.
Fig. 2.

Particle sizes distribution of Eudragit®L100 nanoparticles and Eudragit®/PLGA nanoparticles by Malvern Zetasizer. The mean particles size of the nanoparticles (a 20:80, b 30:70, c 50:50, d Eudragit®L100) obtained was 241, 247.4, 263, and 274 nm with a polydispersity index of 0.18, 0.13, 0.12, and 0.19, respectively (mean ± SD, n = 3, p < 0.05)
In this study, Fig. 3 shows in vitro release of diclofenac sodium from Eudragit®L100 nanoparticles and Eudragit®/PLGA nanoparticles of different compositions of Eudragit®L100 and PLGA in pH 6.8 buffer solutions. The pattern of drug release depends on various factors, such as initial drug loading ratio, polymer concentration, the specific properties of the network of polymer chains (e.g., the chain length, their flexibility, and mobility, their water uptake and swelling behavior) or potential interactions between polymer and drug. Generally, fast drug release is attributed to more water uptake, swelling ratio, and/or polymer degradation (36,37). All formulations showed initial burst release of diclofenac sodium. The nanoparticles formulations produced the release profiles with an initial burst effect in which diclofenac sodium release ranged between 38% and 47% within 4 h. This fast release was related to diclofenac sodium adsorbed on the nanoparticles surface and/or to the release of the drug encapsulated near to nanoparticles surface (38). Probably, a higher surface adsorption of the drug was observed for nanoparticles showed the better entrapment efficiency (38). In addition to this explanation, in a previous study, sodium diclofenac-loaded microspheres were prepared by using three low-molecular weight polyesters, (PLGA, poly(L-lactic acid) (PLA), and poly(δ-valerolactone) (PV)) by Lin et al. (39) who indicated that, the first-order release rate was also found in all the microspheres after an initial drug burst and ranked in the order of PLGA > PLA > PV microspheres.
Fig. 3.

In vitro release profile of diclofenac sodium in phosphate buffer of pH 6.8 at 37°C from Eudragit®L100 nanoparticles and Eudragit®/PLGA nanoparticles (mean ± SD, n = 6, p < 0.05)
In the present study, statistically significant difference was observed using Student’s t test among all formulations (Fig. 3). It was confirmed due to fact that P value is <0.05. The nanoparticles prepared using Eudragit®L100 released 92% of the drug content within 12 h. However, the release of diclofenac sodium from Eudragit®/PLGA nanoparticles showed a biphasic pattern. Following this initial phase, the release of diclofenac sodium was slow. The cumulative release at 72 h was 56%, 69%, and 81% for Eudragit®/PLGA (20:80), Eudragit®/PLGA (30:70) and Eudragit®/PLGA (50:50) nanoparticles, respectively. In this case, increasing the amount of Eudragit® L100 resulted in an increase in the diclofenac sodium released, because, Eudragit® L100 has a pH-dependent solubility and its swelling and erosion increase as the pH increases. Acrylic polymers such as Eudragit® L100 and Eudragit® L100-55 are commonly used for coating of tablets and preparation of matrix tablets, controlled-release formulations. These polymers can dissolve rapidly upon deprotonation of carboxylic acid groups at specific pH values. Thereby, the release profiles of these nanoparticles exhibit significant pH-sensitivity (40). Dai et al. (40) described that cyclosporine A-loaded pH sensitive nanoparticles using Eudragit® L 100, Eudragit® S 100 and Eudragit® L100-55. They reported that the nanoparticles exhibited perfect pH-dependent release profiles (40). In another study, pH-dependent swellable and erodable-buffered matrices made of hydroxypropyl methylcellulose (HPMC) and pH-dependent solubility polymer (Eudragit® L100-55) was developed and studied the effect of the microenvironment pH on the release pattern of diclofenac sodium by Al-Taani and Tashtoush (41). It has been reported that the swelling and erosion occurred simultaneously from matrices made up of HPMC and Eudragit L100-55. The increase in matrix erosion and swelling with increase of the pH was due to the increase in ionization of methacrylic acid moiety present in Eudragit L100-55. This created electrostatic repulsion forces between Eudragit polymer chains, which disrupt the matrix and increase both swelling and erosion as the pH increased. The drug release from the matrices was directly related to percentage swelling and percentage erosion (41). Moreover, Basavaraj et al. (42) prepared Eudragit® S microspheres containing diclofenac sodium and reported that a controlled drug release of 60–84% was obtained over a period of 8 h.
The obtained data in this study indicate that appropriate combinations of Eudragit® L100 (pH dependent) and PLGA might be suitable for adequately controlling diclofenac sodium release and assure more reproducible drug release behavior.
CONCLUSION
Diclofenac sodium-loaded nanoparticles, which have not been reported yet, were prepared easily and successfully by using blends of biodegradable polymer with non-biodegradable polymer. The encapsulation efficiency and the drug release behavior were influenced by the amount of Eudragit in the blend. The drug release experiments demonstrated the efficacy of these nanoparticles in sustaining the diclofenac sodium release profile. Hence, the nanoparticles showed potential use for controlling the release of NSAIDs.
References
- 1.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Contr Release. 2001;70(1–2):1–20. doi: 10.1016/S0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 2.Serda RE, Godin B, Blanco E, Chiappini C, Ferrari M. Multi-stage delivery nano-particle systems for therapeutic applications. Biochim Biophys Acta. 2010 (in press). [DOI] [PMC free article] [PubMed]
- 3.Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75(1):1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61(2):158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galindo-Rodriguez SA, Allemann E, Fessi H, Doelker E. Polymeric nanoparticles for oral delivery of drugs and vaccines: a critical evaluation of in vivo studies. Crit Rev Ther Drug Carrier Syst. 2005;22(5):419–464. doi: 10.1615/critrevtherdrugcarriersyst.v22.i5.10. [DOI] [PubMed] [Google Scholar]
- 6.Yamanaka YJ, Leong KW. Engineering strategies to enhance nanoparticle-mediated oral delivery. J Biomater Sci Polym Ed. 2008;19(12):1549–1570. doi: 10.1163/156856208786440479. [DOI] [PubMed] [Google Scholar]
- 7.Warner TD, Vojnovic I, Bishop-Bailey D, Mitchell JA. Influence of plasma protein on the potencies of inhibitors of cyclooxygenase-1 and -2. FASEB J. 2006;20(3):542–544. doi: 10.1096/fj.05-4434fje. [DOI] [PubMed] [Google Scholar]
- 8.Mitchell JA, Warner TD. Cyclo-oxygenase-2: pharmacology, physiology, biochemistry and relevance to NSAID therapy. Br J Pharmacol. 1999;128(6):1121–1132. doi: 10.1038/sj.bjp.0702897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sena MM, Chaudhry ZF, Collins CH, Poppi RJ. Direct determination of diclofenac in pharmaceutical formulations containing B vitamins by using UV spectrophotometry and partial least squares regression. J Pharm Biomed Anal. 2004;36(4):743–749. doi: 10.1016/j.jpba.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 10.González M, Rieumont J, Dupeyron D, Perdomo I, Fernandez E, Abdón L, et al. Nanoencapsulation of acetyl salicylic acid within enteric polymer nanopaticles. Rev Adv Mater Sci. 2008;17:71–75. [Google Scholar]
- 11.Piao ZZ, Lee MK, Lee BJ. Colonic release and reduced intestinal tissue damage of coated tablets containing naproxen inclusion complex. Int J Pharm. 2008;350(1–2):205–211. doi: 10.1016/j.ijpharm.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 12.Moustafine RI, Margulis EB, Sibgatullina LF, Kemenova VA, Van den Mooter G. Comparative evaluation of interpolyelectrolyte complexes of chitosan with Eudragit L100 and Eudragit L100-55 as potential carriers for oral controlled drug delivery. Eur J Pharm Biopharm. 2008;70(1):215–225. doi: 10.1016/j.ejpb.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Saffari M, Shahbazi M, Ardestani MS. Formulation and in vitro evaluation of Eudragit L100® microspheres of piroxicam. Nat Precedings. 2008;1544(1):1–5. [Google Scholar]
- 14.Andrews GP, Jones DS, Diak OA, McCoy CP, Watts AB, McGinity JW. The manufacture and characterisation of hot-melt extruded enteric tablets. Eur J Pharm Biopharm. 2008;69(1):264–273. doi: 10.1016/j.ejpb.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Devarajan PV, Sonavane GS. Design and evaluation of pH modulated controlled release matrix systems for colon specific delivery of indomethacin. Pharmazie. 2007;63(10):736–742. [PubMed] [Google Scholar]
- 16.Devarajan PV, Sonavane GS. Preparation and in vitro/in vivo evaluation of gliclazide loaded Eudragit nanoparticles as sustained release carriers. Drug Dev Ind Pharm. 2007;33(2):101–111. doi: 10.1080/03639040601096695. [DOI] [PubMed] [Google Scholar]
- 17.Rowe RC, Sheskey PJ, Owen SC. Handbook of pharmaceutical excipients. 5. London: Pharmaceutical Press and American Pharmacists Association; 2006. [Google Scholar]
- 18.Mohamed F, van der Walle CF. Engineering biodegradable polyester particles with specific drug targeting and drug release properties. J Pharm Sci. 2008;97(1):71–87. doi: 10.1002/jps.21082. [DOI] [PubMed] [Google Scholar]
- 19.Dillen K, Bridts C, Van der Veken P, Cos P, Vandervoort J, Augustyns K, et al. Adhesion of PLGA or Eudragit/PLGA nanoparticles to staphylococcus and pseudomonas. Int J Pharm. 2008;349(1–2):234–240. doi: 10.1016/j.ijpharm.2007.07.041. [DOI] [PubMed] [Google Scholar]
- 20.Jawahar N, Eagappanath T, Nagasamy V, Jubie S, Samanta MK. Preparation and characterisation of PLGA-nanoparticles containing an Anti-hypertensive agent. Int J Pharm Tech Res. 2009;1(2):390–393. [Google Scholar]
- 21.Song X, Zhao Y, Hou S, Xu F, Zhao R, He J, et al. Dual agents loaded PLGA nanoparticles: systematic study of particle size and drug entrapment efficiency. Eur J Pharm Biopharm. 2008;69(2):445–453. doi: 10.1016/j.ejpb.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 22.Cetin M, Aktas Y, Vural I, Capan Y, Dogan LA, Duman M, et al. Preparation and in vitro evaluation of bFGF-loaded chitosan nanoparticles. Drug Deliv. 2007;14(8):525–529. doi: 10.1080/10717540701606483. [DOI] [PubMed] [Google Scholar]
- 23.International Conference on Harmonization (ICH), Q2b: Validation of analytical procedures: methodology. US FDA Federal Register; 1997 (Vol. 62): p. 27463
- 24.Hombreiro Pérez M, Zinutti C, Lamprecht A, Ubrich N, Astier A, Hoffman M, et al. The preparation and evaluation of poly(epsilon-caprolactone) microparticles containing both a lipophilic and a hydrophilic drug. J Contr Release. 2000;65(3):429–438. doi: 10.1016/S0168-3659(99)00253-9. [DOI] [PubMed] [Google Scholar]
- 25.Cetin M, Capan Y, Vural I, Dogan AL, Guc D, Hincal AA, et al. Preparation and characterization of bFGF and BSA loaded microspheres. J Drug Deliv Sci Tech. 2005;15(5):371–375. [Google Scholar]
- 26.Mundargi RC, Srirangarajan S, Agnihotri SA, Patil SA, Ravindra S, Setty SB, et al. Development and evaluation of novel biodegradable microspheres based on poly(d, l-lactide-co-glycolide) and poly(epsilon-caprolactone) for controlled delivery of doxycycline in the treatment of human periodontal pocket: in vitro and in vivo studies. J Control Release. 2007;119(1):59–68. doi: 10.1016/j.jconrel.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 27.D'Souza SS, DeLuca PP. Methods to assess in vitro drug release from injectable polymeric particulate systems. Pharm Res. 2006;23(3):460–474. doi: 10.1007/s11095-005-9397-8. [DOI] [PubMed] [Google Scholar]
- 28.Kouchak M, Atyabi F. Ion exchange, an approach to prepare an oral floating drug delivery system for diclofenac. Iran J Pharm Res. 2004;2:93–97. [Google Scholar]
- 29.Tunçay M, Caliş S, Kaş HS, Ercan MT, Peksoy I, Hincal AA. Diclofenac sodium incorporated PLGA (50:50) microspheres: formulation considerations and in vitro/in vivo evaluation. Int J Pharm. 2000;195(1–2):179–188. doi: 10.1016/S0378-5173(99)00394-4. [DOI] [PubMed] [Google Scholar]
- 30.Kilic AC, Capan Y, Vural I, Gursoy RN, Dalkara T, Cuine A, et al. Preparation and characterization of PLGA nanospheres for the targeted delivery of NR2B-specific antisense oligonucleotides to the NMDA receptors in the brain. J Microencapsul. 2005;22(6):633–641. doi: 10.1080/02652040500162766. [DOI] [PubMed] [Google Scholar]
- 31.Saxena V, Sadoqi M, Shao J. Indocyanine green-loaded biodegradable nanoparticles: preparation, physicochemical characterization and in vitro release. Int J Pharm. 2004;278(2):293–301. doi: 10.1016/j.ijpharm.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 32.Sahoo SK, Labhasetwar V. Nanoparticles interface: an important determinant in nanoparticle-mediated drug/gene delivery. In: Gupta RB, Kompella UB, editors. Nanoparticle technology for drug delivery. New York: Taylor & Francis Group; 2006. pp. 139–154. [Google Scholar]
- 33.Bala I, Bhardwaj V, Hariharan S, Kharade SV, Roy N, Ravi Kumar MN. Sustained release nanoparticulate formulation containing antioxidant-ellagic acid as potential prophylaxis system for oral administration. J Drug Target. 2006;14(1):27–34. doi: 10.1080/10611860600565987. [DOI] [PubMed] [Google Scholar]
- 34.Konan YN, Cerny R, Favet J, Berton M, Gurny R, Allémann E. Preparation and characterization of sterile sub-200 nm meso-tetra(4-hydroxylphenyl)porphyrin-loaded nanoparticles for photodynamic therapy. Eur J Pharm Biopharm. 2003;55(1):115–124. doi: 10.1016/S0939-6411(02)00128-5. [DOI] [PubMed] [Google Scholar]
- 35.Fattal E, Quaglia F, Gupta P, Brazeau G. Biodegradable microparticles for the development of less-painful and less-irritating parenterals. In: Gupta P, Brazeau G, editors. Injectable Drug Development: Techniques to Reduce Pain and Irritation. Informa Health Care; 1999. p. 355–78.
- 36.Wischke C, Schwendeman SP. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int J Pharm. 2008;364(2):298–327. doi: 10.1016/j.ijpharm.2008.04.042. [DOI] [PubMed] [Google Scholar]
- 37.Choi HS, Seo SA, Khang G, Rhee JM, Lee HB. Preparation and characterization of fentanyl-loaded PLGA microspheres: in vitro release profiles. Int J Pharm. 2002;234(1–2):195–203. doi: 10.1016/S0378-5173(01)00968-1. [DOI] [PubMed] [Google Scholar]
- 38.Musumeci T, Ventura CA, Giannone I, Ruozi B, Montenegro L, Pignatello R, et al. PLA/PLGA nanoparticles for sustained release of docetaxel. Int J Pharm. 2006;325(1–2):172–179. doi: 10.1016/j.ijpharm.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 39.Lin SY, Chen KS, Teng HH, Li MJ. In vitro degradation and dissolution behaviours of microspheres prepared by three low molecular weight polyesters. J Microencapsul. 2000;17(5):577–586. doi: 10.1080/026520400417630. [DOI] [PubMed] [Google Scholar]
- 40.Dai J, Nagai T, Wang X, Zhang T, Meng M, Zhang Q. pH-sensitive nanoparticles for improving the oral bioavailability of cyclosporine A. Int J Pharm. 2004;280(1–2):229–240. doi: 10.1016/j.ijpharm.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 41.Al-Taani BM, Tashtoush BM. Effect of microenvironment pH of swellable and erodable buffered matrices on the release characteristics of diclofenac sodium. AAPS J Pharm Sci Tech. 2003;4(3):E43. doi: 10.1208/pt040343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basavaraj BV, Deveswaran R, Bharath S, Abraham S, Furtado S, Madhavan V. Hollow microspheres of diclofenac sodium—a gastroretentive controlled delivery system. Pak J Pharm Sci. 2008;21(4):451–454. [PubMed] [Google Scholar]