

Abstract
Inhaled recombinant secretory leukocyte protease inhibitor (rSLPI) has shown potential for the treatment of inflammatory lung conditions. Rapid inactivation of rSLPI by cathepsin L (Cat L) and rapid clearance from the lungs has limited clinical efficacy to date. Previous studies by us have shown that encapsulation of rSLPI within1,2-dioleoyl-sn-glycero-3-[phospho-L-serine]/cholesterol (DOPS/Chol) liposomes protects rSLPI against Cat L inactivation in vitro. Liquid DOPS–rSLPI preparations were found to be unstable upon long-term storage and nebulisation. The aim of this study was therefore to develop a method of manufacture for preparing DOPS–rSLPI liposomes as a dry powder for inhalation. DOPS–rSLPI dry powders were lyophilised and subsequently micronised with a novel micronisation aid. The effects of formulation and processing on rSLPI stability, activity, and uniformity of content within the powders were characterised. Using D-mannitol as the micronisation aid, dry powder particles in the inhalable size range (<5 μm) were prepared. By optimising process parameters, up to 54% of rSLPI was recovered after micronisation, of which there was no significant loss in anti-neutrophil elastase activity and no detectable evidence of protein degradation. Aerosolisation was achieved using a dry powder inhaler, and mass median aerodynamic diameter (MMAD) was evaluated after collection in a cascade impactor. Aerosolisation of the DOPS–rSLPI dry powder yielded 38% emitted dose, with 2.44 μm MMAD. When challenged with Cat L post-aerosolisation, DOPS–rSLPI dry powder was significantly better at retaining a protective function against Cat L-induced rSLPI inactivation compared to the aqueous DOPS–rSLPI liposome dispersion and was also more stable under storage.
KEY WORDS: liposome, powder, protein, pulmonary, rSLPI
INTRODUCTION
Local drug delivery to the lungs is an effective means of treating a range of pulmonary diseases such as asthma, cystic fibrosis, bronchitis, and emphysema. Inhalation therapy is traditionally reserved for delivery of smaller molecules, such as corticosteroids and bronchodilators; however, recent developments in aerosol science have made possible the pulmonary administration of proteins and peptides for both systemic and local therapeutic effect, and a range of peptides and proteins is currently being investigated for inhalation including recombinant secretory leukocyte protease inhibitor (rSLPI). rSLPI has been investigated as a potential therapeutic for inflammatory lung disease (1–5). Current anti-inflammatory treatments such as inhaled corticosteroids and sodium cromoglycate are used to alleviate inflammation in the lungs; however, studies have demonstrated that use of corticosteroids in particular shows no reduction in disease progression (6,7).
Delivery of rSLPI directly to the lungs via inhalation increases targeting and minimises systemic effects. This method of delivery has previously been demonstrated to exhibit an increased half-life over intravenous administration (8,9). rSLPI has been administered via inhalation and intratracheal instillation in a number of in vivo animal and human studies (4,5,10,11), but the success of inhaled rSLPI therapy has been limited by rapid clearance (12) and extensive degradation by proteases, particularly cathepsins (13), necessitating repeated dosages every 12 h in order to maintain therapeutic effectiveness (4).
Liposomes have advantages over other potential vehicles for lung targeting, including their ability to facilitate sustained release of their cargo in the lungs, to increase drug residence time in the lungs (14–16), improve stability of the drug both in vitro and in vivo, biocompatibility with lung surfactant components of which 85% is phospholipid-based (17,18), local targeting providing increased potency, and reduced toxicity (19–22). In addition, the high drug loading capacity of liposomes and the low excipient to drug ratio of lipid-based carriers result in less excipient accumulation in the lungs after repeated administration compared to polymer-based carriers (23). Their application in the delivery of peptides or proteins to the lungs, however, has yet to be fully explored. Liposome-encapsulated rSLPI for inhalation was prepared by us in order to protect rSLPI from cathepsin degradation and to prolong its residence time in the lungs. It was demonstrated that encapsulation of rSLPI within liposomes composed of 1,2-dioleoyl-sn-glycero-3-[phospho-L-serine]/cholesterol (DOPS/Chol) protects rSLPI against cathepsin L (Cat L) inactivation in vitro (24). We have tested this delivery system and assessed it to be biocompatible in vitro using the Calu-3 airway epithelial cell line and U937 monocytes (24) and bioactive in vivo (unpublished data). The aqueous dispersion of DOPS–rSLPI liposomes was found to be unstable during long-term storage and nebulisation; therefore in this current study, we aimed to prepare a DOPS–rSLPI dry powder for inhalation in order to overcome the failings of the aqueous dispersion.
Pulmonary delivery has seen a recent surge in interest in dry powder inhaler (DPI) technology, motivated primarily by the phase-out of ozone-depleting chlorofluorocarbons in metered dose inhalers, and benefits associated with DPIs such as high patient compliance (25,26), high dose carrying capabilities (27), and increased drug stability (26,28). Dry powders are of particular interest for the inhalation of proteins. Classic nebulisation methods such as jet nebulisation and ultrasonic nebulisation can lead to protein degradation (29). A dry powder formulation offers a means of improving protein drug stability during aerosolisation by using a dry powder inhaler device that would not subject the protein formulation to the stresses commonly associated with nebulisation. In addition, proteins are more stable in a dry powder form, and so a dry powder product allows for long-term storage (30–32).
As well as maintaining a high level of rSLPI activity, a dry powder preparation can also be of benefit in maintaining the quality of liposome vesicular carrier systems during storage. Liposomes are prone to instability when stored as aqueous dispersions due to lipid degradation (33–35) and/or liposome aggregation and fusion that can ultimately lead to drug leakage, particularly of water-soluble drugs such as rSLPI (36,37). Previous studies carried out by us on the DOPS–rSLPI aqueous dispersion have indicated that there is a degree of liposome instability and rSLPI leakage overtime (24). Reformulating aqueous liposome dispersion as a dry powder can improve the storage stability of liposome dispersions by decreasing the tendency for liposome aggregation and fusion and thereby significantly increase shelf life (37–43). Nebulisation of aqueous liposome dispersions can also prove problematic. For example, jet nebulisation often results in disruption of the liposomal bilayers culminating in the leakage of encapsulated drug (24,44,45). Liposome formulations containing cholesterol and high concentrations of phospholipids tend to be quite viscous (46,47) and as such are not nebulised reproducibly using ultrasonic technology (48). Therefore, aerosolisation of liposomes in a powder form avoids the disruption of liposome bilayers often observed during nebulisation. Another important advantage associated with the use of a dry powder is the higher pulmonary deposition attainable with dry powder inhalers, typically 50–75%, compared to 20–25% pulmonary deposition achievable with nebulisation (18).
The aims of this study were therefore to develop a method of manufacture for the preparation of DOPS–rSLPI liposome dry powder for inhalation. DOPS–rSLPI dry powder was prepared by lyophilisation of the liposome-encapsulated rSLPI and subsequent micronisation of the powder with a micronisation aid. The effects of formulation and processing on uniformity of content, rSLPI activity, liposome integrity, rSLPI leakage, and liposome size stability were characterised. Aerosolisation was carried out using a dry powder inhaler, and in vitro mass median aerodynamic diameter (MMAD) after collection in a cascade impactor was evaluated. The benefits of the DOPS–rSLPI DPI formulation compared to the DOPS–rSLPI aqueous dispersion under conditions of storage and aerosolisation were evaluated.
MATERIALS AND METHODS
Materials
The following were the materials used: cholesterol and DOPS (Avanti Polar Lipids® Inc.), recombinant human rSLPI was a gift from Amgen® (Thousand Oaks, California, USA), human sputum leukocyte elastase (Elastin Products Company®, Missouri, USA), rabbit anti-SLPI polyclonal IgG and goat anti-rabbit IgG-HRP antibody (Santa Cruz Biotechnology® Inc.), and SuperSignal West Pico chemiluminescent substrate kit (Medical Supply Company®, Dublin, Ireland); cathepsin L, N-(methoxysuccinyl)-Ala-Ala-Pro-Val 4-nitroanilide, and all other reagents were obtained from Sigma-Aldrich® (Tallaght, Dublin, Ireland); D-mannitol, Sigma and Freezone 6 l bench-top freeze dryer (Labconco); MC One C0386 Fluid Jet Mill (Jet Pharma SA, Switzerland); Malvern Mastersizer 2000, Sirocco, version 5.22 software and high performance particle sizer (HPPS®) (Malvern), Intal Spinhaler® (Rhône-Poulenc Rorer); and the Copley Glass Twin Stage Impinger, Copley Digital Flowmeter Model DFM2, Andersen cascade impactor, critical flow controller (TPK), and 90 l/min cascade impactor conversion kit (Copley Scientific Ltd, UK).
Liposome Preparation and Characterisation
DOPS/Chol liposomes were prepared by the conventional thin film hydration procedure. Briefly DOPS was mixed with cholesterol at a ratio of 7:3 and dissolved in chloroform/methanol (2:1). Solvent was removed by evaporation using a rotary evaporator. rSLPI was incorporated into the formulation in a rehydrating buffer (phosphate buffered saline (PBS), pH 7.4). Size reduction of the liposome suspension was achieved by extrusion using a mini-extruder (Avanti Polar Lipids Inc.) through 200 nm pore size polycarbonate membranes. Non-entrapped protein was removed by centrifugation at 45,000 rpm at 4°C for 40 min. The supernatant was removed and the pellet washed with PBS and re-centrifuged. This step was repeated for a further two washes.
The size distribution of the liposomes was determined by dynamic light scattering (HPPS®, Malvern Instruments) and the encapsulation efficiency of rSLPI in DOPS/Chol liposomes was determined, after disruption of the liposomes, by reverse phase-HPLC (RP-HPLC). Briefly, liposomes were disrupted via 0.5% Triton X and loaded onto a Vydac narrow bore C18 column (#218TP5205, Vydac®, Hesperia, California) for RP-HPLC analysis. Gradient elution occurred over 40 min using a mobile phase of water and acetonitrile with 0.1% trifluoroacetic acid and rSLPI. Area under the curve was analysed at 214 nm. The supernatant samples were also analysed by RP-HPLC. % EE was defined as the rSLPI encapsulated in liposomes as a percentage of loading dose. The Stewart assay was used to determine the concentration of phospholipid present in the liposomal formulations. Briefly 2 ml chloroform and 2 ml ferrothiocyanate reagent and 0.1 ml of liposome sample were vortexed vigorously for 1 min. The resultant mixture was centrifuged at 1,000 rpm for 5 min and the lower chloroform layer removed by glass pipette and measured at 485 nm. Based on a standard curve for the appropriate lipid, the concentration of phospholipid present in the sample was calculated.
rSLPI Stability and In Vitro Activity
To ensure that rSLPI had not been cleaved during processing western blot analysis was used to ensure that the molecular weight of rSLPI had not been altered. Samples and standards containing 125 ng rSLPI were electrophoresed on 15% polyacrylamide gel and blotted onto nitrocellulose. After blocking in I-block®, rSLPI was detected using affinity purified rabbit anti-SLPI polyclonal IgG (1:1,000 in I-block) for 1 h followed by incubation with goat anti-rabbit IgG-HRP antibody (1:7,500) for 1 h. Development was carried out using SuperSignal West Pico® chemiluminescent substrate kit (13).
Activity of rSLPI was assayed by measuring its inhibition of human neutrophil elastase (NE) activity on the substrate N-methoxy-succinyl-Pro-Ala-Ala-Val-p-nitroanilide. rSLPI was incubated with NE at room temperature for 5 min. Upon addition of the substrate, the change in absorbance (ΔAbs) at 405 nm was measured from T0 to T5 min (11).
Dry Powder Liposome Preparation
Liposome Lyophilisation
Liposomes were snap-frozen in liquid nitrogen and lyophilised in a bench-top freeze dryer (Labconco®) for approximately 48 h at −50°C and 0.035 mBar. Lyophilised liposomes were stored in a desiccator at room temperature until required. Residual moisture content of lyophilised DOPS–rSLPI liposomes was assessed using Karl Fisher analysis.
Liposome Milling
Jet milling was used to micronise lyophilised liposome powders. Micronisation was carried out under nitrogen gas using the MC One® jet mill (Jet Pharma Group®) operated at seven bar venturi pressure and five bar ring pressure. Optimisation of liposome micronisation was facilitated by the addition of D-mannitol prior to milling as the micronisation aid.
Powder Characterisation
Phospholipid Assay
The Stewart assay was used to determine the concentration of phospholipid present in the liposomal formulations (49).
rSLPI Quantification
To assess the uniformity of rSLPI distribution within the liposome powder the European Pharmacopoeial method (Ph. Eur. method 2.9.6; B.P. Vol II 1998 Appendix XII H A201) was applied. Ten random samples of each powder were taken. Liposome powders were rehydrated with PBS, disrupted using Triton X 0.5% v/v and assayed for rSLPI content by RP-HPLC as before.
Particle Size Analysis of Powders
Particle size analysis was carried out both before and after micronisation by laser diffraction using the Scrirocco® dry dispersion chamber (Mastersizer 2000®, Malvern Instruments). Particle size distribution was expressed as the span. Liposome powders were imaged by scanning electron microscopy (SEM) before and after micronisation. The sample was mounted onto aluminium stubs and sputter coated with a 50–100-A layer of gold. The liposomes were imaged using a Hitachi S4300 scanning electron microscope at an accelerating voltage of 10 kV.
Activity and Stability of rSLPI after Powder Preparation
Leakage of Protein from Dry Powder Preparations after Resuspension
DOPS–rSLPI liposome powder was rehydrated with PBS and ultracentrifuged at 45,000 rpm for 40 min at 4°C. The concentration of rSLPI present in the supernatant (non-encapsulated fraction) was determined by RP-HPLC. The concentration of rSLPI encapsulated in DOPS/Chol liposomes was assayed by first disrupting the liposomes using Triton X 0.5% v/v followed by RP-HPLC analysis as before. The percentage of rSLPI leaked from liposomes was calculated as a percentage of that originally encapsulated.
Western Blot Analysis of rSLPI
To ensure the molecular weight of rSLPI had not been altered by lyophilisation and micronisation, western blot analysis was carried out as before.
Anti-Neutrophil Elastase Activity and Cathepsin Challenge of DOPS/rSLPI Dry Powder
Before testing rSLPI activity, liposome powders were rehydrated with PBS and disrupted with Triton X detergent (0.5% v/v) to release rSLPI from liposome encapsulation, and anti-neutrophil elastase (anti-NE) activity was determined as described previously. Inactivation of rSLPI by Cat L was assessed by measuring rSLPI’s anti-NE activity following incubation of rSLPI (alone or encapsulated in liposomes) with Cat L, compared to its activity when incubated with PBS (negative control). DOPS/rSLPI dry powder was suspended in PBS with or without Cat L and incubated for 2 h at 37°C in 0.1 M sodium acetate buffer pH 5.5, containing 1 mM EDTA and 10 mM dithiothreitol. The reaction was stopped by the addition of 0.2 M Tris, pH 8.5 containing 1 μM E-64. The rSLPI samples and controls were then assayed for the remaining anti-NE activity as outlined above.
Aerosolisation of DOPS–rSLPI
Aerosolisation of the micronised dry powder formulations was achieved using a standard low resistance dry powder inhaler, the Spinhaler®. Forty-milligramme quantities of liposome powder were placed in ‘size 2’ gelatin capsules. The liposome powder was mixed with large carrier D-mannitol particles (sieve size 45–63 μm) in the ratio 1:1 (liposome/D-manntol) directly prior to loading into the gelatin capsule. The capsule was pierced and the contents entrained into a vacuum pump-supplied airflow connected to either a twin stage impinger (TSI) or an Andersen cascade impactor (ACI) for aerodynamic assessment of the DOPS–rSLPI formulations.
TSI Analysis
After aerosolisation of liposome powder into a TSI using a flow rate of 60 ± 5 L/min, samples from the upper and lower stages of the TSI were removed for analysis. Liposomes present in the collected samples were disrupted using Triton X-100 0.5% v/v, and the quantity of rSLPI present was assayed by RP-HPLC. The emitted dose was calculated as the sum of protein assayed in both the upper and lower impinger parts and expressed as a percentage of protein loaded into the capsule prior to aerosolisation. The fine particle fraction (FPF) was determined by assaying the lipid and protein content in the lower stages of the impinger chamber (parts E–G, as per the British Pharmacopoeia Apparatus A) and expressed as a percentage of the emitted dose. Non-respirable fraction (% NRF) is defined as sum of deposition in the upper stages of the impinger (parts B–D, as per the British Pharmacopoeia Apparatus A) and is expressed as a percentage of the emitted dose.
Andersen Cascade Impactor Analysis
The eight-stage ACI was adapted for use with the Spinhaler® DPI by operating at a flow rate of 90 l/min, with the addition of the necessary extra stages to the ACI and coating of the plates with a solution of silicon oil in hexane (1% v/v) to allow attachment of particles depositing on these plates. After assembly of the ACI apparatus, a critical flow controller (TPK) was attached and the flow adjusted until P1 = 4.0 kPa. The test time used was determined such that 4 l of air would be drawn through the inhaler, this is considered to be the normal inspiratory capacity of an average size adult male of approximately 70 kg, and flow rate stability was assured by testing for sonic (critical) flow. After aerosolisation, the apparatus was carefully dismantled, and in each of the stages, filter, throat, and preseparator were removed, rinsed in a fixed volume of PBS, the liposomes were disrupted with Triton X 0.5% v/v, and the samples were assayed for rSLPI and lipid content as described previously. The MMAD and geometric standard deviation (GSD) were subsequently calculated. The FPF was defined as the sum of depositions on stages 2 through to the filter and expressed as a percentage of the emitted dose.
Storage Stability
A storage stability study was carried out where DOPS–rSLPI liposome powder was stored in sealed glass vials in a desiccator at room temperature for 5 months. In a previous study by us (24), the storage stability study of an aqueous DOPS–rSLPI liposome dispersion was carried out by storing the dispersion in sealed glass vials at 4°C for 7 weeks. For both forms of preparation, samples were removed at weekly intervals and analysed for rSLPI content, rSLPI activity and liposome size, and rSLPI leakage due to liposome disruption. The ability of the liposomes to protect rSLPI against degradation by Cat L after storage was also examined as an indicator of rSLPI leakage.
Statistical Analysis of Results
Results were expressed as mean ± standard deviation. Where appropriate, the unpaired t test was used to determine the significance of results. In all cases, a probability value of less than 0.05 was considered to be significant.
RESULTS AND DISCUSSION
Liposome Encapsulation of rSLPI
rSLPI was encapsulated in DOPS/Chol liposomes with an average encapsulation efficiency of 74.1 ± 2.97%. The high encapsulation efficiency achieved was due to the process of ‘active encapsulation’, where a charged substance is encapsulated into liposomes of opposite charge (50). Post-extrusion, the DOPS–rSLPI liposomes were 153.6 ± 2.47 nm in size. RP-HPLC and western blot analysis were carried out to verify that rSLPI had not been cleaved during the encapsulation process. To confirm that the molecule also retained its anti-protease activity, the ability of rSLPI to inhibit NE activity in vitro was also assessed. No degradation of rSLPI was evident after encapsulation and rSLPI retained 100% anti-NE activity. Previous studies by us have shown that encapsulation of rSLPI into DOPS/Chol liposomes is also a highly efficient means of protecting rSLPI from Cat L degradation (24). However, storage and nebulisation of aqueous liposome dispersions often lead to leakage of their cargo (17,51), and in our studies, leakage of rSLPI from the liposomes did occur after jet nebulisation and during their storage at 4°C rendering the leaked rSLPI vulnerable to inactivation by Cat L (24). In order to overcome these delivery and storage issues, a dry powder for inhalation of DOPS–rSLPI was prepared.
Proteins and peptides are often stored as dry powders prior to reconstitution in order to extend their shelf life, for example, enzymes, antibodies, and many other proteins used in research as well as therapeutic proteins such as insulin, have been dried to improve their long-term stability (30,52–55). Studies have also examined the potential of drying to enhance liposome stability. Storage of liposomes in the dry state extends their shelf life by minimising oxidation of lipids (41), while also preventing leakage of the entrapped material (39,40,56). More often, liposomes are formulated for inhalation as an aqueous dispersion due to ease of manufacture and nebulisation; however, the stability benefits associated with dry liposome powders has increased the number of studies investigating liposomes as dry powders for inhalation and many have achieved dry powder systems with aerodynamic properties suitable for efficient delivery to the lungs (56).
Optimisation of DOPS–rSLPI Powder Manufacture
DOPS–rSLPI powder was prepared by lyophilising the preparation followed by jet milling of the lyophilised powder. To determine drug and excipient losses due to processing, samples of powder were collected after each step and weighed and the total weight expressed as a percentage of the sum of all the components used to prepare DOPS–rSLPI liposomes up to that point. The yield of powder obtained after the lyophilisation step was calculated to be 86.7 ± 2.69% (n = 9). Lyophilisation of DOPS–rSLPI produced a white powder with a large median diameter of 19.2 ± 8.6 μm (Table I) and poor flowability.
Table I.
Particle Size and Particle Size Distribution of DOPS–rSLPI Powder Before and After Micronisation with a Micronisation Aid by Jet Mill (Venturi Pressure 7 and Ring Pressure 5 Bar) Using Different Ratios of DOPS–rSLPI/D-mannitol 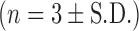
| d (0.5 μm) | Span | |
|---|---|---|
| Before micronisation | ||
| DOPS–rSLPI | 19.2 ± 8.6 | 7.9 ± 8.6 |
| After micronisation | ||
| DOPS–rSLPI/D-mannitol 1:10 | 1.6 ± 0.1 | 1.98 ± 0.1 |
| DOPS–rSLPI/D-mannitol 1:3.5 | 3.3 ± 0.3 | 2.1 ± 0.2 |
| DOPS–rSLPI/D-mannitol 1:2 | 3.5 ± 0.6 | 17.8 ± 2.8 |
Note: DOPS-rSLPI powder micronised alone, i.e. without micronisation aid, could not be harvested from the jet mill.
Micronisation of the lyophilised powder alone caused aggregation of the powder particles with no reduction in particle size to the extent that the liposome powder could not even be harvested from the jet mill. At the temperatures reached during the jet milling process, it is highly probable that phase transitions were reached and exceeded, leading to aggregation of the lipid material. Liposomes undergo various changes in their physical state depending on the lipids used in their preparation, and the temperatures they are exposed to. DOPS has quite a low transition temperature (Tm, the temperature at which the lipid changes from an ordered gel phase to a liquid crystalline phase) of −11°C. Cholesterol is frequently added to liposome formulations to limit membrane phase transition and also to strengthen and improve the fluidity of phospholipid membranes (57,58). However, at the temperatures reached during the jet milling process, which is variable depending on the type of material being micronised, it is highly possible that phase transitions would be reached and exceeded, leading to aggregation of liposome particles. Since the main transition temperature of D-mannitol is significantly higher at 10.7°C (59), D-mannitol is not as susceptible to the heats of jet milling as DOPS and thus can be used to facilitate micronisation of DOPS-based liposome powders.
The inclusion of inert powders for the micronisation of dried liposome powders for inhalation has only been investigated in another study (60). D-mannitol was used for this purpose in our study. DOPS–rSLPI and D-mannitol mixtures were prepared in ratios of 1:2, 1:3.5, and 1:10 DOPS–rSLPI/D-mannitol and subsequently micronised by jet milling. DOPS–rSLPI powder micronised with the D-mannitol micronisation aid flowed more freely, did not aggregate easily, and particle sizes within the respirable size range were readily produced. It was clear that the higher the ratio of micronisation aid to liposome included in the formulation pre-micronisation, the smaller the particle size and the narrower the associated size distribution that could be achieved upon micronisation. SEM images of DOPS–rSLPI liposome powders (DOPS–rSLPI/D-mannitol 1:10) before and after micronisation with D-mannitol are shown in Fig. 1. A significant reduction in particle size after micronisation with micronisation aid is clearly evident from the SEM images, as well as an improved and more even size distribution.
Fig. 1.

rSLPI in DOPS/Chol liposomes a freeze dried and b jet-milled with micronisation aid (DOPS–rSLPI/D-mannitol 1:10) and shown at two levels of magnification where i is low magnification (scale bar of 50 μm) and ii is high magnification (scale bar of 20 μm)
The yield of rSLPI recovered after micronisation was calculated as a percentage of that initially used in the liposome formulation and found to be 44.4 ± 7.79%, 36.1 ± 3.46%, and 54.7 ± 8.27% for DOPS–rSLPI/D-mannitol 1:10, 1:3.5, and 1:2 powders, respectively. The formulations were also tested for uniformity of rSLPI content and were found to comply with the Eur Pharm specifications for uniformity of content. Moisture content is an important determinant of the storage stability of powders since moisture levels greater than 2% can lead to particle aggregation during storage (61). This moisture-driven aggregation leads to increased particle size and results in suboptimal and inconsistent dose delivery upon aerosolisation. The moisture content of the jet-milled powder (DOPS–rSLPI/D-mannitol 1:2) was assessed using the Karl Fisher apparatus and observed to be low at 0.93 ± 0.12% water, indicating a low tendency to aggregate.
The addition of D-mannitol, as a micronisation aid, is a novel method for micronising non-friable powders, such as liposome powders. It is hypothesised that the micronisation aid coats the lyophilised liposome particles, thus causing the powder particles to take on physical properties of the dominant powder in the mix, in this case D-mannitol, which has a higher phase transition temperature. The addition of a micronisation aid such as D-mannitol may reduce the overall plasticity of the powder particles thus minimising plastic deformation and interparticlulate aggregation, improving powder flow and enhancing particle susceptibility to fracturing upon collision. The positive influence of D-mannitol inclusion prior to micronisation was immediately apparent from the quality as well as quantity of powder collected from the jet mill, since without D-mannitol as a micronisation aid, the liposome particles could not be harvested from the jet mill. The positive effects of micronisation aid inclusion were later substantiated by more in-depth characterisation of the powder based on size analysis, morphological analysis, and aerodynamic assessment.
Effect of Processing on the Stability and Activity of DOPS–rSLPI Powder
The processes of lyophilisation and jet milling can cause instability to proteins and peptides. No evidence of rSLPI degradation was seen after lyophilisation and milling, as assessed by RP-HPLC and western blot analysis.
Any deterioration in rSLPI activity after formulating DOPS–rSLPI as a powder was assessed by testing remaining activity against NE and comparing this to the activity of an equivalent amount of rSLPI formulated as DOPS–rSLPI prior to lyophilisation (Fig. 2). Percentage anti-NE activity assayed was 95.9 ± 3.59% for rSLPI in micronised DOPS–rSLPI powder, compared to 92.6 ± 10.1% for rSLPI in DOPS/Chol prior to lyophilisation. There was therefore no significant reduction in activity due to any step in the formulation process (p > 0.05).
Fig. 2.

Anti-NE activity (for equivalent amounts of rSLPI) remaining after formulation of DOPS–rSLPI as a dry powder for inhalation  Also shown is the ‘Total percentage of rSLPI encapsulated’ in the liposomes, expressed as the percentage rSLPI initially added to the formulation. % Activity is the anti-NE activity of rSLPI and is normalised back to standard non-encapsulated rSLPI. Double asterisks indicate a significant difference between two groups (p < 0.01)
Also shown is the ‘Total percentage of rSLPI encapsulated’ in the liposomes, expressed as the percentage rSLPI initially added to the formulation. % Activity is the anti-NE activity of rSLPI and is normalised back to standard non-encapsulated rSLPI. Double asterisks indicate a significant difference between two groups (p < 0.01)
Cathepsin L Challenge of DOPS–rSLPI Powder
Despite the ability of lyophilisation to improve the stability of the formulation long-term, liposomes may be compromised during the lyophilisation and micronisation processes themselves, leading to leakage of entrapped material (57,62). Therefore, the ability of the lyophilised liposomes to protect rSLPI against inactivation by Cat L was assessed as evidence of rSLPI retention within the liposomes (Fig. 3). Anti-NE activity of rSLPI remaining after incubation of resuspended DOPS–rSLPI powder with Cat L for 2 h at 37°C was 95.8 ± 0.28%. The activity of DOPS–rSLPI powder was comparable to that of the DOPS–rSLPI liposome dispersion that had not undergone powder formulation steps (92.6 ± 4.4%). This demonstrates that the protection offered to rSLPI by encapsulation in liposomes did not diminish after formulating DOPS–rSLPI liposomes as a dry powder for inhalation. It also confirms that leakage of rSLPI did not occur due to dry powder processing steps since Cat L would have inactivated any free rSLPI that had leaked out of compromised liposomes.
Fig. 3.

The retained anti-NE activity of rSLPI before and after formulation of DOPS–rSLPI as a dry powder for inhalation and compared to non-encapsulated rSLPI. All samples were incubated with Cat L at 37°C for 2 h 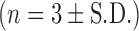 . % Activity is the anti-NE activity of rSLPI. Triple asterisks indicate a highly significant difference between rSLPI Alone and each of the other two groups (p < 0.0001)
. % Activity is the anti-NE activity of rSLPI. Triple asterisks indicate a highly significant difference between rSLPI Alone and each of the other two groups (p < 0.0001)
Aerosolisation of DOPS–rSLPI Powder
The influence that the ratio of liposome powder to micronisation aid had on the aerodynamics of the DOPS–rSLPI dry powder for inhalation was tested using the two mixtures that had displayed encouraging particle size characteristics, DOPS–rSLPI/D-mannitol (1:10) and DOPS–rSLPI/D-mannitol (1:3.5). After the micronisation step, both powder mixtures were also mixed 1:1 with large D-mannitol carrier particles (45–63 μm sieve size) known to promote efficient powder deaggregation upon aerosolisation. The powders were filled into size two gelatin capsules and aerosolised using a low resistance dry powder inhaler device, the Spinhaler®. The Spinhaler® device was actuated into the TSI. The TSI apparatus was used to study the aerodynamic properties of the formulation by assessing the emitted dose (% ED), fine particle fraction (% FPF), and non-respirable fraction (% NRF). A significantly higher emitted dose of approximately 38% was achieved for the 1:10 mixture compared to the 1:3.5 mixture (p < 0.01, Table II). However, the more concentrated liposome powder (1:3.5) appeared to achieve higher % FPFs of close to 60% compared to approximately 40% for the 1:10 mixture (p < 0.005, Table II).
Table II.
Aerosolisation of DOPS–rSLPI Powder by the Spinhaler® Dry Powder Inhaler into a Twin Stage Impinger 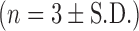
| Formulation | % rSLPI deposited | ||
|---|---|---|---|
| DOPS–rSLPI/D-mannitol powder | % ED | % FPF | % NRF |
| 1:10 | 37.7 ± 2.0 | 38.7 ± 3.6 | 61.3 ± 3.6 |
| 1:3.5 | 31.5 ± 0.5 | 59.5 ± 5.4 | 40.5 ± 5.4 |
% FPF and % NRF are expressed as a percentage of the emitted dose
% ED percent emitted dose, % FPF percent fine particle fraction, % NRF percent non-respirable fraction
It is possible that the difference in % FPFs obtained from the two DOPS–rSLPI dry powder formulations may have been influenced by particle size differences (Table II) and/or the amount of powder fines (particles <10 μm) present in the formulations. When mixed with the liposome powder and placed in the jet mill, D-mannitol present here as the micronisation aid is itself micronised to a fine particle size during the jet milling process. The presence of these fine D-mannitol particles influences the aerodynamic properties of the formulation. Therefore, D-mannitol acts not only to facilitate micronisation of DOPS–rSLPI powder, but the fine D-mannitol particles present also affect the aerodynamic properties of the end product. It appears from the % FPF achieved for both DPI products, 1:3.5 and 1:10, that the extent to which aerodynamic properties of the formulations are influenced is proportional to the amount of fines present. Many studies have considered the effect of including small quantities of fine particles (‘fines’) on the performance of DPI products. At certain percentages, it has been shown that fines can increase the % FPF of dry powders by preferentially binding to the strongest binding sites on the large carrier particles (D-mannitol with particle size >40 μm), thus preventing strong adhesion of drug to the carrier or possibly by forming mixed agglomerates that are more easily deaggregated and dispersed during aerosolisation (61,63,64). It can be surmised that the quantity of fines present in the 1:3.5 DPI formulation was beneficial in increasing % FPF. However, a higher amount of fines, such as in the 1:10 DPI formulation may prove detrimental to the aerodynamic properties by causing increased cohesive forces between the powder particles, potentially leading to ineffective deaggregation and hence poor dispersion of the powder particles upon aerosolisation. Based on these findings, it can be concluded that once the ratio of D-mannitol used in the formulation is optimised, this inert powder can play multiple beneficial roles in a DPI; its customary role in providing large carrier particles (>40 μm) with which to improve powder flow and facilitate effective deaggregation of active drug particles upon inspiratory flow, a novel role as the micronisation aid in facilitating the micronisation process, and also a role in further improving the aerosolisation of a DPI by providing fines to aid in effective deaggregation.
The ACI was used to determine the MMAD of DOPS–rSLPI powder (DOPS–rSLPI/D-mannitol, 1:10). The deposition profile of rSLPI and lipid on each stage of the cascade impactor is shown in Fig. 4. The presence of non-encapsulated rSLPI could play a role in the deposition difference seen between lipid and active proteins since the smaller molecular size of the rSLPI alone compared with DOPS–rSLPI will have a significant effect on their deposition pattern, as previously seen for liposomal dry powders of β-glucuronidase prepared for pulmonary delivery by Lu et al. 2005 (65). The MMAD was calculated to be 2.44 ± 0.12 μm with an associated GSD of 2.03 ± 0.10. The % FPF obtained was similar to that seen for TSI analysis at 33.3 ± 6.46%. DPI devices of a higher resistance than the Spinhaler® device used would be expected to generate greater air turbulence sufficient to cause deaggregation of the powder and achieve even higher % FPFs. However, the % FPFs achieved by both DOPS–rSLPI powder formulations were still satisfactory with % FPFs comparable to those achieved by liposomal powders for inhalation documented elsewhere (56,66,67).
Fig. 4.

DOPS–rSLPI powder jet-milled with D-mannitol (1:10), mixed 1:1 with D-mannitol (sieve size 45–63 μm) and aerosolised using the Spinhaler® device into an Andersen cascade impactor (ACI). Data presented as the percentage rSLPI and DOPS deposited on each stage expressed as a percentage of the sum of rSLPI and DOPS in all stages 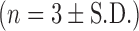
Finally, a post-aerosolisation comparison between the DOPS–rSLPI powder and aqueous dispersion in terms of their respective abilities in retaining rSLPI encapsulation within the liposomes, thus protecting rSLPI from enzymatic inactivation, was carried out using a cathepsin L challenge. Samples to be assessed were removed from the lower stage of the Twin Stage Impinger. Non-encapsulated rSLPI, aqueous DOPS–rSLPI dispersion post-nebulisation, and DOPS–rSLPI dry powder post-aerosolisation were all challenged with Cat L for 2 h, and the level of anti-NE activity remaining was determined. rSLPI anti-NE activity after aerosolisation of DOPS–rSLPI powder by a DPI and challenged with Cat L was 95.84 ± 0.28% compared to 46.8 ± 4.8% for nebulised DOPS–rSLPI aqueous dispersion (Fig. 5). These results indicate that the dry powder offers significantly more protection for rSLPI compared to the nebulised dispersion due to leakage of rSLPI from the aqueous liposome dispersion during the nebulisation process (p < 0.0001).
Fig. 5.

Percentage anti-NE activity of non-encapsulated rSLPI and rSLPI encapsulated in DOPS–rSLPI liposomes and nebulised and DOPS–rSLPI dry powder for inhalation after incubation with Cat L for 2 h at 37°C 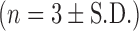 . % Activity is the anti-NE activity of rSLPI and is normalised back to standard non-encapsulated rSLPI. Triple asterisks indicate a highly significant difference between each of the three groups (p < 0.0001)
. % Activity is the anti-NE activity of rSLPI and is normalised back to standard non-encapsulated rSLPI. Triple asterisks indicate a highly significant difference between each of the three groups (p < 0.0001)
Storage Stability of DOPS–rSLPI Powder
Samples of DOPS–rSLPI powder (1:10 DOPS–rSLPI/D-mannitol) were stored in sealed glass vials in a desiccator at room temperature for 4 months as a long-term storage study. Samples were removed from storage periodically and the stability of rSLPI tested. RP-HPLC and western blot analysis indicated that rSLPI remained uncleaved and anti-NE assays indicated that rSLPI remained active throughout the storage period. The ability of the liposome carrier to protect rSLPI against inactivation by Cat L after storage was also assessed. The rSLPI activity remaining for DOPS–rSLPI powder stored for 5 months at room temperature and incubated with Cat L was 85.1 ± 5.8% and not statistically different from that of freshly prepared, aqueous DOPS–rSLPI incubated with Cat L that retained 92.6 ± 10.1% activity (Fig. 6). In a previous study by us, storage of aqueous DOPS–rSLPI liposome dispersion at 4°C for just 7 weeks rendered rSLPI anti-NE activity after incubation with Cat L at 80.4 ± 3.5% (24), but this was not a statistically significant difference from the anti-NE activity retained by freshly prepared aqueous DOPS–rSLPI after incubation with Cat L of 92.6 ± 10.1%; however, when size analysis and size distributions were compared in both DOPS–rSLPI aqueous dispersion and powder after storage, a significant difference was observed in the change in particle size and size distribution over the storage period. The liposomes stored in aqueous dispersion increased in size and size distribution by a factor of 1.35 and 3, respectively, compared to values at the beginning of the storage study (24), while the liposome powder formulation did not alter in size or size distribution throughout the 5 months of storage, suggesting that the powder formulation is a more stable formulation in terms of these measured physical properties and remains so for at least 5 months at room temperature compared to the aqueous dispersion stored at 4°C for 7 weeks.
Fig. 6.

Remaining anti-NE activity of rSLPI in DOPS–rSLPI powder incubated with cathepsin L (Cat L) for 2 h at 37°C, freshly prepared; after storage for 5 months at room temperature; data for aqueous dispersion stored for 7 weeks at 4°C taken from reference (24) 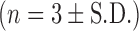 . % Activity is the anti-NE activity of rSLPI and is normalised back to standard non-encapsulated rSLPI
. % Activity is the anti-NE activity of rSLPI and is normalised back to standard non-encapsulated rSLPI
CONCLUSION
A novel method for the preparation of liposomal dry powders for protein delivery via the lungs was developed, which offers the convenience of dry powder inhalation with the additional benefits of improved liposome integrity during aerosolisation and size stability during storage.
Using D-mannitol as a micronisation aid, dry powder particles in the inhalable size range <5 μm were prepared by jet milling lyophilised DOPS–rSLPI powder. Aerosolisation of the DOPS–rSLPI dry powder mixed in a ratio of 1:10 with micronisation aid (DOPS–rSLPI/D-mannitol) produced an emitted dose of 38%, with an MMAD of 2.44 μm. When challenged with cathepsin L, the liposome dry powder was significantly superior at retaining its protective role against enzymatic inactivation of rSLPI compared to the aqueous dispersion post-nebulisation. In addition, the liposome powder was more stable physically in terms of retaining liposome size stability after storage at room temperature for 5 months. Formulation of DOPS–rSLPI liposomes as a dry powder for inhalation significantly enhances the overall stability of this liposome product during both aerosolisation and storage, when compared to the aqueous liposome dispersion.
Acknowledgements
This study was funded by the Irish Research Council for Science, Engineering and Technology (SC/2004/B0419) and the Health Research Board (HRB). The authors gratefully acknowledge the supply of rSLPI from Amgen®.
Abbreviations
- ACI
Andersen cascade impactor
- Anti-NE
Anti-neutrophil elastase
- Cat L
Cathepsin L
- CFCs
Chlorofluorocarbons
- Chol
Cholesterol
- DOPS
1,2-Dioleoyl-sn-glycero-3-[phospho-L-serine]
- DOPS–rSLPI
rSLPI encapsulated in DOPS/Chol liposomes
- DPI
Dry powder inhaler
- ED
Emitted dose
- FPF
Fine particle fraction
- GSD
Geometric standard deviation
- MMAD
Mass median aerodynamic diameter
- NE
Neutrophil elastase
- NRF
Non-respirable fraction
- SEM
Scanning electron microscopy
- rSLPI
Recombinant secretory leukocyte protease inhibitor
- SLPI
Secretory leukocyte protease inhibitor
- TSI
Twin stage impinger
References
- 1.Barrios VE, Middleton SC, Kashem MA, Havill AM, Toombs CF, Wright CD. Tryptase mediates hyperresponsiveness in isolated guinea pig bronchi. Life Sci. 1998;63(26):2295–303. doi: 10.1016/S0024-3205(98)00518-9. [DOI] [PubMed] [Google Scholar]
- 2.Forteza RM, Ahmed A, Lee T, Abraham WM. Secretory leukocyte protease inhibitor, but not alpha-1 protease inhibitor, blocks tryptase-induced bronchoconstriction. Pulm Pharmacol Ther. 2001;14(2):107–10. doi: 10.1006/pupt.2000.0276. [DOI] [PubMed] [Google Scholar]
- 3.Gillissen A, Birrer P, McElvaney NG, Buhl R, Vogelmeier C, Hoyt RF, Jr, et al. Recombinant secretory leukoprotease inhibitor augments glutathione levels in lung epithelial lining fluid. J Appl Physiol. 1993;75(2):825–32. doi: 10.1152/jappl.1993.75.2.825. [DOI] [PubMed] [Google Scholar]
- 4.McElvaney NG, Doujaiji B, Moan MJ, Burnham MR, Wu MC, Crystal RG. Pharmacokinetics of recombinant secretory leukoprotease inhibitor aerosolized to normals and individuals with cystic fibrosis. Am Rev Respir Dis. 1993;148(4 Pt 1):1056–60. doi: 10.1164/ajrccm/148.4_Pt_1.1056. [DOI] [PubMed] [Google Scholar]
- 5.Wright CD, Havill AM, Middleton SC, Kashem MA, Lee PA, Dripps DJ, et al. Secretory leukocyte protease inhibitor prevents allergen-induced pulmonary responses in animal models of asthma. J Pharmacol Exp Ther. 1999;289(2):1007–14. [PubMed] [Google Scholar]
- 6.Keatings VM, Jatakanon A, Worsdell YM, Barnes PJ. Effects of inhaled and oral glucocorticoids on inflammatory indices in asthma and COPD. Am J Respir Crit Care Med. 1997;155(2):542–8. doi: 10.1164/ajrccm.155.2.9032192. [DOI] [PubMed] [Google Scholar]
- 7.Adcock IM. Steroid resistance in asthma. Molecular mechanisms. Am J Respir Crit Care Med. 1996;154(2 Pt 2):S58–61. doi: 10.1164/ajrccm/154.2_Pt_2.S58. [DOI] [PubMed] [Google Scholar]
- 8.Bergenfeldt M, Bjork P, Ohlsson K. The elimination of secretory leukocyte protease inhibitor (SLPI) after intravenous injection in dog and man. Scand J Clin Lab Invest. 1990;50(7):729–37. doi: 10.3109/00365519009091066. [DOI] [PubMed] [Google Scholar]
- 9.Stolk J, Camps J, Feitsma HI, Hermans J, Dijkman JH, Pauwels EK. Pulmonary deposition and disappearance of aerosolised secretory leucocyte protease inhibitor. Thorax. 1995;50(6):645–50. doi: 10.1136/thx.50.6.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gast A, Anderson W, Probst A, Nick H, Thompson RC, Eisenberg SP, et al. Pharmacokinetics and distribution of recombinant secretory leukocyte proteinase inhibitor in rats. Am Rev Respir Dis. 1990;141(4 Pt 1):889–94. doi: 10.1164/ajrccm/141.4_Pt_1.889. [DOI] [PubMed] [Google Scholar]
- 11.McElvaney NG, Nakamura H, Birrer P, Hebert CA, Wong WL, Alphonso M, et al. Modulation of airway inflammation in cystic fibrosis. In vivo suppression of interleukin-8 levels on the respiratory epithelial surface by aerosolization of recombinant secretory leukoprotease inhibitor. J Clin Invest. 1992;90(4):1296–301. doi: 10.1172/JCI115994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogelmeier C, Gillissen A, Buhl R. Use of secretory leukoprotease inhibitor to augment lung antineutrophil elastase activity. Chest. 1996;110(6 Suppl):261S–6S. doi: 10.1378/chest.110.6_Supplement.261S. [DOI] [PubMed] [Google Scholar]
- 13.Taggart CC, Lowe GJ, Greene CM, Mulgrew AT, O’Neill SJ, Levine RL, et al. Cathepsin B, L, and S cleave and inactivate secretory leucoprotease inhibitor. J Biol Chem. 2001;276(36):33345–52. doi: 10.1074/jbc.M103220200. [DOI] [PubMed] [Google Scholar]
- 14.Couvreur P, Fattal E, Andremont A. Liposomes and nanoparticles in the treatment of intracellular bacterial infections. Pharm Res. 1991;8(9):1079–86. doi: 10.1023/A:1015885814417. [DOI] [PubMed] [Google Scholar]
- 15.McCullough HN, Juliano RL. Organ-selective action of an antitumor drug: pharmacologic studies of liposome-encapsulated beta-cytosine arabinoside administered via the respiratory system of the rat. J Natl Cancer Inst. 1979;63(3):727–31. doi: 10.1093/jnci/63.3.727. [DOI] [PubMed] [Google Scholar]
- 16.Taylor KM, Taylor G, Kellaway IW, Stevens J. The influence of liposomal encapsulation on sodium cromoglycate pharmacokinetics in man. Pharm Res. 1989;6(7):633–6. doi: 10.1023/A:1015917918130. [DOI] [PubMed] [Google Scholar]
- 17.Niven RW, Schreier H. Nebulization of liposomes. I. Effects of lipid composition. Pharm Res. 1990;7(11):1127–33. doi: 10.1023/A:1015924124180. [DOI] [PubMed] [Google Scholar]
- 18.Misra A, Jinturkar K, Patel D, Lalani J, Chougule M. Recent advances in liposomal dry powder formulations: preparation and evaluation. Expert Opin Drug Deliv. 2009;6(1):71–89. doi: 10.1517/17425240802652309. [DOI] [PubMed] [Google Scholar]
- 19.Clark JM, Whitney RR, Olsen SJ, George RJ, Swerdel MR, Kunselman L, et al. Amphotericin B lipid complex therapy of experimental fungal infections in mice. Antimicrob Agents Chemother. 1991;35(4):615–21. doi: 10.1128/aac.35.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freise CE, Liu T, Hong K, Osorio RW, Papahadjopoulos D, Ferrell L, et al. The increased efficacy and decreased nephrotoxicity of a cyclosporine liposome. Transplantation. 1994;57(6):928–32. doi: 10.1097/00007890-199403270-00027. [DOI] [PubMed] [Google Scholar]
- 21.Gruber SA, Venkataram S, Canafax DM, Cipolle RJ, Bowers L, Elsberry D, et al. Liposomal formulation eliminates acute toxicity and pump incompatibility of parenteral cyclosporine. Pharm Res. 1989;6(7):601–7. doi: 10.1023/A:1015905615404. [DOI] [PubMed] [Google Scholar]
- 22.Wyde PR, Six HR, Wilson SZ, Gilbert BE, Knight V. Activity against rhinoviruses, toxicity, and delivery in aerosol of enviroxime in liposomes. Antimicrob Agents Chemother. 1988;32(6):890–5. doi: 10.1128/aac.32.6.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhavane R, Karathanasis E, Annapragada AV. Agglomerated vesicle technology: a new class of particles for controlled and modulated pulmonary drug delivery. J Control Release. 2003;93(1):15–28. doi: 10.1016/S0168-3659(03)00359-6. [DOI] [PubMed] [Google Scholar]
- 24.Gibbons AM, McElvaney NG, Taggart CC, Cryan SA. Delivery of rSLPI in a liposomal carrier for inhalation provides protection against cathepsin L degradation. J Microencapsul. 2009;26:513–522. doi: 10.1080/02652040802466535. [DOI] [PubMed] [Google Scholar]
- 25.Hansen NC, Evald T, Ibsen TB. Terbutaline inhalations by the Turbuhaler as replacement for domiciliary nebulizer therapy in severe chronic obstructive pulmonary disease. Respir Med. 1994;88(4):267–71. doi: 10.1016/0954-6111(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 26.Chougule MB, Padhi BK, Jinturkar KA, Misra A. Development of dry powder inhalers. Recent Pat Drug Deliv Formul. 2007;1:11–21. doi: 10.2174/187221107779814159. [DOI] [PubMed] [Google Scholar]
- 27.Zhu J, Wen J, Ma Y, Zhang H, Zhu J, Wen J, Ma Y, Zhang H, Zhu J, Wen J, Ma Y, Zhang Hs. Dry Powder Inhaler. USA. 2007.
- 28.Newman SP. Dry powder inhalers for optimal drug delivery. Expert Opin Biol Ther. 2004;4(1):23–33. doi: 10.1517/14712598.4.1.23. [DOI] [PubMed] [Google Scholar]
- 29.Niven RW. Delivery of biotherapeutics by inhalation aerosol. Crit Rev Ther Drug Carrier Syst. 1995;12(2–3):151–231. doi: 10.1615/critrevtherdrugcarriersyst.v12.i2-3.20. [DOI] [PubMed] [Google Scholar]
- 30.Salnikova MS, Middaugh CR, Rytting JH. Stability of lyophilized human growth hormone. Int J Pharm. 2008;358(1–2):108–13. doi: 10.1016/j.ijpharm.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 31.DePaz RA, Dale DA, Barnett CC, Carpenter JF, Gaertner AL, Randolph TW. Effects of drying methods and additives on the structure, function, and storage stability of subtilisin: role of protein conformation and molecular mobility. Enzyme Microb Technol. 2002;31(6):765–74. doi: 10.1016/S0141-0229(02)00173-4. [DOI] [Google Scholar]
- 32.Franks F. Freeze-drying of bioproducts: putting principles into practice. Eur J Pharm Biopharm. 1998;45(3):221–9. doi: 10.1016/S0939-6411(98)00004-6. [DOI] [PubMed] [Google Scholar]
- 33.Kensil CR, Dennis EA. Alkaline hydrolysis of phospholipids in model membranes and the dependence on their state of aggregation. Biochemistry. 1981;20(21):6079–85. doi: 10.1021/bi00524a025. [DOI] [PubMed] [Google Scholar]
- 34.Grit M, Zuidam NJ, Underberg WJ, Crommelin DJ. Hydrolysis of partially saturated egg phosphatidylcholine in aqueous liposome dispersions and the effect of cholesterol incorporation on hydrolysis kinetics. J Pharm Pharmacol. 1993;45(6):490–5. doi: 10.1111/j.2042-7158.1993.tb05585.x. [DOI] [PubMed] [Google Scholar]
- 35.Grit M, Crommelin DJ. The effect of aging on the physical stability of liposome dispersions. Chem Phys Lipids. 1992;62(2):113–22. doi: 10.1016/0009-3084(92)90089-8. [DOI] [PubMed] [Google Scholar]
- 36.Vemuri S, Rhodes CT. Preparation and characterization of liposomes as therapeutic delivery systems: a review. Pharm Acta Helv. 1995;70(2):95–111. doi: 10.1016/0031-6865(95)00010-7. [DOI] [PubMed] [Google Scholar]
- 37.Ausborn M, Nuhn P, Schreier H. Stabilization of liposomes by freeze-thaw- and lyophilization techniques: problems and opportunities. Eur J Pharm Biopharm. 1992;38(4):133–8. [Google Scholar]
- 38.Lloyd AW, Olliff CJ, Rutt KJ. A comparison of carboxylate salts as liposomal cryoprotectants. Int J Pharm. 1996;131(2):257–62. doi: 10.1016/0378-5173(95)04361-6. [DOI] [Google Scholar]
- 39.Glavas-Dodov M, Fredro-Kumbaradzi E, Goracinova K, Simonoska M, Calis S, Trajkovic-Jolevska S, et al. The effects of lyophilization on the stability of liposomes containing 5-FU. Int J Pharm. 2005;291(1–2):79–86. doi: 10.1016/j.ijpharm.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 40.Zhang JA, Xuan T, Parmar M, Ma L, Ugwu S, Ali S, et al. Development and characterization of a novel liposome-based formulation of SN-38. Int J Pharm. 2004;270(1–2):93–107. doi: 10.1016/j.ijpharm.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 41.Shulkin PM, Seltzer SE, Davis MA, Adams DF. Lyophilized liposomes: a new method for long-term vesicular storage. J Microencapsul. 1984;1(1):73–80. doi: 10.3109/02652048409031539. [DOI] [PubMed] [Google Scholar]
- 42.Changsan N, Chan HK, Separovic F, Srichana T. Physicochemical characterization and stability of rifampicin liposome dry powder formulations for inhalation. J Pharm Sci. 2009;98:628–39. doi: 10.1002/jps.21441. [DOI] [PubMed] [Google Scholar]
- 43.van Bommel EMG, Crommelin DJ. Stability of doxorubicin-liposomes on storage: as an aqueous dispersion, frozen or freeze-dried. Int J Pharm. 1984;22:299–310. doi: 10.1016/0378-5173(84)90030-9. [DOI] [PubMed] [Google Scholar]
- 44.Taylor KMG, Taylor G, Kellaway IW, Stevens J. The stability of liposomes to nebulisation. Int J Pharm. 1990;58(1):57–61. doi: 10.1016/0378-5173(90)90287-E. [DOI] [Google Scholar]
- 45.Niven RW, Speer M, Schreier H. Nebulization of liposomes. II. The effects of size and modeling of solute release profiles. Pharm Res. 1991;8(2):217–21. doi: 10.1023/A:1015896121377. [DOI] [PubMed] [Google Scholar]
- 46.Bridges PA, Taylor AJ, McCallion ON. Nebulisation of liposomes: the effect of formulation variables. Bristol: The Aerosol Society; 1995. [Google Scholar]
- 47.Bridges PA, Taylor AJ, McCallion ON. The effects of lipid concentration on the nebulisation of liposomes. Proc Int Symp Control Release Bioact Mater. 1995;22:2021–2. [Google Scholar]
- 48.McCallion ON, Taylor KM, Thomas M, Taylor AJ. Nebulization of fluids of different physicochemical properties with air-jet and ultrasonic nebulizers. Pharm Res. 1995;12(11):1682–8. doi: 10.1023/A:1016205520044. [DOI] [PubMed] [Google Scholar]
- 49.Stewart JC. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem. 1980;104(1):10–4. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 50.Mayer LD, Bally MB, Hope MJ, Cullis PR. Techniques for encapsulating bioactive agents into liposomes. Chem Phys Lipids. 1986;40(2–4):333–45. doi: 10.1016/0009-3084(86)90077-0. [DOI] [PubMed] [Google Scholar]
- 51.Niven RW, Carvajal TM, Schreier H. Nebulization of liposomes. III. The effects of operating conditions and local environment. Pharm Res. 1992;9(4):515–20. doi: 10.1023/A:1015844430695. [DOI] [PubMed] [Google Scholar]
- 52.Patton JS, Bukar J, Nagarajan S. Inhaled insulin. Adv Drug Deliv Rev. 1999;35(2–3):235–47. doi: 10.1016/S0169-409X(98)00074-X. [DOI] [PubMed] [Google Scholar]
- 53.Rave KM, Nosek L, de la Pena A, Seger M, Ernest CS, 2nd, Heinemann L, et al. Dose response of inhaled dry-powder insulin and dose equivalence to subcutaneous insulin lispro. Diab Care. 2005;28(10):2400–5. doi: 10.2337/diacare.28.10.2400. [DOI] [PubMed] [Google Scholar]
- 54.Hollander PA, Blonde L, Rowe R, Mehta AE, Milburn JL, Hershon KS, et al. Efficacy and safety of inhaled insulin (exubera) compared with subcutaneous insulin therapy in patients with type 2 diabetes: results of a 6-month, randomized, comparative trial. Diab Care. 2004;27(10):2356–62. doi: 10.2337/diacare.27.10.2356. [DOI] [PubMed] [Google Scholar]
- 55.Codrons V, Vanderbist F, Verbeeck RK, Arras M, Lison D, Preat V, et al. Systemic delivery of parathyroid hormone (1–34) using inhalation dry powders in rats. J Pharm Sci. 2003;92(5):938–50. doi: 10.1002/jps.10346. [DOI] [PubMed] [Google Scholar]
- 56.Schreier H, Mobley WC, Concessio N, Hickey AJ, Niven R. Formulation and in vitro performance of liposome powder aerosol. STP Pharm Sci. 1994;4:38–44. [Google Scholar]
- 57.Mobley WC. The effect of jet-milling on lyophilized liposomes. Pharm Res. 1998;15(1):149–52. doi: 10.1023/A:1011929626769. [DOI] [PubMed] [Google Scholar]
- 58.Bloom M, Evans E, Mouritsen OG. Physical properties of the fluid lipid-bilayer component of cell membranes: a perspective. Q Rev Biophys. 1991;24(3):293–397. doi: 10.1017/S0033583500003735. [DOI] [PubMed] [Google Scholar]
- 59.Yu L, Mishra DS, Rigsbee DR. Determination of the glass properties of D-mannitol using sorbitol as an impurity. J Pharm Sci. 1998;87(6):774–7. doi: 10.1021/js970224o. [DOI] [PubMed] [Google Scholar]
- 60.Desai TR, Hancock RE, Finlay WH. Delivery of liposomes in dry powder form: aerodynamic dispersion properties. Eur J Pharm Sci. 2003;20(4–5):459–67. doi: 10.1016/j.ejps.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 61.Shah SP, Misra A. Liposomal amikacin dry powder inhaler: effect of fines on in vitro performance. AAPS PharmSciTech. 2004;5(4):e65. doi: 10.1208/pt050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Desai TR, Wong JP, Hancock RE, Finlay WH. A novel approach to the pulmonary delivery of liposomes in dry powder form to eliminate the deleterious effects of milling. J Pharm Sci. 2002;91(2):482–91. doi: 10.1002/jps.10021. [DOI] [PubMed] [Google Scholar]
- 63.Jones MD, Price R. The influence of fine excipient particles on the performance of carrier-based dry powder inhalation formulations. Pharm Res. 2006;23(8):1665–74. doi: 10.1007/s11095-006-9012-7. [DOI] [PubMed] [Google Scholar]
- 64.Shah SP, Misra A. Liposomal amphotericin B dry powder inhaler: effect of fines on in vitro performance. Pharmazie. 2004;59(10):812–3. [PubMed] [Google Scholar]
- 65.Lu D, Hickey AJ. Liposomal dry powders as aerosols for pulmonary delivery of proteins. AAPS PharmSciTech. 2005;6(4):E641–8. doi: 10.1208/pt060480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joshi M, Misra A. Dry powder inhalation of liposomal Ketotifen fumarate: formulation and characterization. Int J Pharm. 2001;223(1–2):15–27. doi: 10.1016/S0378-5173(01)00705-0. [DOI] [PubMed] [Google Scholar]
- 67.Joshi MR, Misra A. Liposomal budesonide for dry powder inhaler: preparation and stabilization. AAPS PharmSciTech. 2001;2(4):25. doi: 10.1208/pt020425. [DOI] [PMC free article] [PubMed] [Google Scholar]