

SUMMARY
Small molecules stabilize specific protein conformations from a larger ensemble, enabling molecular switches that control diverse cellular functions. We show here that the converse also holds true, where the conformational state of the estrogen receptor can direct distinct orientations of the bound ligand. “Gain of allostery” mutations that mimic the effects of ligand in driving protein conformation allowed crystallization of the partial agonist ligand WAY-169916 with both the canonical active and inactive conformations of the estrogen receptor. The intermediate transcriptional activity induced by WAY169916 is associated with the ligand binding differently to the active and inactive conformations of the receptor. Analyses of a series of chemical derivatives demonstrated that altering the ensemble of ligand binding orientations changes signaling output. The coupling of different ligand binding orientations to distinct active and inactive protein conformations defines a novel mechanism for titrating allosteric signaling activity.
INTRODUCTION
Defining the principles of ligand binding and allostery are important for understanding small molecule signaling in cellular physiology, and for developing improved therapeutic agents. In their folded state, proteins display an ensemble of conformations that encompass an energy landscape (Figure 1)1-5. Within this framework, current ligand binding theory holds that a given ligand will interact with a subset of conformations that are competent for binding, thus redistributing the native state population and allowing allosteric changes in other binding sites 6-13. Small molecule selection of protein conformation is fundamental to many cellular processes, including signal transduction 14, molecular movement 15, allostery 16,17, and nucleotide polymerization 18. A critical feature of these events is that the selection of protein conformation drives an on-off type switch in function. However, allosteric modulators, including ligands for nuclear receptors 19-21, kinases, and GPCRs 22, can induce graded signaling outcomes, through mechanisms that are as yet poorly understood.
Figure 1. An Energy Landscape Model of Ligand Binding.
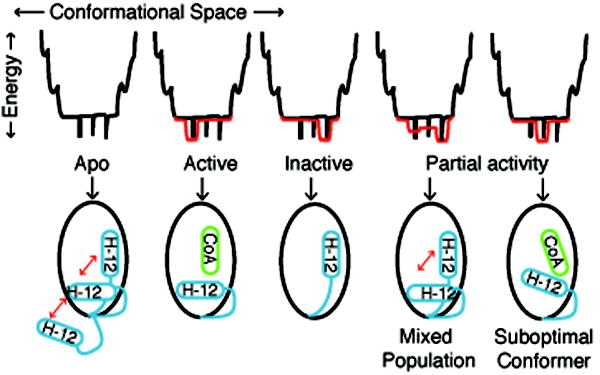
The relationship between protein conformations and energy is depicted as an energy well, with folded proteins populating the bottom regions of the well. In the statistical ensemble model of ligand binding, the relative population of different protein conformers is determined by the statistical sum of the thermodynamic energy of each protein-ligand substate. In the absence of ligand, nuclear receptors are thought to be conformationally dynamic, shown as black spikes at the bottom of the energy well. Full agonists or antagonists productively interact with a subset of lower energy conformations, shown in red. For nuclear receptors, agonist or antagonist ligands stabilize specific conformations of helix 12 (H-12). Partial agonists could either stabilize a unique conformer with suboptimal coactivator binding, or induce a mixed population of active and inactive conformers.
Nuclear steroid hormone receptors, such as estrogen receptor-α (ERα), are allosterically regulated transcription factors that undergo ligand-driven conformational selection 9,10,23. The ER ligand-binding domain (LBD) is comprised of twelve alpha helices and a beta sheet, which are organized into a hydrophobic core, a ligand-binding pocket, and a transcriptional coregulator binding site. The most C-terminal helix, helix 12, is stabilized by full agonist ligands to dock against helices 3 and 11, a conformation where it forms part of the binding site for transcriptional coactivator proteins that carry forward the agonist signal 10,23. In contrast, antagonists such as tamoxifen stabilize an inactive conformation of the receptor, because their bulky and basic side chain obstructs the active conformation of helix 12, repositioning this helix into the coactivator-binding pocket, blocking coregulator interactions and interrupting the signal 10,24. This helix 12 allosteric switch is highly conserved in the nuclear receptor superfamily 23,25, allowing ligand selection of either the active or inactive conformations. However, nuclear receptors can also direct ligand-specific, graded activity through poorly understood mechanisms. The impact of conformational dynamics on the bound ligand is also poorly understood.
In order to analyze the effects of different conformations on the bound ligand, we used surface mutations to stabilize the canonical active and inactive conformations of ERα. Here we show that distinct active and inactive conformations of ERα bind the ligand in different orientations. Remarkably, the partial ER agonist WAY-169916 (Table 1, 1) bound differently to the active and inactive conformers of ERα, suggesting a novel mechanism for titrating activity. We synthesized new derivatives of WAY-166916 that crystallized with ERα in different binding orientations from the parent compound, which are consistent with the distinct activity profiles of these new compounds. These findings suggest a new mechanism for generating graded allosteric signaling, whereby an ensemble of bound ligand orientations determines the percent of active and inactive protein conformers in solution.
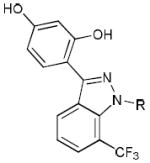
Table 1.
Chemical Derivatives of Way-169916
| Substitution | ERα RBA* | |
|---|---|---|
 |
|
0.076 |
|
|
0.32 | |
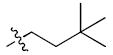 Dimethylbutyl, 3 Dimethylbutyl, 3
|
0.36 | |
|
|
1.46 | |
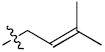 Methylbutenyl, 5 Methylbutenyl, 5
|
0.25 | |
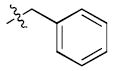 Benzyl, 6 Benzyl, 6
|
0.20 | |
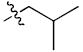 Isobutyl, 7 Isobutyl, 7
|
0.32 |
Relative Binding Affinity as a percentage of estradiol affinity (0.2nM)
RESULTS
Characterization of WAY-169916-driven ER activities
The ERα partial agonist ligand, WAY-169916 (Table 1), represents the first example of an ER ligand that has broad anti-inflammatory activity in vivo, but that lacks the proliferative effects associated with strong transcriptional activation 26,27. As expected, WAY-169916 demonstrated limited activation of an estrogen response element (ERE)-driven luciferase reporter (Figure 2a-b) and endogenous estrogen- responsive genes, pS2/Trefoil and Greb1 (Figure 2c). However, WAY-169916 strongly inhibited TNFα-induced expression of the inflammatory gene, Monocyte Chemoattractant Protein-1 (MCP-1, Figure 2d), demonstrating that this ERα ligand is active in cells and that it suppresses this mechanistically distinct, NF-κB dependent signaling pathway 26. Further, estradiol-dependent interaction between ERα and the Grip1 transcriptional coactivator was completely blocked by the full antagonist, ICI182,780 (Fulvestrant), but was only partially blocked by WAY-169916 (Figure 2e). Therefore, WAY-169916 displays partial agonist transcriptional activity and intermediate coactivator recruitment.
Figure 2. WAY-169916 is an ERα partial agonist.
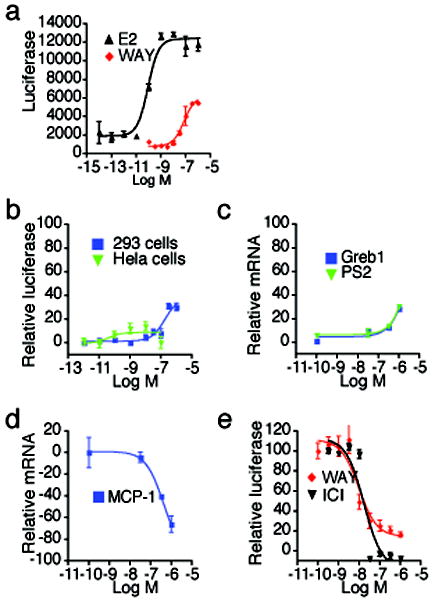
(a) Estrogen responsive luciferase assays were performed by transfection of a 3xERE-luc reporter in ER-positive MCF-7 cells. After 24 hr, cells were treated with the indicated doses of ligands overnight, and then assessed for luciferase activity.
(b) The indicated cells were transfected as in (a), but with the addition of an ERα expression plasmid. Shown are dose responses to WAY-169916, relative to estradiol treatment.
(c) Estrogen responsive gene expression was assayed in MCF-7 cells treated for 2 hr with the indicated doses of WAY-169916. Cells were then processed for qRT-PCR analysis of mRNA, which was normalized to 18S rRNA levels.
(d) The NFκB responsive gene, MCP-1 was induced by treating MCF-7 cells for 2 hr with 15 ng/ml of TNFα, and increasing doses of WAY-169916, and then processed for qRT-PCR analysis of mRNA levels.
(e) A mammalian two-hybrid assay was used to define the ligand dependent interaction of the Gal4-Grip1 coactivator and VP16-ERα LBD, using a Gal4 response element luciferase reporter. Data represent mean + SEM for (a-e), and are plotted as a percentage of the maximal response seen with estradiol for (b-e). ICI = ICI182,780 (faslodex, Fulvestrant)
Validation of gain of allostery ERα mutants
Partial agonists have proven difficult to crystallize, which we surmise relates to their induction of a mixed population of active and inactive conformers, interfering with crystallization. To overcome this, we previously demonstrated that ERα Y537S stabilizes helix 12 in the agonist conformation by adding an additional hydrogen bond between helices 3 and 12 28, allowing us to crystallize a series of partial agonists. Importantly, the Y537S mutation is on the surface of the ERα LBD and does not affect the interaction of the receptor with its ligands, as shown previously by the crystallization of genistein with both wild type and Y537S mutant 28. Further, the mutation can be fully antagonized by tamoxifen in cells 29, demonstrating that a single hydrogen bond can only stabilize conformers that are reasonably well populated in solution. Thus this gain of allostery approach allowed us to interrogate ligand specific reorganizations inside the ligand binding pocket, within the context of the active conformation 28,30. We used this mutant to obtain the crystal structure of WAY-166916 bound to ERα Y537S, in the active conformation (Supplemental Figure 1a, Supplemental Table 1).
To define the interaction of WAY-169916 bound to the inactive conformation of the receptor, we crystallized it with ERα LBD L536S/L372R (Supplemental Figure 1b, Supplemental Table 1), a receptor with mutations that stabilize helix 12 in the conformation seen with eleven published antagonist-bound ERα structures. These mutations were designed to add hydrogen bonds and stabilize helix 12 in the canonical inactive conformation. This 2.3 Å structure revealed an overall fold, and location of helix 12, identical to that seen with the previously published structures of ERα with antagonists (Supplemental Figure 2a). We also obtained a crystal structure of Raloxifene with ERα L536S (Supplemental Table 1), which also displayed the canonical inactive conformation of helix 12 (Supplemental Figure 2b), and identical ligand binding compared to a published wild-type structure24 (Supplemental Figure 2c). Thus, the mutations stabilize the canonical inactive conformation without altering ligand interactions with the receptor.
As an additional functional test, we examined corepressor association with inactive conformation mutants using a mammalian two-hybrid assay. A stabilization of helix 12 in the coregulator-binding cleft should block recruitment of corepressors to the coregulator binding site 9, and indeed this was manifest for both the L536S and L372R mutations (Supplemental Figure 2d). Though all mutants were expressed at levels lower than wild type ERα (Supplemental Figure 2e), deletion of helix 12 (531stop) allowed full interaction with corepressor. Thus, the failure of the 372R and 536S mutants to bind to NCoR is not due to reduced expression, and helix 12 blocks the NCoR interaction. Structure and coregulator binding thus establish that these mutants indeed stabilize the well-characterized inactive conformation of ERα.
Crystallization with the active conformation of ERα
The structure of WAY-169916 when bound to the active conformation of Y537S ERα LBD was solved at 1.8 Å resolution (Supplemental Table 1), and displays very clear electron density for the ligand (Figure 3a). As with other partial agonist ligands crystallized with the active conformation of the ERα LBD 28,30, WAY-169916 shifted helix 11 towards helix 12 (Figure 3b), mediated by ligand-specific repositioning of L525 and H524 in helix 11 (Figure 3c). This shift in helix 11 pushes the C-terminal half of helix 12 away from the core of the receptor, relative to full agonist structures (Figure 3b-c). This suboptimal packing of helix 12 suggests that partial agonists induce a “strain” in the active conformation, leading to helix 12 adopting the active conformation only part of the time in solution. Here, the mutant hydrogen bond stabilizes the strained active conformation, allowing it to be visualized.
Figure 3. The structure of WAY-169916 bound to the active conformation of ERα.
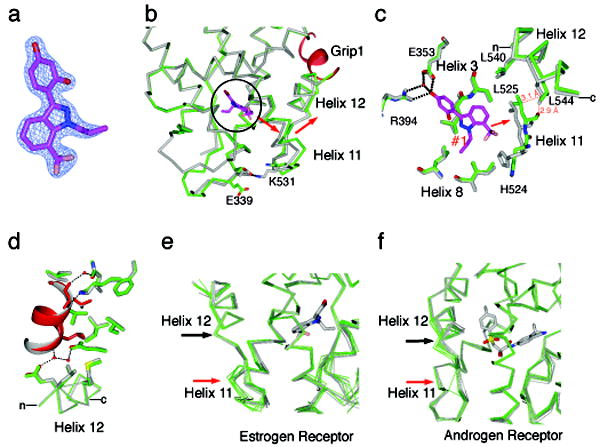
(a) A 2Fo-Fc map shows the electron density for the ligand, and is contoured to 2.0 σ.
(b) A portion of the structure of WAY-169916 bound to ERα in the active conformation (green) is shown superimposed with the structure of ERα bound to a full agonist (gray) 31. The ligand is oriented in the Z-axis of the image and is circled. The Grip1 coactivator peptide is colored red. The red arrow indicates the shift in the positioning of helix 11, and the associated shift in the C-terminus of helix 12 in the WAY-169916 structure.
(c) Selected residues in the ligand-binding pocket are shown, illustrating how the positioning of WAY-169916 (magenta) induces a shift in helix 11 (green), relative to the full agonist structure (gray). The red dashed lines show clashes between the WAY-169916 induced position of helix 11 and helix 12 in the superimposed full agonist structure.
(d) The WAY-169916 (green) and full agonist structure (gray) 31 were superimposed on the coactivator binding site, including helices 3-5 and 12. Shown are the amino acids that directly contact the Grip1 peptide, which is colored red in the WAY-169916 structure.
(e) The WAY-169916 structure and 6 published partial agonist structures (green) 28,30 were superimposed with 7 full agonist structures (gray) 28,31, and shown as c-α traces.
(f) A series of 17 androgen receptor structures were superimposed, and colored as in (e). Here, partial agonists distort the helix 12 portion of the coactivator binding cleft.
Generally, allosteric modulators could induce intermediate activity by stabilizing a specific conformation of the second binding site that is distorted (Figure 1). If this “suboptimal conformation model” were operative, it should be evident by changes in the coactivator binding site that would lead to reduced affinity for coactivators. Alternatively, intermediate activity could derive from a “mixed population model” consisting of distinct active and inactive conformers that coexist.
We superimposed the coactivator binding sites of the WAY-169916 and a full agonist structure 31, including helices 3-5 and helix 12. An R.M.S.D. of 0.24 Å for the superimposed coactivator binding sites suggests no differences, and visualization of this region shows identical conformations for the coactivator peptide, and the amino acids contacting the peptide (Figure 3d). While the C-terminus of helix 12 is shifted indirectly by WAY-169916, the N-terminal portion that contacts the Grip1 coactivator peptide is identical to the full agonist structure. Further analysis of seven partial agonist structures demonstrates that the N-terminus of helix 12 is not changed (Figure 3e, black arrow), even though helix 11 is distorted (Figure 3e, red arrow). Strikingly, the androgen receptor shows the opposite pattern (Figure 3f), where partial agonists grossly distort the coactivator binding site by repositioning the N-terminus of helix 12. Thus, for ERα, partial agonist activity is not associated with a distorted coactivator binding site, but rather with control of helix 12 dynamics by helix 11, suggesting that both active and inactive conformers are significantly populated in solution.
Protein conformation determines ligand orientation
In the active conformation structures, helix 12 constrains the mobility of helices 3 and 11. This suggested that WAY-169916 could induce a further widening of the pocket when helix 12 relocates in the inactive conformer mutant (Supplemental Figure 1b). We measured the intra-molecular distances of helix 12 L525 to either A350 in helix 3, or F404 in the beta-sheet, for all four molecules in the asymmetric unit of the WAY-169916 inactive ERα conformer structure. Compared to eleven published inactive conformation structures bound to various antagonists, the widening of the pocket by more than 2 Å caused by WAY-196619 is highly significant (Student’s t-test, p< 1×10-6)(Figure 4a).
Figure 4. An unconstrained inactive conformation of ERα shows two non-overlapping orientations of WAY-169916.
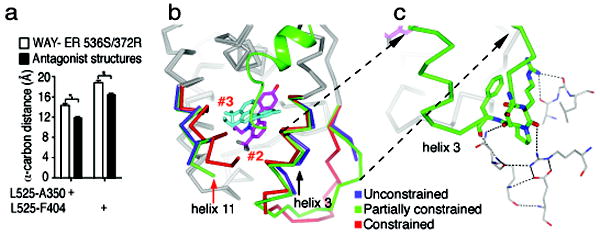
(a) The width of the ligand-binding pocket was measured between the main chain carbon of the indicated amino acids. Shown is the distance for the 4 chains of the WAY-169916 inactive conformation structure compared with 11 published ERα structures bound to different antagonists.
(b) The active conformer structure (red) was superimposed with the two chains of the dimer in the inactive conformation structure. Both of these chains (blue and green) show similar large shifts in helix 11 (red arrow), but differential shifts in helix 3 (black arrow). Two molecules of bound WAY-169916 are colored magenta and cyan, defining ligand orientations #2 and #3, respectively.
(c) The chain with a partially constrained conformer of helix 3 shows stabilization of the preceding loop by crystal packing. The symmetry related model is show as thin sticks, and the dashed lines represent hydrogen bonds. The partially constrained conformer is shows as c-alpha trace, with helix 3 and the preceding loop colored green, along with selected amino acids that participate in crystal packing.
This widening is largely due to shifts in the positioning of helix 11 in both chains in the WAY-166916-bound inactive conformation dimer (Figure 4b, red arrow). Helix 11 forms part of the dimer interface, where the C-terminal ends of the paired helices 11 move 3.1 Å closer to each other, compared to the agonist conformation structure of WAY-169916 (Supplemental Figure 2). Importantly, the nearest crystal contact is more than 12 Å from the C-terminus of helix 11, indicating that the shift is indeed ligand induced, and not a result of crystal packing. In contrast to helix 11, the two chains in the dimer show differences in the degree of movement in helix 3 (Figure 4b, black arrow). Specifically, in one molecule of the dimer, helix 3 has no crystal contacts, allowing unconstrained motion of both helix 3 and helix 11. In the other monomer, the loop preceding helix 3 is stabilized by significant crystal packing interactions (Figure 4c), which limit its flexibility, resulting in a partially constrained conformation that lies between the fully constrained (agonist conformation) and fully unconstrained positions (Figure 4b).
The fully, partially, and un-constrained conformations of helices 3 and 11 define three unique binding modes for WAY-169916. We coined the ligand orientation in the active conformer as orientation #1 (Figure 5). Remarkably, the unconstrained conformer demonstrates simultaneous binding of two non-overlapping molecules of the ligand, in orientations #2 and #3 (Figure 4b, Figure 5, and Supplemental Figures 3 and 4), a novel finding for the steroid hormone receptors. The slight narrowing of the pocket in the partially constrained conformer is further associated with an additional ligand orientation #4, which overlaps with orientations #2 and #3, and is described in more detail in the supplemental results and Supplemental Figures 3 and 4.
Figure 5. Protein substates define a pathway of ligand dynamics.
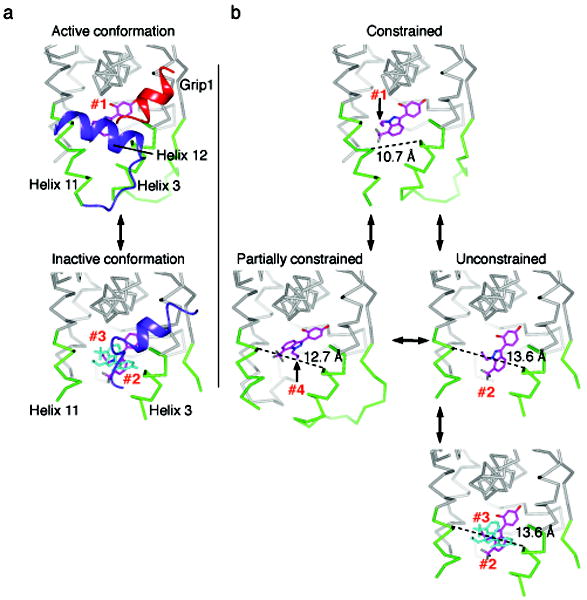
(a) A comparison of the active and inactive conformation structures, with helix 12 colored blue and the Grip1 coactivator peptide is colored red. In the active conformation, helix 12 packs against helices 3 and 11, constraining their movement. The ligand is bound in orientation #1, and is colored magenta. As the strained active conformation results in the relocation of helix 12 into the inactive conformation, the pocket widens, allowing binding of multiple ligands in orientations #2 and #3, colored blue.
(b) A model of ligand dynamics associated with different protein substates. Helix 12 is not shown to allow visualization of the different ligand orientations associated with distinct protein conformers. The width of the ligand binding pocket supports distinct ligand binding orientations, #1-4.
Collectively these findings suggest a revised model of the dynamics of the ligand-protein ensemble, whereby protein dynamics are coupled to different ligand binding orientations (Figure 5). The ligand binds to the agonist conformation in a strained fashion, enabling productive coactivator recruitment and transcriptional activity. As helix 12 switches from the active conformation, the strain is relieved, allowing helices 3 and 11 to move further apart, and the ligand to rotate into ligand orientation #4. As the pocket continues to widen, the ligand flips into orientation #2, allowing the docking of the second ligand in orientation #3 (Figure 5). Thus, the trapping of conformational substates with targeted mutations, and fortuitous crystal packing, demonstrates that protein conformation is coupled to and is instructive in directing changes in ligand orientation and binding stoichiometry.
The ligand binding ensemble directs graded signaling
If an ensemble of ligand binding modes is physiologically relevant to producing graded signaling responses, then one should be able to produce chemical derivatives that favor specific orientations having distinct activity profiles. We synthesized a series of derivatives (Table 1), in which the allyl group of WAY-169916 was replaced with bulkier substitutions (2-7), designed to block the dual binding of orientations #2 and #3. These compounds show at least a two-fold higher affinity for ERα than WAY-169916 (Table 1), and were tested in luciferase activity assays (Figure 6). Strikingly, the three derivatives with two rotatable bonds (2-4) in the side group showed greater agonist activity than WAY-169916, while the derivatives with one rotatable bond induced less transcriptional activity (Figure 6, 5-7). Thus small changes in the side chain elicit graded signaling output.
Figure 6. Activity profiles of WAY-169916 derivatives.
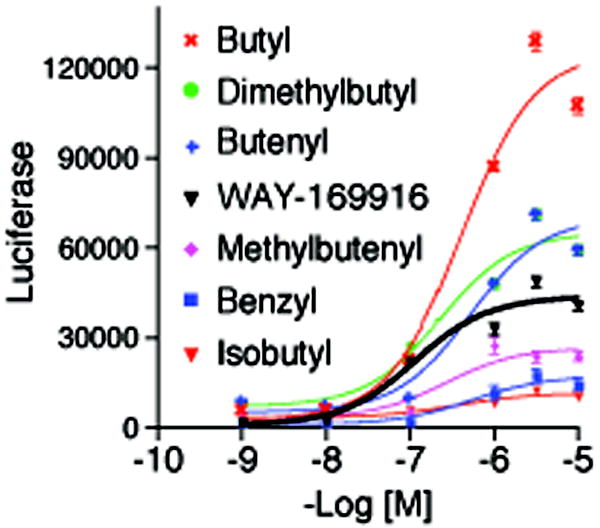
Estrogen responsive luciferase assays were performed by transfection of a 3xERE-luc reporter in ERα-positive MCF-7 cells. After 24 hr, cells were treated with the indicated doses of ligands overnight, and then assessed for luciferase activity.
Figure 7. Ligand binding ensembles direct ERα activity profiles.
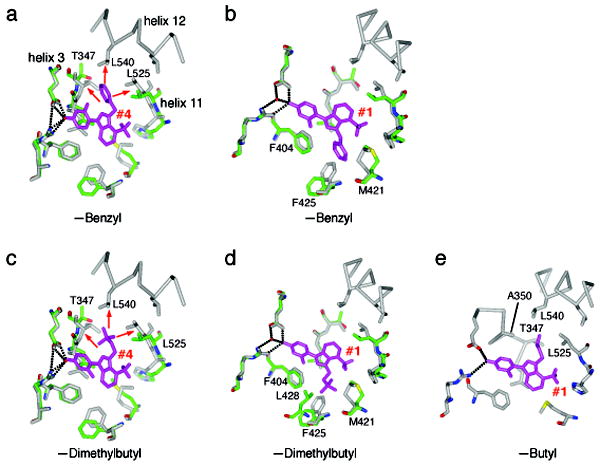
(a) The structure of the benzyl derivative with ERα in the inactive conformation has one molecule in the dimer with an unconstrained conformer, and one molecule with a partially constrained conformer. The unconstrained conformer is shown in green, bound to the benzyl derivative, which is colored magenta. The ligand adopts an orientation identical to orientation #4 of WAY-169916. The superimposed structure of WAY-169916 in the active conformation is colored gray. The red arrows indicate clashes with the superimposed active conformation structure, and widening of the pocket to accommodate this ligand orientation.
(b) Same as (a), but with the partially constrained inactive conformer of ERα. The ligand adopts an orientation identical to orientation #1 of WAY-169916. Here, there are no clashes with the superimposed active conformation.
(c) The dimethylbutyl substitution was modeled onto the structure of the benzyl derivative in the unconstrained conformer in orientation #4, and shows similar clashes with superimposed active conformation structure.
(d) The dimethylbutyl derivative was modeled onto ligand orientation #1, in the partially constrained conformer. As with the benzyl derivative, there are no clashes with the active conformation. However, the dimethylbutyl shows additional favorable VDW contacts with L428 and F425, suggesting that this orientation is more highly populated in solution.
(e) The butyl derivative was modeled into ligand orientation #4 on the active conformation structure. The butyl group forms favorable VDW interactions with the indicated amino acids.
From this series, we obtained crystal structures of the methylbutenyl 5 and benzyl 6 derivatives bound to the inactive conformation of ERα: these derivatives that have reduced efficacy compared to WAY-169916. These structures are in the same space group as the WAY-169916 structure (Supplemental Table 1), with identical crystal packing, and again produce an unconstrained and partially constrained conformer in each dimer. Both derivatives bound in orientation #4 in the unconstrained conformer, but adopted conformation #1 in the partially constrained conformer (electron density maps surrounding the ligand are shown in Supplemental Figure 5). In the unconstrained conformer, with orientation #4, the benzyl (Figure 7a) or methylbutenyl group (Supplemental Figure 5) extends between helices 3 and 11, forcing them apart in a conformation that is incompatible with docking of helix 12 in the agonist conformation. When superimposed on the agonist WAY-169916 structure, the side chains also directly clashed with the agonist conformation of helix 12 (Figure 7a).
In the partially constrained chains of ERα bound to the benzyl or methylbutenyl derivatives, the ligands adopted conformation # 1 (Figure 7b, Supplemental Figure 5). Importantly, there was no widening of the pocket relative to the agonist conformation structure of WAY-169916, suggesting that this ligand orientation is instructive in directing the limited transcriptional activity of these compounds through stabilizing the active conformer. These data further demonstrate that targeted changes in the ensemble of binding orientations change the activity profile of the compounds.
If bulkier derivatives can adopt two distinct binding orientations, what determines their relative activity profile? One possibility is that the compounds differ in their population of the two orientations, only one of which supports an active receptor conformer. This hypothesis predicts that compounds with very low efficacy, such as the benzyl derivative, favor orientation #4 in solution, with the bulky group disrupting the agonist conformation of helix 12, while compounds with higher efficacy favor orientation #1. Indeed, modeling demonstrates this for the dimethylbutyl derivative 3, which has greater agonist activity than WAY-169916. In orientation #4, this bulky side group has strong clashes with helices 3, 11, and 12, and cannot be rotated into a favorable orientation, just as observed with the benzyl derivative (Figure 7c). However, in orientation #1, the side group displays additional close van der Waals interactions with L428 and F425 that are not seen with the benzyl or methylbutenyl derivatives (Figure 7d). These favorable interactions suggest that the increased efficacy of this compound derives from a change in the population of ligand orientations, with this derivative showing greater affinity for the protein in orientation #1, which supports the agonist conformation.
A second determinant of activity could be that compounds differ in their distortion of the agonist conformation with ligand orientation #4. Modeling of the butyl derivative 2, which has the highest transcriptional activity of the WAY-169916 derivatives, supports this mechanism. Here, the butyl group can adopt packing interactions that are readily accommodated in the agonist conformation (Figure 7e), and include additional favorable stabilizing interactions with helix 12 not seen with the parent compound. Thus, activity is regulated both by the ensemble of ligand orientations, and the specific ERα substructures that they stabilize. These additional structures demonstrate: 1) that protein conformation and ligand orientation can adopt coupled ensembles; and 2) that differential ligand binding to active and inactive conformations regulate graded signaling output, which can be used for rational drug design.
DISCUSSION
This work demonstrates that protein conformation determines ligand binding orientation in a dynamic fashion. Current theories of ligand binding suggest that proteins display an ensemble of conformations, and that small molecules select for or bind to a subset of these. This is shown quite dramatically in the therapeutic targeting of specific protein conformations. For example, tamoxifen binding is specific for the inactive conformation of ERα, as this agent physically obstructs the active conformation 10. In this context, switching conformation induces the dissociation of ligands that are highly selective for a specific conformation. In contrast, our findings establish that conformational change can also direct adaptable interactions, where different conformers of a receptor are instructive in selecting for and/or binding to different orientations—or even stoichiometry—of a given ligand, giving a mixed population of protein conformation/ligand orientation states that collectively determine biological activity.
Several examples support the notion that the differential binding of a small molecule to distinct protein conformations is a general phenomenon. For example, Imatinib binds differently to the active conformation of Syk 32 versus to the inactive conformation of Abl 8. This effect is also seen with a compound that flips in its binding mode between ERα and ERβ 33, which have highly conserved binding pockets. The differential binding of one ligand to two such closely related proteins suggests that differential binding of a ligand to distinct conformers of one protein can also occur, as shown here. We have also observed flipped ligand binding modes of two very closely related compounds when bound to PPARγ 34, and closely related compounds also bind in a distinct fashion to p38 in the DFG-in and –out binding modes 35. Notably, the binding epitopes for the flipped ligand orientations #1 and #4 of the partial agonist WAY-169916 are highly conserved in the steroid receptor family. Indeed, superposition of our benzyl derivative-ERα structure with other steroid receptor crystal structures demonstrates many examples of ligand side groups in both orientations (Supplemental Figure 6). Collectively, these observations suggest that ligand dynamics should be explored in other allosteric signaling systems, and as a novel approach to drug design.
Defining how allosteric modulators elicit graded signaling is broadly important for understanding basic mechanisms of protein dynamics and allostery, their impact on physiology, as well as therapeutic development. The development of allosteric modulators with intermediate, or partial agonist activity are widely sought for improved specificity and side effect profiles for a number of nuclear receptors26,27,36-38, and more broadly for kinases and GPCRs22.
In general, allosteric mechanisms that produce graded responses could stabilize a specific conformer that reduces the affinity of the second binding site for its substrate or ligand (Figure 1). This occurs with the androgen receptor, where partial agonists directly perturb the helix 12 portion of the coactivator binding cleft. Alternatively, an allosteric modulator may change the proportion and/or population of active versus inactive conformers. We show here that WAY169916 destabilizes the helix 11-helix 12 interface. This shift, however, does not alter the coactivator-binding site, supporting a model where activity levels reflect the population of conformers having helix 12 in the active conformation. Thus, graded responses occur through changes in the ensemble of protein conformations. NMR studies have established that protein conformational dynamics and higher energy substates mediate allostery and enzyme activity 2-4. Here, we show that the same concept applies to small molecule ligands, where different ligand conformations control signaling output. This suggests that ligand dynamics should be explored in other allosteric signaling systems, and as a novel approach to drug design.
The conformational trapping approach applied here was designed to mimic the allosteric effects of ligands in stabilizing the well-characterized active and inactive conformations of ERα, through addition of one or two surface mutations. There are many examples of mutations that alter binding of a ligand. While such mutations alter structure, our approach differs by stabilizing well characterized, pre-existing conformers. Crystallization of wild-type and mutant proteins in both active 28 and inactive conformers (Supplemental Figures 1-2), bound to identical ligands, clearly demonstrates that our conformation trapping does not perturb ERα structure or ligand interactions, but rather changes the equilibrium between pre-existing conformers. While other inactive conformations may exist in solution, we have shown that the key structural feature is the removal of the constraining effects of helix 12 on helices 3 and 11, allowing the widening of the pocket.
One potential caveat is that the higher energy substate of WAY-169916 bound to ER in the active conformation might be an artifact of the Y537S point mutation. However, the shift in helix 11 with partial agonists has now been seen with six distinct chemical scaffolds (Figure 3, 28,30). Moreover, these compounds, and WAY-169916, do not have the larger side groups seen with androgen receptor antagonists that distort AR helix 12. Thus, there is no physical basis to suggest how WAY-169916 could induce a single suboptimal conformation, as seen with AR. Most importantly, we tested our hypothesis with targeted chemistry and demonstrated, with two different scaffolds, that simply removing a single methyl group rendered new derivatives as stronger agonists, and eliminated the shift in helix 11 28,30. Further, a shifted position of helix 11 has been seen with partial agonists bound to wild-type ERβ 39, the retinoid X receptor 40, and the constitutive androstane receptor 41. Thus ligand control of helix 12 via positioning of helix 11 is a general phenomenon among at least a subset of nuclear receptors. To support the active conformation, only a certain degree of helix 11 shift can be tolerated. This supports a model where helices 3 and 11 show dynamic motions coupled to ligand orientation, and as they come closer together, helix 12 can adopt the active conformer. The gain of allostery approach presented here thus allows one to visualize a higher energy conformer that can be gently trapped by one or two surface mutations.
METHODS
Protein Purification and Crystallization
The ERα LBD (amino acids 298-554) was mutated (Tyr-537-Ser or Tyr-536-Ser) with the Stratagene Quickchange Mutagenesis kit, and cloned into a modified PET vector with a ligation independent cloning site, His6-tag, and TEV protease site 42. The protein was induced in BL21 (DES) cells, and purified as described 28. Chemical synthesis of the ligands will be described elsewhere. For each receptor-ligand complex, both the Emerald Biosciences Wizard I&II and Hampton Research Index I&II screens were probed. Initial hits were optimized using a grid screen around pH, precipitant concentration, and protein concentration. The crystals were cryoprotected in either glycerol, PEG, or sucrose added to the mother liquor.
Data was collected at SER-CAT at the Advanced Photon Source, Argonne, Illinois, as well as at SSRL beamline 11-1 and scaled with HKL2000. The structures were solved with molecular replacement using Phaser. Compound pdb and cif files were generated with PRODRG 43. Refinement and rebuilding were performed with CCP4 44, Phenix 45, and Coot 46. Molecular graphics images were generated with CCP4 Molecular Graphics 47.
The omit maps for the WAY-169916 ERα 372S/536S structure were generated following refinement with a bias removal protocol. After deleting the ligands from the final model, three cycles of refinement were performed in PHENIX including NCS (grouping chain B with A, and chain D with C), 1 Å shake of the atomic coordinates, starting B factors reset to 30, and simulated annealing. This model was sent to the TLS Motion Determination server to define TLS groups 48. Three cycles of refinement were then carried out with NCS, TLS (20 groups/chain), ordered solvent, and geometry optimization.
Plasmid DNA and Transient Transfections
The ERE and NFκB luciferase assays, and native gene analysis were performed in MCF-7 cells as described 31. The GAL4-NCoR expression construct encodes NCoR residues 2220-2295, which harbors receptor interaction domain 1 (NCoR-ID1). The fragment was generated by PCR using gene specific primers that incorporated SalI and MluI restriction sites and subcloned into the pBIND GAL4-DNA binding domain expression vector (Z. Jin & CL Smith). Mammalian two-hybrid assays were performed essentially as described previously (1). Briefly, HeLa cells were transfected with 100 ng of expression vector for VP16, VP16-ERα wt, VP16-ERα 536S, 372R, or 531Stop), as well as 100 ng of either the GAL4 or GAL4-NCoR expression plasmids, and 1 mg of the pG5-Luc reporter construct, using 4 ml Lipofectamine reagent (Invitrogen, Carlesbad, CA) per well. Approximately 16 hr thereafter, cells were treated with 100 nM raloxifene or ethanolic vehicle for 24 hr and then harvested for measurement of luciferase activity. Relative luciferase measurements were normalized to total cellular protein determined using the Bio-Rad Protein Assay system (Bio-Rad, Hercules, CA).
RNA isolation and qRT-PCR
Total RNA was isolated from MCF7 cells using RNeasy (Qiagen), which was used to generate cDNA. PCR analysis was performed on an ABI PRISM 7900HT. Values are normalized with 18S rRNA content.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health PHS R37 DK15556 (J.A.K.), DK53002 (C. L. S.), CA132022 and DK077085 (K.W.N) and the Frenchman’s Creek Women for Cancer Research (G.G.). We would like to thank John Cleveland for comments on the manuscript, Liz Potterton and Stuart McNichols for help with CCP4MG, and Kathy Carlson for performing the ERα ligand binding assays.
Footnotes
Contributions A.A.P. synthesized compounds, G.G. and M.C.P. performed cell based assays, J.B.B., A.A.P., M.Z., J.N., and P.V.A. performed x-ray crystallography and data analysis, A.A.P., J.B.B., C.L.S., P.D.A., J.A.K., and K.W.N designed and supervised experiments, and K.W.N. wrote the manuscript with input from all the authors.
Competing financial interests The authors declare no competing financial interests.
Chemical Synthesis and Characterization are described in Supplemental Methods
References
- 1.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 2.Boehr DD, Dyson HJ, Wright PE. An NMR perspective on enzyme dynamics. Chem Rev. 2006;106:3055–3079. doi: 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- 3.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisenmesser EZ, et al. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 5.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 6.Luque I, Leavitt SA, Freire E. The linkage between protein folding and functional cooperativity: two sides of the same coin? Annu Rev Biophys Biomol Struct. 2002;31:235–256. doi: 10.1146/annurev.biophys.31.082901.134215. [DOI] [PubMed] [Google Scholar]
- 7.Nagar B, et al. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 2003;112:859–871. doi: 10.1016/s0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 8.Schindler T, et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 9.Xu HE, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature. 2002;415:813–817. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- 10.Shiau AK, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 11.Weber G. Ligand binding and internal equilibria in proteins. Biochemistry. 1972;11:864–878. doi: 10.1021/bi00755a028. [DOI] [PubMed] [Google Scholar]
- 12.Seeliger MA, et al. c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure. 2007;15:299–311. doi: 10.1016/j.str.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Margarit SM, et al. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 14.Milburn MV, et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science. 1990;247:939–945. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 15.Sablin EP, Fletterick RJ. Nucleotide switches in molecular motors: structural analysis of kinesins and myosins. Curr Opin Struct Biol. 2001;11:716–724. doi: 10.1016/s0959-440x(01)00265-2. [DOI] [PubMed] [Google Scholar]
- 16.Sondermann H, et al. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell. 2004;119:393–405. doi: 10.1016/j.cell.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Velyvis A, Yang YR, Schachman HK, Kay LE. A solution NMR study showing that active site ligands and nucleotides directly perturb the allosteric equilibrium in aspartate transcarbamoylase. Proc Natl Acad Sci U S A. 2007;104:8815–8820. doi: 10.1073/pnas.0703347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steitz TA. Visualizing polynucleotide polymerase machines at work. EMBO J. 2006;25:3458–3468. doi: 10.1038/sj.emboj.7601211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bramlett KS, et al. A natural product ligand of the oxysterol receptor, liver X receptor. J Pharmacol Exp Ther. 2003;307:291–296. doi: 10.1124/jpet.103.052852. [DOI] [PubMed] [Google Scholar]
- 20.Yamasaki K, et al. Comparison of reporter gene assay and immature rat uterotrophic assay of twenty-three chemicals. Toxicology. 2002;170:21–30. doi: 10.1016/s0300-483x(01)00505-4. [DOI] [PubMed] [Google Scholar]
- 21.Bhavnani BR. Pharmacokinetics and pharmacodynamics of conjugated equine estrogens: chemistry and metabolism. Proc Soc Exp Biol Med. 1998;217:6–16. doi: 10.3181/00379727-217-44199. [DOI] [PubMed] [Google Scholar]
- 22.Lewis JA, Lebois EP, Lindsley CW. Allosteric modulation of kinases and GPCRs: design principles and structural diversity. Curr Opin Chem Biol. 2008;12:269–280. doi: 10.1016/j.cbpa.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 23.Darimont BD, et al. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brzozowski AM, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 25.Renaud JP, et al. Crystal structure of the RAR-gamma ligand-binding domain bound to all-trans retinoic acid. Nature. 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- 26.Chadwick CC, et al. Identification of pathway-selective estrogen receptor ligands that inhibit NF-kappaB transcriptional activity. Proc Natl Acad Sci U S A. 2005;102:2543–2548. doi: 10.1073/pnas.0405841102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steffan RJ, et al. Synthesis and activity of substituted 4-(indazol-3-yl)phenols as pathway-selective estrogen receptor ligands useful in the treatment of rheumatoid arthritis. J Med Chem. 2004;47:6435–6438. doi: 10.1021/jm049194+. [DOI] [PubMed] [Google Scholar]
- 28.Nettles KW, et al. NFkappaB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat Chem Biol. 2008;4:241–247. doi: 10.1038/nchembio.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol. 1996;10:1388–1398. doi: 10.1210/mend.10.11.8923465. [DOI] [PubMed] [Google Scholar]
- 30.Zhou HB, et al. Elemental isomerism: a boron-nitrogen surrogate for a carboncarbon double bond increases the chemical diversity of estrogen receptor ligands. Chem Biol. 2007;14:659–669. doi: 10.1016/j.chembiol.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Nettles KW, et al. Structural plasticity in the oestrogen receptor ligand-binding domain. EMBO Rep. 2007 doi: 10.1038/sj.embor.7400963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atwell S, et al. A novel mode of Gleevec binding is revealed by the structure of spleen tyrosine kinase. J Biol Chem. 2004;279:55827–55832. doi: 10.1074/jbc.M409792200. [DOI] [PubMed] [Google Scholar]
- 33.Norman BH, et al. Benzopyrans are selective estrogen receptor beta agonists with novel activity in models of benign prostatic hyperplasia. J Med Chem. 2006;49:6155–6157. doi: 10.1021/jm060491j. [DOI] [PubMed] [Google Scholar]
- 34.Bruning JB, et al. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure. 2007;15:1258–1271. doi: 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 35.Angell RM, et al. Biphenyl amide p38 kinase inhibitors 4: DFG-in and DFG-out binding modes. Bioorg Med Chem Lett. 2008;18:4433–4437. doi: 10.1016/j.bmcl.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 36.Gelman L, Feige JN, Desvergne B. Molecular basis of selective PPARgamma modulation for the treatment of Type 2 diabetes. Biochim Biophys Acta. 2007;1771:1094–1107. doi: 10.1016/j.bbalip.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 37.Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159–191. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- 38.Schacke H, et al. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci U S A. 2004;101:227–232. doi: 10.1073/pnas.0300372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shiau AK, et al. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat Struct Biol. 2002;9:359–364. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 40.Love JD, et al. The structural basis for the specificity of retinoid-X receptor-selective agonists: new insights into the role of helix H12. J Biol Chem. 2002;277:11385–11391. doi: 10.1074/jbc.M110869200. [DOI] [PubMed] [Google Scholar]
- 41.Shan L, et al. Structure of the murine constitutive androstane receptor complexed to androstenol: a molecular basis for inverse agonism. Mol Cell. 2004;16:907–917. doi: 10.1016/j.molcel.2004.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stols L, et al. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 43.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 44.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 46.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 47.Potterton E, McNicholas S, Krissinel E, Cowtan K, Noble M. The CCP4 molecular-graphics project. Acta Crystallogr D Biol Crystallogr. 2002;58:1955–1957. doi: 10.1107/s0907444902015391. [DOI] [PubMed] [Google Scholar]
- 48.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.