

Abstract
The ability of p53 to induce apoptosis in cells with damaged DNA is thought to contribute greatly to its tumour suppressor function. P53 has been proposed to induce apoptosis via numerous transcriptional targets or even by direct cytoplasmic action. Two transcriptional targets shown to mediate its apoptotic role in several cell types encode Noxa and Puma, BH3-only members of the Bcl-2 family. To test if their functions in p53-dependent apoptosis overlap, we generated mice lacking both. These mice develop normally and no tumours have yet arisen. In embryonic fibroblasts, the absence of both Noxa and Puma prevented induction of apoptosis by etoposide. Moreover, following whole body γ-irradiation, the loss of both proteins protected thymocytes better than loss of Puma alone. Indeed, their combined deficiency protected thymocytes as strongly as loss of p53 itself. These results indicate that, at least in fibroblasts and thymocytes, p53-induced apoptosis proceeds principally via Noxa and Puma, with Puma having the predominant role in diverse cell types. The absence of tumours in the mice suggests that tumour suppression by p53 requires functions in addition to induction of apoptosis.
Keywords: apoptosis, DNA damage, p53, Puma, Noxa
DNA damage can result in cell cycle arrest or apoptosis, and both outcomes require the tumour suppressor p53.1 Defects in the cellular response to DNA damage can promote tumour development and impair the response of tumour cells to anti-cancer therapy.1 Thus, the p53 gene is mutated in the majority of human cancers and tumours, lacking p53 function, often respond poorly to γ-radiation and chemotherapy. Moreover, individuals with Li Fraumeni syndrome, who have germ-line heterozygous mutations in p53, are highly prone to diverse cancers at a young age. Similarly, mutant mice heterozygous or homozygous for a p53 deletion are highly predisposed to develop tumours, particularly lymphomas or sarcomas.2,3
The p53 protein is activated in response to diverse stress stimuli and regulated by multiple mechanisms that affect its stability.1 This transcription factor is thought to mediate its diverse functions largely by activating distinct target genes.1 It is well established that p53 triggers apoptosis through the ‘Bcl-2-regulated’ (also called ‘intrinsic’ or ‘mitochondrial’) pathway, because p53-dependent apoptosis can be inhibited by overexpression of Bcl-2 or its pro-survival homologues.4-6 How p53 triggers apoptosis, however, is still not fully resolved. Most evidence suggests that it functions through its ability to activate transcription of various pro-apoptotic target genes,1 including certain members of the Bcl-2 family (see below). However, several groups have reported evidence that p53 can trigger apoptosis through direct binding to either pro- or anti-apoptotic members of the Bcl-2 family on the outer mitochondrial membrane.7-11 It has for example been argued that, following DNA damage, p53 induces a rapid transcription-independent apoptosis of thymocytes that precedes the induction of p53 target genes.8 In that model, p53-induced death should proceed normally in the absence of the critical pro-apoptotic p53 target genes.
The Bcl-2 family of proteins, which regulates developmentally programmed cell death and cytotoxic stress-induced apoptosis,12 contains three structurally and functionally distinct subgroups: Bcl-2-like pro-survival proteins, which share up to four (BH) regions of homology; pro-apoptotic Bax/Bak-like proteins, which contain the BH1, BH2 and BH3 regions; and the pro-apoptotic BH3-only proteins, which share only the short (16–25 residue) BH3 domain. The BH3-only proteins are activated transcriptionally and/or post-translationally by death stimuli and initiate apoptosis signalling, whereas Bax/Bak-like proteins play an essential role further downstream.13,14 Recently, two BH3-only proteins, Noxa and Puma, have been shown to be critical for p53-mediated apoptosis. Both the noxa and puma genes are direct transcriptional targets of p53,15-17 but they can also be induced by p53-independent mechanisms.18 Studies with knockout mice have shown that Puma plays a major role in p53-mediated and p53-independent apoptosis in a broad range of cell types,19-21 whereas Noxa has a more restricted role in p53-mediated apoptosis of fibroblasts.19,22
Although cycling non-transformed lymphocytes and lymphoma cells can die in a p53-independent manner in response to DNA damage,6 non-cycling cells, such as (most) CD4+8+ (double-positive: DP) thymocytes and pre-B cells are completely dependent on p53 for cell killing following this insult.6,23,24 Although loss of Puma strongly protects against DNA damage-induced apoptosis, in several cell types, this protection was significantly weaker than that afforded by loss of p53.19-21 Since Noxa and Puma are both regulated by p53, it appears likely that these two BH3-only proteins have overlapping functions. To test this hypothesis, we have generated Noxa/Puma doubly deficient (DKO) mice, and we report here the characterisation of their phenotype. The results suggest that, at least in certain cell types, the apoptotic function of p53 relies almost exclusively on Noxa and Puma or, in some cases, on Puma alone.
Results
Mice lacking both Noxa and Puma develop normally and are not tumour prone
Mice lacking either Noxa or Puma are normal in appearance, body weight and weight of major organs.19-22 To investigate whether Noxa and Puma overlap in function, we crossed noxa−/− and puma−/− mice to generate mice lacking both. As expected, no puma and noxa RNA transcripts appeared in thymocytes or spleen cells from the noxa−/−puma−/− mice, whereas normal levels of puma mRNA were seen in noxa−/− cells (data not shown) and normal levels of noxa mRNA in puma−/− cells (Figure 1a). In addition, the levels of Bim, Bid and Bad protein were similar in cells from wildtype (wt), noxa−/−, puma−/− and noxa−/−puma−/− mice (Figure 1b). As expected, no Puma protein could be detected in dexamethasone-treated or γ-irradiated puma−/− or noxa−/−puma−/− thymocytes (Figure 1b), but Puma protein levels increased over time in γ-irradiated wt but not in p53−/− thymocytes (Figure 1c). These results document that we have generated noxa−/−puma−/− mice and that loss of Noxa, Puma or both does not cause a compensatory upregulation in the level of any other BH3-only protein tested.
Figure 1.

noxa and puma are not expressed in noxa−/−puma−/− double knockout mice. (a) RT-PCR analysis on cDNAs generated with total RNA from thymocytes and spleen cells from mice of the indicated genotypes. Wt thymocyte cDNA, which was used in a reaction without reverse transcriptase enzyme, is included as a negative control. The identity of the PCR products was confirmed by Southern blotting using an internal oligonucleotide specific for noxa cDNA as a probe (top panel). RT-PCR analysis with primers specific for hprt was used as a loading control. Sizes of DNA size standards (in bp) are shown on the left hand side (bottom panel). (b) Western blot analysis of thymocytes from wt, noxa−/−, puma−/− and noxa−/−puma−/− mice cultured for 7 h in the presence or absence of 1 μM dexamethasone or following 2.5 Gy γ-irradiation. Western blots were probed for Bim, Bid, Bad or Puma protein levels. Probing for β-actin was included as a loading control. The molecular weight (in kDa) of protein size standards is shown on the left hand side. (c) Western blot analysis of Puma protein expression in wt or p53−/− thymocytes cultured without treatment (Unt) or following treatment with the indicated doses of γ-irradiation. Time in culture is indicated in hours. The molecular weight (in kDa) of protein size standards is shown on the left hand side
Noxa/Puma doubly deficient mice were born at a normal frequency from inter-crosses of noxa−/−puma+/− mice (36 expected out of 144 offspring, 34 observed) and had a normal appearance, behaviour and health up to at least 1 year of age. Mice deficient for p53 are highly prone to spontaneous tumours, with a particularly high incidence of thymic lymphomas, most dying by 6 months of age.2,3 In contrast, only one of 22 puma+/− mice that have been monitored for a year or longer has developed a tumour (a CD4+8+ thymoma at 37 weeks of age). Among 14 puma−/− mice monitored for at least 11 months, one was found dead at 15 weeks with an enlarged spleen and thymus, but due to the deteriorated state of the mouse we could not determine whether this was indeed a tumour. Furthermore, no tumours arose in 10 noxa−/−puma−/− mice monitored for more than 12 months and these animals remained healthy.
Inter-crosses of noxa−/−puma−/− mice produced litters of normal size and comparable numbers of male and female progeny. Moreover, the noxa−/−puma−/− females reared their pups normally. The weights of noxa−/−puma−/− males and females at 3 and 6–8 weeks were comparable to those of wt littermates, as was the appearance and weight of major organs (thymus, spleen, lung, heart, kidneys, liver and testes).
BH3-only proteins play a critical role in the programmed death of haemopoietic cells. We therefore investigated the effect of loss of both Noxa and Puma on haemopoiesis by comparing thymus, spleen, lymph node, bone marrow and peripheral blood of 6–11 week-old noxa−/−puma−/− mice with those of wt and single knockout animals. We found that all these tissues from noxa−/−puma−/−mice had normal size, weight and cellularity (see below). Moreover, the blood contained normal numbers of B and T lymphocytes, neutrophils, monocytes, eosinophils, basophils and platelets as well as a normal hematocrit (data not shown).
Together, these results demonstrate that combined deficiency of Noxa and Puma does not affect embryogenesis, haemopoiesis, behaviour or reproduction in mice, nor does it predispose them to tumour development.
Response of Noxa/Puma-deficient lymphocytes to apoptotic stimuli in vitro
Although loss of Puma can protect lymphocytes against a range of death stimuli, the protection from p53-dependent stimuli (e.g. etoposide or γ-radiation), and p53-independent insults (e.g. glucocorticoids or cytokine deprivation), is not always complete.19-21 Hence, Noxa might well contribute to the death because, like Puma,16-18 its expression can be upregulated by both p53-independent and p53-dependent mechanisms.15 We therefore compared the death of lymphocytes from the DKO and single knockout mice with that of p53-deficient cells in response to p53-independent stimuli (incubation in simple medium or treatment with dexamethasone, PMA, ionomycin, staurosporine or tunicamycin), and the p53-dependent apoptosis induced by etoposide or γ-irradiation.
We found that thymocytes from noxa−/−puma−/− mice were slightly more refractory to high doses of γ-radiation (5 Gy) than those from puma−/− mice (Figure 2a; P=0.032). In response to all the other insults tested, however, thymocytes from noxa−/−puma−/− and puma−/− mice behaved indistinguishably (Supplementary Figure 1a provides an extended kinetic analysis). Both exhibited higher resistance than wt thymocytes to spontaneous apoptosis and death induced by treatment with etoposide (Supplementary Figure 1a), low doses of γ-radiation, dexamethasone or PMA but were normally sensitive to ionomycin or tunicamycin (Figure 2a and data not shown). Similarly, noxa−/−puma−/− pro-B/pre-B cells from bone marrow (Figure 2b), and mature T and B cells from lymph nodes (Figure2c and data not shown) were no more resistant to these cytotoxic stimuli than their puma−/− counterparts (Supplementary Figure 1a).
Figure 2.

Sensitivity of lymphocytes from noxa−/−puma−/− mice to apoptotic stimuli in culture. (a) Purified immature CD4+8+ thymocytes from wt, noxa−/−, puma−/−, noxa−/−puma−/− or p53−/− mice were cultured for 8 h in the presence of dexamethasone (Dex, 1 μM) or for 24 h with phorbol 12-myristate 13-acetate (PMA, 10 ng/ml) or following γ-irradiation (1.25 or 5 Gy). Immature pro-B/pre-B cells (B220+sIg−) sorted from the bone marrow (b) or mature T cells (Thy-1+) sorted from the lymph nodes (c) of wt, noxa−/−, puma−/−, noxa−/−puma−/− or p53−/− mice were cultured for 24 h following γ-irradiation (5 Gy). Viability of unstimulated cells of each type did not differ significantly between genotypes at these times. Data points represent means±S.D. of cells from 3–5 mice of each genotype. The percentage of viable noxa−/−puma−/− thymocytes remaining after treatment with 5Gy γ-radiation was significantly greater than for puma−/− thymocytes treated with the same dose (P=0.032) and was not significantly different to the number of viable p53−/− thymocytes. For a detailed kinetic analysis see Supplementary Figure 1
Activated T cells require IL-2 (or certain other cytokines) for survival, and apoptosis following cytokine deprivation can be inhibited by Bcl-2 overexpression5 or loss of the BH3-only protein Bim.25 Since loss of Puma partially protects resting lymphocytes and myeloid progenitors from cytokine withdrawal, 19,20 we investigated the impact of combined Noxa/Puma loss on IL-2 deprivation in T cell blasts. Puma-deficient T cell blasts were significantly, albeit incompletely, protected from IL-2 withdrawal, γ-irradiation or treatment with dexamethasone, etoposide, PMA or tunicamycin, and the noxa−/−puma−/− T cells blasts responded identically (data not shown).
Notably, comparison with p53-deficient cells revealed the major role of Puma, alone or together with Noxa, in p53-mediated death. The noxa−/−puma−/− and the puma−/− thymocytes were as refractory as the p53−/− ones to etoposide or 1.25 Gy γ-irradiation and at least 70% as resistant to 5Gy γ-irradiation (Figure 2a and Supplementary Figure 1a). Thus, in thymocytes, Noxa and Puma are clearly essential for the death elicited by p53, with Puma having the major role. Similarly, in the pro-B/pre-B cells, loss of Puma, alone or together with Noxa, appeared to account for at least 60% of the protection provided by p53 loss (Figure 2b). In the mature T cells, however, even their combined loss provided much less protection than p53 loss (Figure 2c).
Noxa and Puma act together in etoposide-induced killing of E1A-transformed MEF
Expression of the adenovirus oncoprotein E1A sensitises mouse embryonic fibroblasts (MEF) to DNA damage-induced apoptosis, in part by stabilising p53.26 Since loss of Noxa, or to an even greater extent loss of Puma, inhibited etoposide-induced apoptosis of E1A-expressing MEF,19,22 we tested whether Noxa and Puma cooperate in this death response. Wildtype, noxa−/−, puma−/−, noxa−/− puma−/− and p53−/− MEF expressing E1A were either cultured in medium with serum (unstimulated) or without serum, or γ-irradiated or treated with etoposide (Figure 3). Both the puma−/− and p53−/− MEF survived better than the wt or the noxa−/− cells in the unstimulated cultures, where overcrowding and limiting growth factors in the serum likely both contribute to apoptosis (Supplementary Figure 2a provides a kinetic analysis). Consistent with this, the p53−/− MEF were almost completely refractory to serum withdrawal (Figure 3b), as were the noxa−/−puma−/− and puma−/− MEF, even after several days in culture (Supplementary Figure 2b). Thus, in E1A-transformed MEF, p53-induced activation of Puma is critical for the death provoked by overcrowding and serum deprivation, and Noxa appears not to be required for this process.
Figure 3.

Sensitivity of embryonic fibroblasts from noxa−/−puma−/− mice to apoptotic stimuli. E1A-expressing MEF from wt, noxa−/−, puma−/−, noxa−/−puma−/− or p53−/− embryos were cultured for 24 h in simple medium with (a) or without serum (b), or were exposed to 10 μg/ml (c) or 100 μg/ml (d) etoposide or γ-irradiation (50 Gy) and analysed after 24 (e) or 72h (f). Data points represent means±S.D. of cells from 3-5 independent embryos of each genotype. The percentage of viable noxa−/−puma−/− E1A-MEF remaining after treatment with 10 μg/ml etoposide was significantly greater than for puma−/− E1A-MEF treated with the same dose (P=0.012). For a detailed kinetic analysis see Supplementary Figure 2
By contrast, upon exposure to etoposide, Noxa and Puma clearly are both critical, because the noxa−/−puma−/− MEF survived better than puma−/− MEF. At a lower dose of etoposide (10 μg/ml), loss of Puma conferred greater resistance than loss of Noxa alone but not as much as their combined loss (P = 0.012; Figure 3c). Similarly, at a higher dose of etoposide (100 μg/ml), ~50% of noxa−/−puma−/− MEF remained viable at 24 h, when only ~20% of the noxa−/− or puma−/− cells remained viable and almost all wt cells had died (Figure 3d). Nevertheless, by 48 h, most MEF of all genotypes treated with etoposide were dead (Supplementary Figure 2c and d), perhaps due to killing by a non-apoptotic process.
Although MEF of all genotypes were initially profoundly resistant to high doses of γ-irradiation (Figure 3e), at later time points, only noxa−/−puma−/− and puma−/− MEF survived (Figure 3f; see Supplementary Figure 2e for a kinetic analysis), indicating that Puma is the major initiator of cell death in this setting. Remarkably, puma−/− and/or noxa−/−puma−/− MEF were more resistant to etoposide (Figure 3d) and γ-radiation (Figure 3f) than cells from p53−/− embryos. A likely explanation for this finding is the observation that upon DNA damage, E1A-expressing MEF lacking Noxa, Puma or both will arrest at the G1/S cell-cycle checkpoint, whereas those lacking p53 are unable to do so and may therefore undergo a p53-independent death within the S orG2 M phase of the cell cycle (22 and our unpublished observations).
The greatly enhanced survival of noxa−/−puma−/− MEF questions models in which p53 can directly induce apoptosis at the mitochondrial level (see Discussion). To rule out the possibility that p53 transcriptional activation function was compromised in our knockout MEF, we examined the protein levels of p53 and its transcriptional target, p21, in MEF infected with control or E1A-expressing retroviruses. MEF expressing the control retrovirus expressed low levels of p53, even after treatment with etoposide (Supplementary Figure 3a). As expected,26 MEF expressing E1A had higher p53 levels (Supplementary Figure 3b). The E1A-induced rise in the p53 level in the absence of noxa or puma, or both, was comparable to that seen in wt MEF, as was the induction of p21. Moreover, etoposide further increased p53 and p21 levels comparably in wt MEF or MEF deficient for noxa, puma or both (Supplementary Figure 3c). Thus, in the absence of noxa and puma, p53 is activated normally and is functional.
Together, Noxa and Puma account for the γ-irradiation-induced death of thymocytes in vivo
To investigate the individual and combined roles of Noxa and Puma in vivo, we performed whole-body γ-irradiation of wt, noxa−/−, puma−/−, noxa−/−puma−/−and p53−/− mice and analysed their haemopoietic compartments 20 h later. We determined the subcellular composition of haemopoietic tissues by flow cytometric analysis of cells incubated with antibodies to cell subset-specific surface markers.
The thymus of untreated noxa−/−puma−/− mice had normal numbers of CD4−8− pro-T cells, CD4+8+ pre-T and both CD4+8− and CD4−8+ mature T cells (Figure 4a; see untreated controls). The death of thymocytes and pre-B cells following γ-irradiation is dependent on p53.6,23,24 As previously reported, γ-irradiation of wt mice resulted in massive death of thymocytes (Figure 4b), particularly in the highly sensitive immature CD4+8+ thymocyte population, where cell numbers fell ~5-fold following exposure to 2.5 Gy and ~70-fold following 5Gy (Figure 4c). In contrast, γ-irradiation of p53−/− mice at doses as high as 5Gy had little effect on thymocyte numbers, the CD4+8+ cells falling only ~30% (Figure 4c). Consistent with our previous observations,21 loss of Noxa alone did not affect γ-irradiation-induced thymocyte killing in vivo, whereas loss of Puma provided very substantial protection. In puma−/− mice, exposure to 2.5 Gy reduced the CD4+8+ thymocytes only ~20% and even 5Gy produced only a ~50% reduction – resulting in ~50-fold higher survival than in wt mice. At 2.5 Gy, where loss of Puma offered essentially complete protection, concomitant loss of Noxa did not, of course, further augment survival. However, at 5 Gy, significantly more CD4+8+ thymocytes were recovered from noxa−/−puma−/− mice than puma−/− mice (P<0.01). Importantly, at 5Gy the survival of CD4+8+ thymocytes in noxa−/−puma−/− mice was as great as that in p53−/− mice. Thus, essentially all of the pro-apoptotic effects of p53 in this setting rely upon Noxa and Puma. In the case of the mature CD4+8− and CD4−8+ thymocytes; however, loss of Puma afforded as much protection against γ-irradiation as combined loss of Puma and Noxa (Figure 4d and e), indicating that Puma is the dominant effector in these cell types.
Figure 4.
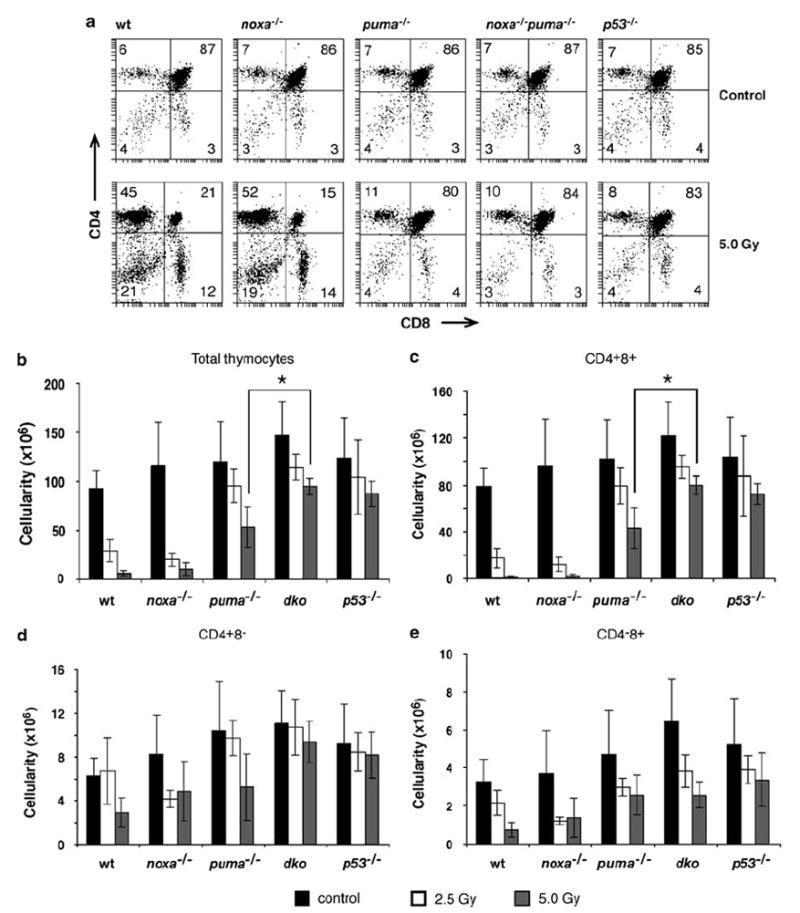
Sensitivity of thymocytes from noxa−/−puma−/− mice to γ-irradiation in vivo. Wt, noxa−/−, puma−/−, noxa−/−puma−/− or p53−/− mice were left untreated or exposed to 2.5 or 5 Gy full body γ-irradiation and the thymus was harvested 20 h later. (a) Representative dot plots of stained thymocytes from control or 5 Gy-irradiated mice of each genotype. Percentages of CD4−8−, CD4−8+ CD4+8− and CD4+8+ cells are indicated in the quadrants. (b) Thymic cellularity was determined for untreated or irradiated mice of each genotype. In (c), (d) and (e), the total number of thymocytes in the indicated subset was determined by multiplying the total thymic cellularity by the percentage of cells in that subset. Bars represent means±S.D. of 4–14 mice of each genotype per treatment in at least three independent experiments. Thymic cellularity and total number of CD4+8+ thymocytes remaining in noxa−/−puma−/− animals treated with 5 Gy γ-radiation was significantly greater than in puma−/− animals treated with the same dose (P<0.01)
To investigate the effect of γ-irradiation in situ, we performed TUNEL staining on thymic sections from wt, noxa−/−, puma−/−, noxa−/−puma−/− and p53−/− mice 20 h after their exposure to 5 Gy. In the wt (Figure 5b and c) and noxa−/− thymus (Figure 5e and f), which had shrunk dramatically, there was extensive loss of the normal architecture and strong TUNEL-positive (brown) staining of the cortex. The sections of puma−/− thymi exhibited dramatically fewer TUNEL-positive cells than those from wt and noxa−/− animals and retained a normal cortical and medullary organisation (Figure 5h and i). Thymic sections from γ-irradiated noxa−/−puma−/− animals (Figure 5k and l) consistently had fewer TUNEL-positive cells than puma−/− sections and looked comparable to those from p53−/− animals (Figure 5n and o), consistent with the cell counting studies (Figure 4). In support of that conclusion, a blinded count confirmed that the noxa−/−puma−/− sections had ~40% less TUNEL-positive cells than the puma−/− ones and almost as few as the p53−/− sections. Moreover, thymi from 5Gy γ-irradiated puma−/− animals (32±4 mg, n=5) were significantly smaller than those from similarly treated noxa−/−puma−/− mice (47±5 mg, n=4; P<0.01).
Figure 5.

In situ analysis of γ-irradiation-induced apoptosis of thymocytes. Thymic sections from wt (a–c), noxa−/− (d–f), puma−/− (g–i), noxa−/−puma−/− (j–l) or p53−/− (m–o) animals that were untreated (top panels) or 20 h after exposure to 5 Gy γ-radiation (middle and bottom panels). The sections were TUNEL-stained using Bio-dUTP to detect nicked DNA in apoptotic cells and nuclei counterstained with hematoxylin. At higher magnification (bottom panels) sections from puma−/− animals (i) exhibit dramatically less apoptotic cells than those from wt and noxa−/− animals (c, f) but more than sections from noxa−/−puma−/− animals (l), which are comparable to those from p53−/− animals (o). The images are representative of three or more independent stains performed on thymi of at least three animals per genotype per treatment
Extent of protection by Noxa/Puma loss varies in different cell types
In the spleen (Figure 6a–c) and the lymph nodes (data not shown), Puma loss potently protected CD4+ and CD8+ mature T cells as well as (B220+sIg+) B cells from γ-irradiation-induced death. In these organs, loss of Noxa alone did not notably protect B or T cells, and combined Noxa/Puma loss did not significantly increase their survival over loss of Puma alone. After 2.5 Gy, wt mice lost ~60% of both their CD4+ and CD8+ T cells in the spleen, and with 5Gy only ~14% of CD4+ and ~6% of CD8+ T cells survived. CD4+ T cells from puma−/− mice were completely protected from death following 2.5 Gy and only a small proportion died following exposure to 5Gy γ-irradiation. The losses of CD4+ T cells in the puma−/− and p53−/− mice were similar, indicating that Puma accounts for most (perhaps all) of the p53-mediated death. Although puma−/− CD8+ T cells and B cells survived far better than the wt cells (~15- or ~10-fold better, respectively), Puma loss provided less protection than loss of p53, but concomitant loss of Noxa did not augment it further (Figure 6). Thus, in these cells, the death mediated by p53 requires Puma plus a pro-apoptotic factor other than Noxa, perhaps another BH3-only protein.
Figure 6.
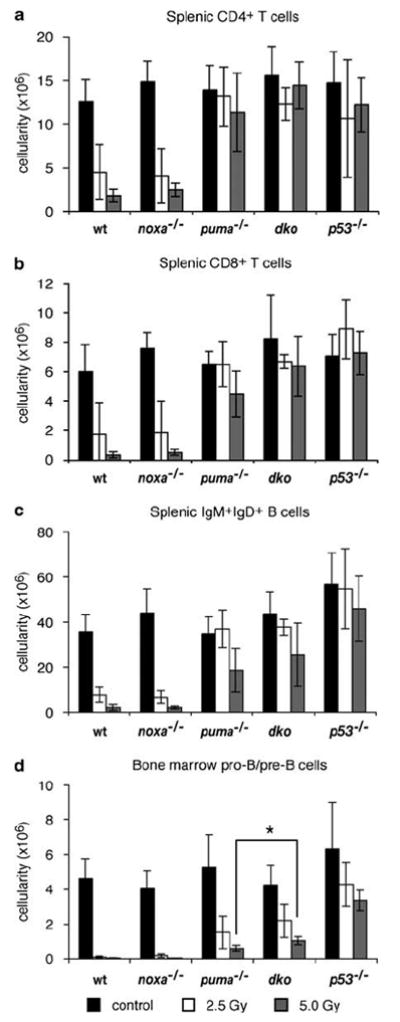
Sensitivity of lymphocytes from noxa−/−puma−/− mice to γ-irradiation in vivo. Wt, noxa−/−, puma−/−, noxa−/−puma−/− or p53−/− mice were left untreated or exposed to 2.5 or 5 Gy full-body γ-irradiation and spleen and bone marrow were harvested 20 h later. (a–c) The number of CD4+ or CD8+ T cells and sIgM+sIgD+ B cells in the spleen from untreated and irradiated mice of each genotype was determined by multiplying the total splenic cellularity with the percentage of the cell subsets. (d) The number of immature B220+sIgM−sIgD− pro-B/pre-B cells in the bone marrow from both femora was calculated using the cellularity and percentage of cell subsets. Bars represent means±S.D. of 4–14 mice of each genotype per treatment in at least three independent experiments. The total number of B220+sIgM−sIgD− pro-B/pre-B cells in the bone marrow of noxa−/−puma−/− animals treated with 5 Gy γ-radiation was significantly greater than in puma−/− animals treated with the same dose (P<0.02)
In the bone marrow, combined Noxa/Puma loss protected B-cell progenitors (pro-B/pre-B cells) from γ-irradiation-induced death modestly better than the loss of Puma alone (Figure 6d). This difference was significant (P<0.02), although the protection was substantially less than that provided by loss of p53, suggesting p53 must activate factors in addition to Noxa and Puma to kill this cell type. In the blood, however, loss of Puma alone or Noxa plus Puma protected T cells and B cells comparably to loss of p53 (data not shown), indicating that Puma is the major inducer of this pathway to their apoptosis.
Following 5Gy γ-irradiation, macrophage numbers in the spleen and peripheral blood of wt mice dropped by ~50%, whereas those in mice lacking p53 or Puma fell only by ~30% (data not shown), indicating that Puma contributes to this death. Likewise, in the bone marrow of wt mice ~75% of macrophages were lost. Again, the increased numbers of macrophages remaining in the marrow of puma−/− and p53−/− mice were similar in magnitude, suggesting that Puma is required for the death of macrophages following γ-irradiation.
In situstaining of splenic sections from untreated wt, noxa−/−, puma−/−, noxa−/−puma−/− and p53−/− mice revealed few TUNEL-positive (brown) cells (Figure 7, top panels), but after exposure to 5Gy γ-radiation many TUNEL-positive cells were evident in sections from the wt and noxa−/− animals (Figure 7b and d). In contrast, sections from puma−/− or noxa−/−puma−/− mice (Figure 7f and h), like those from p53−/− animals (Figure 7j), contained very few TUNEL-positive cells, consistent with our cell counting analysis of the splenic cell populations (Figure 6).
Figure 7.
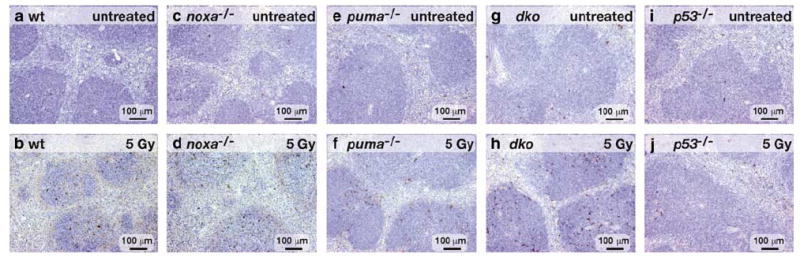
In situ analysis of γ-irradiation-induced apoptosis of splenocytes. Spleen sections from untreated (top panels) or 5 Gy γ-irradiated (20 h earlier, bottom panels) wt (a–b), noxa−/− (c–d), puma−/− (e–f), noxa−/−puma−/− (g–h) or p53−/− (l–j) animals were TUNEL-stained (see Figure 5). Nuclei were counterstained with hematoxylin. Sections from puma−/− and noxa−/−puma−/− animals (f, h) contain very few TUNEL-positive cells, comparable with sections from p53−/− animals (j). The images represent three or more independent stains performed on organs of at least three animals per genotype per treatment
Discussion
We have explored whether Noxa and Puma have overlapping functions in DNA damage-induced apoptosis mediated by p53. Our comparison of apoptosis in mice lacking Puma and/or Noxa with that in p53-deficient animals revealed that DNA damage-induced apoptosis signalling varies with cell type. In accord with previous studies,19-21 we showed that Puma has a major role in every cell type examined. In certain cell types, however, Noxa also clearly contributes, while in yet other cells additional pro-apoptotic factors may also be required.
Cell context-dependent roles of Noxa and Puma
Notably, in certain cell types, such as CD4+8+ thymocytes in vivo, Noxa and Puma accounted for essentially all of the pro-apoptotic activity of p53, and in other cell types, such as mature CD4+ T cells and macrophages, Puma alone suffices. Early work using protein and RNA synthesis inhibitors clearly showed that new gene expression is required for DNA damage-induced lymphocyte apoptosis.27 Our results indicate that the p53-induced expression of both noxa and puma is critical to mediate the full apoptotic response in thymocytes. Notably, loss of Bim has been shown to convey partial protection to CD4+8+ thymocytes treated with γ-radiation both in vitro25 and in vivo.21 This suggests that other BH3-only proteins may also contribute to p53-dependent death in some settings, such as CD8+ T cells and B cells.
Our evidence that Puma is generally more important for cell killing than Noxa is likely due to the fact that Puma binds with high affinity to all pro-survival Bcl-2 family members, whereas Noxa binds only to Mcl-1 and A1.28,29 Most likely, Noxa has a prominent role in p53-mediated apoptosis only in cells that contain higher levels of its targets Mcl-1 and/or A1 than of the other pro-survival proteins.
Although thymocytes and certain other cell types are reportedly completely dependent on p53 for DNA damage-induced apoptosis,23,24 in agreement with another study,30 we found that γ-irradiation of p53−/− (and of noxa−/−puma−/−) mice caused a small but significant reduction in CD4+8+ thymocytes, while no reduction was seen in, for example, splenic T cells. There are two possible, not mutually exclusive, explanations for this reduction. Firstly, since CD4+8+ thymocytes have a much more rapid turnover (~3 days) than mature T cells (several weeks), the small drop may be due to inhibition of new cell production from normally cycling progenitors in the thymus rather than death of CD4+8+ thymocytes per se. Alternatively, DNA damage may lead to activation not only of Noxa and Puma but also of a cell death pathway independent of p53. Since an equivalent small reduction in thymocytes was seen in γ-irradiated bcl-2 transgenic mice,21 that pathway would have to be Bcl-2-insensitive.
Combined Noxa/Puma loss afforded mature B cells and pro-B/pre-B cells less protection against γ-irradiation than p53 deficiency, implicating at least one other death factor. The small but significant role of Bim in DNA damage-induced killing of mature T and B cells21 implicates it. However, since the combined loss of Puma and Bim incompletely protected thymocytes and mature T and B cells from genotoxic damage,31 several BH3-only proteins, for example, Bim, Puma and Noxa, may well have a redundant role. As loss of Bim did not offer any protection to pre-B cells,21 yet another pro-apoptotic protein may figure in their demise. Interestingly, in the absence of Noxa and/or Puma we did not observe any compensatory increase of other BH3-only family members (specifically Bim, Bad or Bid) in untreated thymocytes or thymocytes treated with a p53-dependent or p53-independent death stimulus. We did, however, detect lower levels of Bid cleavage (tBid) in γ-irradiated thymocytes from puma−/− and noxa−/−puma−/− mice than wt thymocytes. We surmise that this is due to the fact that, following genotoxic damage, caspase-8 activation and Bid cleavage are secondary events (downstream of effector caspase activation), and hence neither occurs in cells in which apoptosis is inhibited, for example due to loss of Puma or both Noxa and Puma.
Noxa and Puma clearly have overlapping functions in fibroblasts, since noxa−/−puma−/− MEF expressing E1A survived treatment with etoposide better than noxa−/− or puma−/− MEF, despite similar increases in p53 protein. The transient nature of the protection to MEF offered by their loss is consistent with their temporal expression, which peaks ~6 h following DNA damage and then declines.15,18 Irrespective of their genotype, the fibroblasts all eventually died (by 48–72 h), indicating that etoposide must activate other pro-apoptotic factors or even other modes of cell death at later times. In contrast, the puma−/− and noxa−/−puma−/− MEF expressing E1A were completely refractory to doses of γ-radiation as high as 50 Gy. Unexpectedly, they proved markedly more resistant than the p53−/− MEF to γ-radiation or etoposide. This is probably due to the failure of p53−/− MEF to arrest at the G1/S checkpoint, resulting in catastrophic events in S phase or G2/M. In contrast, upon serum deprivation or overcrowding in culture, the p53−/−, puma−/− and noxa−/−puma−/− MEF expressing E1A all survived very well, indicating that p53-induced activation of Puma mediates apoptosis following these death stimuli. In this situation, a p53-independent process (e.g., p27 induction) probably implements cell cycle arrest, thereby preventing catastrophic events in S phase or G2/M.
Questions regarding the proposed non-transcriptional role of p53
Our results with thymocytes, CD4+ T cells and macrophages argue against an important physiological role, at least in these cell types, for an entirely transcription-independent apoptosis mediated by p53, such as by its direct binding to members of the Bcl-2 family on the outer mitochondrial membrane.8-10 Although cytoplasmic p53 has been proposed to mediate the rapid thymocyte death upon DNA damage,8 if any p53-driven mechanism independent of Puma and Noxa operated in these cells, their loss should not protect against p53-mediated signals. Notably, the apoptosis of colorectal carcinoma cells following DNA damage, but not p53-independent insults that require Puma, was abrogated by removal of the p53-binding sites from the PUMA gene.32
It has been proposed, however, that Puma induces apoptosis specifically by displacing cytoplasmic p53 from Bcl-xL, purportedly allowing the p53 to activate Bax directly.11 It is, however, noteworthy that Puma-induced death does not necessarily require p53, because Puma is critical for several p53-independent apoptotic signals (e.g. treatment with glucocorticoids or phorbol ester),19,20 and its enforced expression readily induces apoptosis in healthy primary cells,28 which should lack active p53, and even in p53-deficient tumour and non-transformed cells.17,33 Moreover, a Puma BH3 peptide readily permeabilises mitochondria from healthy cells, which should lack any active p53, and overcomes the protection mediated by each of the pro-survival family members.34 These results fit much more readily with the model that Puma, when induced by p53 or other transcription factors, provokes apoptosis by engaging all the pro-survival Bcl-2 family members, overcoming their constraint on Bax/Bak activation.28,35 Accordingly, Puma was shown to induce Bax-mediated apoptosis by displacing Bax from Bcl-xL, in a manner independent of p53.36
Implications for development and tumorigenesis
Analysis of the noxa−/−puma−/− mice indicated that Noxa and Puma are dispensable for normal development and haemopoiesis. Since normal numbers of DKO animals of both sexes were born from inter-crosses, combined Noxa/Puma absence does not recapitulate the fatal neural tube closure defect that eliminates ~65% of p53−/− females in utero.3 This developmental defect must therefore involve loss of p53-mediated processes other than or in addition to its apoptotic function. The normal haemopoiesis observed in the Noxa/Puma-deficient animals, which contrasts with the disturbed haemopoiesis in Bimdeficient mice,25 suggests that Noxa and Puma have evolved to mediate stress-induced apoptosis rather than developmentally programmed cell death.
P53-deficient mice stochastically develop a range of neoplasms and all die by 9 months of age.2,3 It has remained unclear whether the cell cycle arrest or apoptotic function of p53 is more critical for tumour suppression. Arrest at the G1/S checkpoint following DNA damage is orchestrated largely by p53-mediated induction of the cyclin-dependent kinase inhibitor p21/Waf1,1 but loss of p21 causes only a low incidence of tumours, arising mostly late in life (>16 months).37 Remarkably, we have not yet observed any tumours in noxa−/−puma−/− animals up to 1 year old, nor have any appeared in Puma-deficient mice (above the low incidence of thymic lymphoma normally observed on the C57BL/6 background). This indicates that loss of the apoptotic function of p53 is insufficient to initiate tumorigenesis. It therefore appears likely that both the apoptotic and cell cycle arrest functions, and perhaps even additional ones, such as induction of cell senescence,38 or perhaps its impact on mitochondrial respiration,39 contribute to the full tumour suppressive action of p53.
Despite little evidence so far in human tumours for abnormalities in expression or loss of Noxa or Puma,40 studies with transgenic mice have implicated Puma in tumour suppression. Knocking down Puma levels by shRNA41 or crosses between puma−/− and Eμ-myc transgenic mice (EM, JMA and AS, unpublished observations) have shown that Puma loss accelerates lymphomagenesis. Perhaps Puma’s tumour suppressor role, like that of Bim,42 requires the context of a pre-existing pro-apoptotic oncogenic alteration, such as Myc overexpression. In accord with that notion, a recent in vivo study suggests that the tumour suppressor function of p53 following irradiation comes into play only in the rare cells that suffer oncogenic mutations (e.g., in Myc or Ras) that induce p19ARF.43 Presumably, extremely few mutated cells contribute to tumorigenesis.43
In conclusion, our analysis of Noxa/Puma-deficient mice has revealed that essentially all of the p53-dependent γ-irradiation-induced death of thymocytes in vivo depends on these two BH3-only proteins. They also cooperate in the p53-dependent apoptosis of certain other cell types, such as MEF expressing E1A, whereas in other cell types, such as mature T and B cells in the spleen, Puma alone is critical. Furthermore, the cell-specific action of Noxa and Puma has important implications for understanding and treating cancer. Since many tumours are defective in p53 function, directly upregulating its apoptotic targets, such as Noxa or Puma, should render the cells sensitive to cytotoxic therapy. Thus, identifying the key mediators of the p53 response in different cell types may help tailor treatments to specific malignancies.
Materials and Methods
Mice
All experiments with mice followed the guidelines of the Melbourne Directorate Animal Ethics Committee. Generation and genotyping of mice deficient for Noxa,19 Puma19 or p533 have been described. All mice were on a C57BL/6 background or in the case of p53−/− had been backcrossed with C57BL/6 mice for >10 generations. To generate mice deficient for both Noxa and Puma, noxa+/− or noxa−/− mice were first crossed with puma+/− mice to produce mice heterozygous at both alleles (noxa+/− puma+/−). These double heterozygotes were crossed with noxa−/− mice to produce noxa−/−puma+/− mice, which were then inter-crossed to produce the double knockout (noxa−/−puma−/− DKO) animals.
Cell culture and cell viability assays
Cells were cultured at 37°C in a humidified 10% CO2 incubator in high-glucose Dulbecco’s Modified Eagle’s medium supplemented with 10% fetal calf serum (JRH Biosciences), 50 μM 2-mercaptoethanol (Sigma) and 100 μM asparagine (Sigma). FACS-sorted primary T and B lymphocytes were cultured at a starting density of 2–5 × 105/ml. Percentage cell viability was determined by FACS analysis as the fraction of cells not stained by either Annexin V-FITC or PI as described.19
Primary MEF were prepared from E14.5 embryos and retrovirally transduced with the Ad5 E1A cDNA or control pHED-puro vector only virus as described previously.19 For cell survival assays, E1A-expressing MEF were plated at 5 × 104/ml and allowed to adhere overnight before challenge with a death stimulus. For analysis of MEF viability, at each time point the adherent cells were trypsinised and collected together with the supernatant, which contains floating dead cells, prior to staining with PI. Percentage cell viability was determined by FACS analysis as the fraction of cells were not stained by PI.
Immunofluorescence staining and flow cytometric analysis
FACS analysis and cell sorting were performed using monoclonal antibodies as described previously.19 Cells were sorted using a MoFlo (Cytomation) or a Diva (Becton Dickinson) high-speed cell sorter. For cell survival assays, immature CD4+8+ thymocytes (pre-T cells) were sorted from the thymus, B220+sIgM−sIgD− pro-B/pre-B cells from bone marrow and mature Thy1+ T cells and B220+ B cells from lymph nodes.
Haemopoietic analysis
Mice were left untreated or exposed to 2.5 or 5 Gy of γ-radiation using a 60Co source and haemopoietic analysis performed 20 h later. Blood was obtained from live mice via the retro-orbital plexus or by cardiac puncture following CO2 anaesthesia. A portion of the blood was analysed using an ADVIA haematology system (Bayer). The remainder was treated with red cell removal buffer prior to immunofluorescent staining with surface marker-specific antibodies and FACS analysis. Single-cell suspensions were prepared from the thymus, lymph nodes (axillary, brachial and inguinal), bone marrow (both femora), and spleen. Viable leukocytes were enumerated using a haemocytometer and trypan blue exclusion. The cell type composition of an organ was determined by immunofluorescent staining with surface marker-specific antibodies and FACS analysis using a FACScan (Becton Dickinson).
Histology and TUNEL assays
Soft tissues and sternum were collected into Bouin’s fixative and formalin, respectively, embedded in paraffin and stained with hematoxylin and eosin. For TUNEL assays, thymus and spleen were fixed in formalin, sectioned onto silane-coated slides, deparaffinised, rehydrated and incubated with 20 μg/ml proteinase K (Roche) for 15 min. The sections were washed in PBS and then incubated for 1 h in a humidified incubator at 37°C with or without terminal deoxynucleotide transferase (Promega) in the presence of biotin-16-deoxyuridine triphosphate (Bio-16-dUTP, Roche). Incorporated Bio-16-dUTP terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling-positive (TUNEL-positive) cells within sections were detected using the VECTASTAIN ABC Elite biotin/avidin system and revealed using the peroxidase and diamonobenzidine (DAB) chromogen kit (Vector Laboratories). Sections were counterstained with hematoxylin and were viewed and photographed using a compound microscope (Zeiss) and a digital camera (Axiocam, Zeiss).
RT-PCR and Southern blot analysis
Total RNA was isolated from thymocytes or spleen cells and reverse transcription-PCR (RT-PCR) performed using the Superscript III First Strand kit (Invitrogen). The cDNA template was used in a PCR reaction using oligonucleotides primers specific for noxa or hprt described previously.19 Southern blot transfer and hybridisation using 32P end-labelled oligonucleotides specific for an internal noxa coding sequence was performed as described previously.19
Western blot analysis
Western blot analysis was performed by standard procedures using protein extracted from thymocytes. Western blots were probed with anti-Bim rabbit polyclonal antibody (Stressgen), anti-Bid rat monoclonal antibody 2D1 (gift of Dr. David Huang, WEHI), anti-Bad rabbit polyclonal antibody (Cell Signalling), anti-Puma rabbit polyclonal antibody (directed to the N terminus of human Puma; ProSci), anti-p53 mouse monoclonal antibody (BD Pharmingen), anti-p21 rabbit polyclonal antibody (Santa Cruz) and anti-E1A mouse monoclonal antibody (BD Pharmingen). Probing with an antibody to β-actin (AC-40, Sigma) was used as a loading control.
Statistical analysis
Statistical analysis was performed using the Student’s t-test (Two-tailed, assuming equal variance). P-values of <0.05 were considered to indicate statistically significant differences.
Supplementary Material
Acknowledgments
We thank N Iannarella, G Siciliano and A Naughton for animal care; Dr. F Battye, C Tarlinton, V Milovac, J Garbe and C Young for cell sorting; T Nikolaou and G Thomas for γ-irradiation; J Corbin for haematological analysis; Dr. S Mihajlovic, A Hasanein, K Weston for histological sections; Drs. M Schuler, S Lowe and G Hannon for expression vectors; and Professor S Cory and Drs. A Harris, D Huang, H Puthalakath, P Bouillet, L O’Reilly, M Pellegrini, L Coultas and C Scott for their input and interesting discussions. This work was supported by fellowships and grants from the NHMRC (Canberra; program #257502), the Leukemia and Lymphoma Society (SCOR grant #7015), the NIH (CA043540-18 and CA80188-6), the JDRF/NHMRC, the Leukemia Research Foundation (LRF), the Cancer Council Victoria Postdoctoral Cancer Research Fellowship and the Austrian Science Fund (FWF).
Abbreviations
- BH
Bcl-2-homology
- DKO
double knockout
- DP
double-positive
- MEF
mouse embryonic fibroblasts
- PMA
phorbol 12-myristate 13-acetate
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling
Footnotes
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
References
- 1.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 2.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CAJ, Butel JS, et al. Mice deficient for p53 are developmentally normal but are susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 3.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Szekely L, Okan I, Klein G, Wiman KG. Wild-type p53-triggered apoptosis is inhibited by bcl-2 in a v-myc-induced T-cell lymphoma line. Oncogene. 1993;8:3427–3431. [PubMed] [Google Scholar]
- 5.Strasser A, Harris AW, Cory S. Bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell. 1991;67:889–899. doi: 10.1016/0092-8674(91)90362-3. [DOI] [PubMed] [Google Scholar]
- 6.Strasser A, Harris AW, Jacks T, Cory S. DNA damage can induce apoptosis in proliferating lymphoid cells via p53-independent mechanisms inhibitable by Bcl-2. Cell. 1994;79:329–339. doi: 10.1016/0092-8674(94)90201-1. [DOI] [PubMed] [Google Scholar]
- 7.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 8.Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol. 2004;24:6728–6741. doi: 10.1128/MCB.24.15.6728-6741.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6:443–450. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- 10.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 11.Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 12.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 13.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang DCS, Strasser A. BH3-only proteins – essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 15.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-only member of the bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 16.Nakano K, Vousden KH. PUMA a novel proapoptotic gene is induced by p53. Molecular Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 17.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Molecular Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 18.Han J, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ, et al. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Natl Acad Sci USA. 2001;98:11318–11323. doi: 10.1073/pnas.201208798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 20.Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 21.Erlacher M, Michalak EM, Kelly PN, Labi V, Niederegger H, Coultas L, et al. BH3-only proteins Puma and Bim are rate-limiting for {gamma} -radiation and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 2005;106:4131–4138. doi: 10.1182/blood-2005-04-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, et al. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003;17:2233–2238. doi: 10.1101/gad.1103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 24.Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, et al. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 25.Bouillet P, Metcalf D, Huang DCS, Tarlinton DM, Kay TWH, Köntgen F, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 26.Lowe SW, Ruley HE. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes and Development. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- 27.Sellins KS, Cohen JJ. Gene induction by g-irradiation leads to DNA fragmentation in lymphocytes. J Immunol. 1987;139:3199–3206. [PubMed] [Google Scholar]
- 28.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 29.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 Domains of BH3-Only Proteins Differentially Regulate Bax-Mediated Mitochondrial Membrane Permeabilization Both Directly and Indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 30.MacFarlane M, Jones NA, Dive C, Cohen GM. DNA-damaging agents induce both p53-dependent and p53-independent apoptosis in immature thymocytes. Mol Pharmacol. 1996;50:900–911. [PubMed] [Google Scholar]
- 31.Erlacher M, Laabi V, Manzl C, Bock G, Tzankov A, Haecker G, et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med. 2006;203:2939–2951. doi: 10.1084/jem.20061552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang P, Yu J, Zhang L. The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc Natl Acad Sci USA. 2007;104:4054–4059. doi: 10.1073/pnas.0700020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Callus BA, Ekert PG, Heraud JE, Jabbour AM, Kotevski A, Vince JE, et al. Cytoplasmic p53 is not required for PUMA-induced apoptosis. Cell Death Differ. 2008;15:213–221. doi: 10.1038/sj.cdd.4402245. [DOI] [PubMed] [Google Scholar]
- 34.Uren RT, Dewson G, Chen L, Coyne SC, Huang DCS, Adams JM, et al. Mitochondrial permeabilization relies on BH3 ligands engaging multiple pro-survival Bcl-2 relatives, not Bak. J Cell Biol. 2007;177:277–287. doi: 10.1083/jcb.200606065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 36.Ming L, Wang P, Bank A, Yu J, Zhang L. PUMA dissociates Bax and Bcl-XL to induce apoptosis in colon cancer cells. J Biol Chem. 2006;281:16034–16042. doi: 10.1074/jbc.M513587200. [DOI] [PubMed] [Google Scholar]
- 37.Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001;61:6234–6238. [PubMed] [Google Scholar]
- 38.Metz T, Harris AW, Adams JM. Absence of p53 allows direct immortalization of hematopoietic cells by the myc and raf oncogenes. Cell. 1995;82:29–36. doi: 10.1016/0092-8674(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 39.Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 40.Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, Siebert R, Climent J, Fresquet V, et al. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007;109:271–280. doi: 10.1182/blood-2006-06-026500. [DOI] [PubMed] [Google Scholar]
- 41.Hemann MT, Zilfou JT, Zhao Z, Burgess DJ, Hannon GJ, Lowe SW. Suppression of tumorigenesis by the p53 target PUMA. Proc Natl Acad Sci USA. 2004;101:9333–9338. doi: 10.1073/pnas.0403286101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci USA. 2004;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature. 2006;443:214–217. doi: 10.1038/nature05077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.