

Abstract
Methamphetamine (METH) is a neurotoxic drug of abuse that damages the dopamine (DA) neuronal system in a highly delimited manner. The brain structure most affected by METH is the striatum where long-term DA depletion and microglial activation are maximal. Endogenous DA has been implicated as a critical participant in METH-induced neurotoxicity, most likely as a substrate for non-enzymatic oxidation by METH-generated reactive oxygen species (ROS). The striatum is also extensively innervated by serotonin (5HT) nerve endings and this neurochemical system is modified by METH in much the same manner as seen in DA nerve endings (i.e., increased release of 5HT, loss of function in tryptophan hydroxylase and the serotonin transporter, long-term depletion of 5HT stores). 5HT can also be modified by ROS to form highly reactive species that damage neurons but its role in METH neurotoxicity has not been assessed. Increases in 5HT levels with 5HTP do not change METH-induced neurotoxicity to the DA nerve endings as revealed by reductions in DA, tyrosine hydroxylase and dopamine transporter levels. Partial reductions in 5HT with p-chlorophenylalanine (PCPA) are without effect on METH toxicity, despite the fact that PCPA largely prevents METH-induced hyperthermia. Mice lacking the gene for brain tryptophan hydroxylase 2 are devoid of brain 5HT and respond to METH in the same manner as wild-type controls, despite showing enhanced drug-induced hyperthermia. Taken together, the present results indicate that endogenous 5HT does not appear to play a role in METH-induced damage to DA nerve endings of the striatum.
Keywords: methamphetamine, serotonin, dopamine, neurotoxicity, tryptophan hydroxylase 2, PCPA, 5HTP
Methamphetamine (METH) is a psychostimulant drug with high abuse potential. METH use continues at high rates and places extreme demands on numerous social, professional and law enforcement networks in response to the consequences associated with its abuse. Perhaps the most dangerous element associated with METH is its ability to cause persistent damage to the central nervous system in humans and in animal models of drug-induced neuronal damage. The mechanisms that mediate METH-induced neurotoxicity are numerous and include elevated oxidative stress (Yamamoto and Bankson 2005, Cadet et al. 2007, Gibb et al. 1990, Fleckenstein et al. 1997b, Volz et al. 2007), mitochondrial dysfunction (Burrows et al. 2000, Brown et al. 2005, Tian et al. 2009) and inflammatory processes associated with microglial activation (Thomas et al. 2004b, Bowyer et al. 2008, Thomas et al. 2004a, LaVoie et al. 2004). The neurotoxic effects of METH are remarkably delineated and are most often associated with reductions in function of dopamine (DA) nerve endings of the striatum. However, more extensive damage to include neuronal death can also occur after high-dose METH intoxication (Cadet et al. 2007, Davidson et al. 2001, Cadet et al. 2003, Krasnova and Cadet 2009).
A great deal of effort has been directed at achieving a better understanding of the mechanisms by which METH damages the CNS. In consideration of the fact that DA nerve endings of the striatum are primary targets of METH-induced damage, and realizing the extreme manner by which METH disrupts DA homeostasis, DA itself has become a primary focus of attention. DA nerve endings comprise the majority of monoamine synaptic input to the striatum. METH leads to a release of DA from vesicle stores into the less protective environments of the cytoplasm and synaptic space where it is exposed to rapid non-enzymatic oxidation to several reactive species such as DA quinones (LaVoie and Hastings 1999, Kuhn et al. 2006). Changes in local presynaptic DA status can have powerful influences on METH toxicity. It is generally observed that treatments that shift DA from vesicles into the cytoplasm (e.g., reserpine) increase toxicity while reductions in the METH-releasable pool of DA (e.g., α-methyl-p-tyrosine) are neuroprotective (Albers and Sonsalla 1995, Thomas et al. 2008, Schmidt et al. 1985, Wagner et al. 1983, Thomas et al. 2009). A role for DA in mediating METH-induced toxicity has been challenged by studies showing that changes in core body temperature (Yuan et al. 2001), more than alterations in intracellular DA status, can explain modifications in METH neurotoxicity. It has also been suggested that norepinephrine (NE) input to the striatum can influence METH-toxicity. Reductions in NE levels have been shown to increase dramatically the damaging effects of METH on DA nerve endings (Weinshenker et al. 2008, Fornai et al. 1995, Fornai et al. 1999, Fornai et al. 1996), despite the fact that NE content of the striatum is extremely low (e.g., about 3–5% of DA).
The striatum contains a predominant input of serotonin (5HT) containing nerve endings from the midbrain raphe nuclei (Michelsen et al. 2007, Steinbusch 1981). Although 5HT synapses represent a significant portion of the monoamine input to the striatum, a role for 5HT in the neurotoxic properties of METH has received relatively little attention. This is surprising because METH is a powerful releaser of 5HT (Berger et al. 1992, Kuczenski et al. 1995, Wall et al. 1995, Cass 2000) and it disrupts presynaptic 5HT status in much the same manner seen for DA. 5HT nerve endings show persistent reductions in function after METH intoxication to include diminished levels of activity for tryptophan hydroxylase and the 5HT transporter (Fleckenstein et al. 2000, Hanson et al. 1998, Fleckenstein et al. 2007, Fleckenstein et al. 1997a). 5HT, like DA, can also serve as a substrate for attack by METH-generated reactive oxygen species (ROS) and the downstream products are known to exert damaging effects on neuronal tissue (Wrona and Dryhurst 1988, Wrona et al. 1986). For example, oxidation of 5HT by superoxide, nitric oxide or peroxynitrite can form tryptamine-4,5-dione (Wrona and Dryhurst 2001, Wrona et al. 1997a), a powerful inhibitor of TPH2 (Wrona and Dryhurst, 2001). Oxidation of 5HT by hydroxyl radical forms 5-hydroxy-3-ethylamino-2-oxindole and 5,6-dihydroxytryptamine (Wrona et al. 1995), both of which are potential 5HT neurotoxins, but these byproducts have not yet been detected in brain after high dose METH treatment (Yang et al. 1997). 5HT can also bind to and activate nitric oxide synthase and increase production of superoxide radical and hydrogen peroxide (Breard and Grillon 2009, Breard et al. 2007). Many of the treatments that profoundly alter METH-toxicity to DA nerve endings of the striatum (e.g., reserpine) cause near-total depletion of 5HT as well, leaving open the possibility that 5HT could react with METH-generated reactive oxygen and nitrogen species to modulate drug-induced neurotoxicity. In light of the precedence implicating endogenous 5HT as a substrate for attack by ROS in the generation of neurotoxic species, the present studies investigated the possibility that 5HT participates in METH-induced neurotoxicity to DA nerve endings of the striatum.
Materials and methods
Materials
(+) methamphetamine hydrochloride, pentobarbital, 5HTP, carbidopa, Triton X-100, Tween 20, methanol, EDTA, all buffers, and HPLC reagents were purchased from Sigma-Aldrich (St. Louis, MO). Bicinchoninic acid protein assay kits were obtained from Fisher Scientific (Rockford, IL). Antibodies against TH (Kuhn and Billingsley 1987) were produced as previously described. Antibodies against DAT were generously provided by Dr. Roxanne Vaughn (University of North Dakota). HRP-conjugated secondary antibodies were obtained from Amersham (Piscataway, NJ) and Jackson ImmunoResearch Laboratories (West Grove, PA).
Animals
Female C57BL/6 mice (Harlan, Indianapolis, IN) and TPH2+/+ and TPH2−/− mice (see below) weighing 20–25 g at the time of experimentation were housed 5 per cage in large shoe-box cages in a light (12 hr light/dark) and temperature controlled room. Mice had free access to food and water. The Institutional Care and Use Committee of Wayne State University approved the animal care and experimental procedures. All procedures were also in compliance with the NIH Guide for the Care and Use of Laboratory Animals.
Generation of mice with null-mutations in the gene for TPH2
A targeting vector was designed that deleted 105 bp of exon 1 coding sequence of TPH2, including the ATG initiation codon. This vector contained two negative selection cassettes (thymidine kinase and diphtheria toxin A) and a neomycin positive selection cassette. The vector was linearized and transfected into ES cells using standard protocols. Positive selection was achieved by incubation of transfected cells with G418 and resistant clones were isolated and screened for homologous recombination. Positive ES cell clones were confirmed by 5’ and 3’ screening (Southern blotting and PCR) and two of these were expanded for injection into C57BL/6J blastocysts. Upon injection, the blastocysts were re-implanted into pseudo-pregnant females and allowed to develop to term. Six highly chimeric males were generated and mated to wild-type C57BL/6J females. Numerous F1 offspring showed transmission of the mutant TPH2 allele to the germline as assessed by agouti coat color. The genotypes of these F1 offspring were confirmed as heterozygous (TPH2+/−) and TPH2+/− males and females were set up as mating pairs to generate homozygous (TPH2−/−), wild-type (TPH2+/+) and TPH2+/− offspring. For further details, see Appendix S1.
Pharmacological and physiological procedures
Mice (wild-type and TPH2−/− mice) were exposed to a neurotoxic regimen of METH comprised of 4 injections of 5 mg/kg i.p. with a 2 hr interval between each injection. This METH regimen is known to cause microglial activation and DA nerve ending damage (Thomas et al. 2004b). To assess the effect of increases in the newly synthesized pool of 5HT on METH toxicity, mice were treated with 5HTP, the immediate precursor to 5HT, 1 hr before the first and third METH injections in a dose of 50 mg/kg along with carbidopa (25 mg/kg) to inhibit peripheral decarboxylase enzymes. PCPA, the irreversible inhibitor of TPH (Koe and Weissman 1966), was used to reduce brain 5HT levels. PCPA was injected in a dose of 300 mg/kg once per day for 3 days prior to the neurotoxic regimen of METH. The last PCPA injection was given 24 hr before the first METH injection. Controls for drug pretreatments and METH received injections of physiological saline on the same schedule used for each respective compound. All injections were given via the i.p. route. Mice were sacrificed 48 hr after the last METH treatment. Body temperature was monitored by telemetry using IPTT-300 implantable temperature transponders from Bio Medic Data Systems, Inc. (Seaford, DE). Temperatures were recorded non-invasively every 20 min starting 60 min before the first METH injection and continuing for 10 hr thereafter using the DAS-5001 console system from Bio Medic.
Determination of striatal monoamine content
The present experiments were restricted to the determination of treatment effects on METH-induced toxicity to the DA system. Thus, striatal tissue was dissected bilaterally from brain 48 hr after treatments and stored at −80°C. Frozen tissues were weighed and sonicated in 10 volumes of 0.16 N perchloric acid at 4°C. Insoluble protein was removed by centrifugation and DA was determined by HPLC with electrochemical detection. In order to characterize the 5HT neurochemical phenotype of TPH2−/− mice, the striatal levels of DA, 5HT and 5-hydroxyindole acetic acid (5HIAA) in TPH2−/− mice and wild-type controls were determined by HPLC (Wolf et al. 1991). TPH2 enzyme activity was determined by measuring the formation of 5HTP from tryptophan by HPLC with fluorescence detection in an in vitro reaction as previously described (Kuhn et al. 2007). For additional information on the TPH2−/− phenotype, see Appendix S1.
Determination of protein levels by immunoblotting
The effects of the pharmacological treatments on striatal TH and DAT levels were determined by immunoblotting as part of the characterization of toxicity to striatal DA nerve endings. Mice were sacrificed by decapitation 48 hr after treatments and striatum was dissected bilaterally. Tissue was stored at −80°C. Frozen tissue was disrupted by sonication in 1% SDS at 95°C and insoluble material was sedimented by centrifugation. Protein was determined by the bicinchoninic acid method and equal amounts of protein (70 μg/lane) were resolved by SDS-polyacrylamide gel electrophoresis and then electroblotted to nitrocellulose. Blots were blocked in Tris buffered saline containing Tween 20 (0.1% v/v) and 5% non-fat dry milk for 1 hr at room temperature. Primary antibodies were added to blots and allowed to incubate for 16 hr at 4°C. Blots were washed 3X in Tris-buffered saline to remove unreacted antibodies and then incubated with HRP-conjugated anti-IgG secondary antibody (1:4000) for 1 hr at room temperature. Immunoreactive bands were visualized by enhanced chemiluminescence and the relative densities of TH- and DAT-reactive bands were determined by scanning films on an Alpha Innotech (San Leandro, CA) AlphaImager HP using automatic spot detection. Immunoblotting results for TH and DAT were normalized to tubulin for quantitation of band densities. For further information on the TPH2−/− phenotype, see Appendix S1.
Data analysis
The effects of drug treatments on striatal DA, TH and DAT content were tested for significance by ANOVA. Individual treatment groups were compared to appropriate controls with a one-way ANOVA followed by Tukey’s Multiple Comparison Test in GraphPad Prism 5. Results from studies of METH-induced hyperthermia in wild-type versus TPH2−/− mice were compared at individual times when core temperatures were maximally increased (i.e., 20–30 min after each drug injection) using a Student’s T-test. Differences were considered significant if p < 0.05.
Results
Effect of increased 5HT content on METH-induced toxicity to striatal DA nerve endings
Mice were treated with 5HTP, the immediate precursor to 5HT, before the first and third injections of METH and animals were sacrificed 2 days later. The results in Fig. 1 show results of these treatments on striatal DA levels. It can be seen that METH lowered DA levels by 60–65%. Treatment of mice with 5HTP alone did not change striatal DA levels. Combined treatment of mice with METH + 5HTP caused the same reduction in striatal DA as seen in mice treated with METH alone. The effects of these same treatments on striatal levels of TH and DAT are presented in Fig. 2. It can be seen that METH caused significant reductions in TH (Fig. 2A) and DAT (Fig. 2B) protein levels (50% reductions for each). 5HTP alone did not change TH or DAT levels by comparison to controls and the combined treatment of 5HTP + METH had effects that were the same as those seen after METH alone. 5HTP alone did not change basal core body temperatures and METH hyperthermia was not altered by 5HTP as shown in Fig. 3.
Fig. 1.
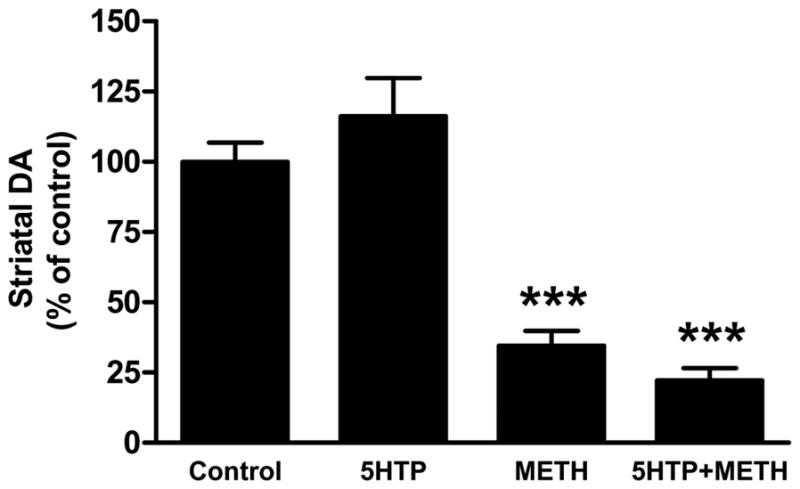
Effects of increases in 5HT on METH-induced depletion of striatal DA. Mice (N = 5–8/group) were treated with saline (control), 5HTP (50 mg/kg) + carbidopa (25 mg/kg), METH alone (4 injections of 5 mg/kg at 2 hr intervals) or with 5HTP + carbidopa 60 min before the first and third injections of METH and sacrificed 48 hr after the last injection. Striatal DA levels were determined by HPLC with electrochemical detection. Results are presented as mean ± SEM relative to controls. Differences between treatment groups were tested for significance by one-way ANOVA followed by Tukey’s multiple comparison test using Graphpad Prism 5. *** indicates significantly different from control (p < 0.001).
Fig. 2.

Effects of increases in 5HT on METH-induced reductions in TH and DAT levels in striatum. Mice (N = 5–8/group) were treated as described in the legend to Fig. 1 and striatal TH (A) and DAT levels (B) were determined by western blot analysis. Representative results from blots are included in each panel and samples for each subject in all groups were scanned and plotted in bar graph format. Results are presented as mean ± SEM. Significant differences were determined by one-way ANOVA followed by Tukey’s multiple comparison test. *** indicates significantly different from control (p < 0.001).
Fig. 3.

Effects of increases in 5HT on METH-induced hyperthermia. Mice (N = 5–8/group) were treated as described in the legend to Fig. 1 and core body temperatures were recorded by telemetry at 20 min intervals for the indicated time period. Results are presented as group means. SEMs are omitted for the sake of clarity and were always < 10% of the respective mean values. Areas under the curve (AUC) for all treatment groups were calculated using GraphPad prism with respect to an arbitrary baseline set to 35°C for all groups. AUC values for the 5HTP + carbidopa group (996) did not differ from controls (985) whereas groups treated with METH (1560) and 5HTP/carbidopa + METH (1433) were not different from one another but differed significantly by comparison to controls (p < 0.001 for each).
PCPA-induced reduction in 5HT content and effects on METH-induced toxicity to striatal DA nerve endings
PCPA was injected once daily at a dose of 300 mg/kg for 3 days prior to the neurotoxic METH regimen. This treatment lowered striatal 5HT content by 60–70% at the time of the first METH injection. Results in Fig. 4 show that PCPA alone did not alter striatal DA content 2 days after treatments. Similarly, the METH-induced reduction in striatal DA (60–65%) was not modified by prior treatment with PCPA (55% reduction). The effects of these same treatments on striatal TH and DAT protein levels are presented in Fig. 5. In agreement with Fig. 2, only METH lowered TH (Fig. 5A) and DAT (Fig. 5B) levels by comparison to controls. PCPA had no effect of its own on TH and DAT and was likewise without effect on the METH-induced reductions in these proteins. PCPA alone did not change basal core body temperature but METH-induced hyperthermia was reduced significantly by PCPA (Fig. 6).
Fig. 4.
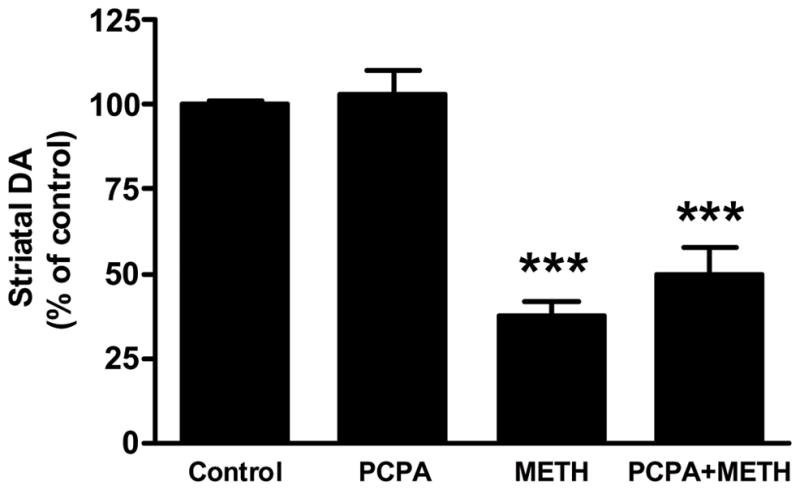
Effects of PCPA on METH-induced depletion of striatal DA. Mice (N = 5–8/group) were treated with saline (control), PCPA (300 mg/kg daily for 3d prior to the first METH injection), METH alone (4 × 5 mg/kg at 2 hr intervals) or with PCPA + METH. Mice were sacrificed 48 hr after the last injection of METH and striatal DA levels were determined by HPLC with electrochemical detection. Results are presented as mean ± SEM relative to controls. Differences between treatment groups were tested for significance by one-way ANOVA followed by Tukey’s multiple comparison test using Graphpad Prism 5. *** indicates significantly different from control (p < 0.001).
Fig. 5.
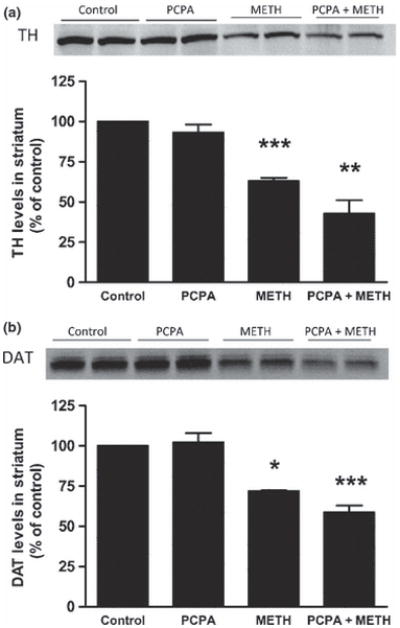
Effects of PCPA on METH-induced reductions in TH and DAT levels in striatum. Mice (N = 5–8/group) were treated as described in the legend to Fig. 4 and striatal TH (A) and DAT levels (B) were determined by western blot analysis. Representative results from blots are included in each panel and samples for each subject in all groups were scanned and plotted in bar graph format. Results are presented as mean ± SEM. Significant differences were determined by one-way ANOVA followed by Tukey’s multiple comparison test. Symbols (*, p < 0.05; ***, p < 0.001) indicate significant differences relative to the respective controls.
Fig. 6.

Effects of PCPA on METH-induced hyperthermia. Mice (N = 5–8/group) were treated as described in the legend to Fig. 4 and core body temperatures were recorded by telemetry at 20 min intervals. Results are presented as group means. SEMs are omitted for the sake of clarity and were always < 10% of the respective mean values. Areas under the curve (AUC) for all treatment groups were calculated using GraphPad Prism 5 with respect to an arbitrary baseline set to 35°C for each group. AUC values for the PCPA (1017) and PCPA + METH (928) groups did not differ from controls (944) whereas METH (1340) treated mice showed significant increases in core temperature by comparison to controls (p < 0.001).
Mice lacking brain TPH2 and 5HT show the same METH-induced toxicity to striatal DA nerve endings as wild-type controls
Mice with a null mutation in the gene for TPH2 (Fig. S1) were devoid of brain TPH2, 5HT and 5HIAA and they lacked the ability to hydroxylate tryptophan (Table S1). Immunoblotting (Fig. S2) and immunohistochemical (Fig. S3) analyses confirmed a lack of TPH2 in brains of homozygous mice and established that 5HT neuronal processes remained intact. TPH2−/− and wild-type controls were treated with the standard METH neurotoxic regimen and the response of the striatal DA system was assessed. The results in Fig. 7 show that METH reduced striatal DA levels to same extent in both genotypes, lowering wild-types to 32% of control and TPH2−/− mice to 28% of control. The effect of METH on DA levels in wild-type and TPH2−/− mice was not different (p > 0.05). Fig. 8 presents immunoblotting results for TH and DAT and show that METH-induced reductions in these proteins were the same in wild-type controls (Figs. 8A & B) and TPH2−/− mice (Figs. 8C & D), respectively. Heterozygous mice (i.e., TPH2+/−) were not different from wild-type with regard to striatal neurochemical status and their response to METH was likewise the same as wild-type controls (data not shown). Resting core body temperatures were slightly higher in TPH2−/− mice by comparison to wild-type mice prior to initiation of drug treatments. TPH2−/− controls showed a gradual reduction in core temperature (from approximately 38°C to 36°C over the 8 hour period of measurement), an effect that was slightly greater than seen in wild-type controls. METH caused significant hyperthermia in both wild-type and TPH2−/− mice. The overall effect of METH on body temperature did not differ between wild-type and TPH2−/− groups, but TPH2−/− mice did show significant elevations (approximately 1°C higher) by comparison to controls at the 20, 140 and 380 min time points (p < 0.05). These results are presented in Fig. 9.
Fig. 7.
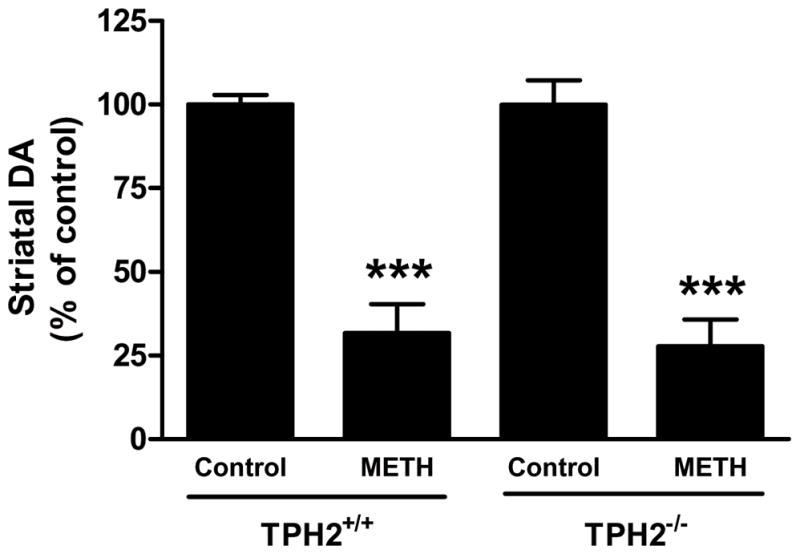
Effects of METH on striatal DA levels in TPH2−/− mice. Wild-type mice or mice lacking the gene for TPH2 (TPH2−/−) were treated with 4 injections of METH (5 mg/kg at 2 hr intervals). Mice were sacrificed 48 hr after the last METH injection and striatal DA levels were determined by HPLC with electrochemical detection. Results are presented as mean ± SEM relative to controls. Differences between treatment groups were tested for significance by one-way ANOVA followed by Tukey’s multiple comparison test using GraphPad Prism 5. *** indicates significantly different from the respective control (p < 0.001).
Fig. 8.

Effects of METH on striatal TH and DAT levels in TPH2−/− mice. Wild-type (A & B) or TPH2−/− (C & D) mice (N = 5–8/group) were treated with METH as described in the legend to Fig. 7 and striatal TH (A & C) and DAT levels (B & D) were determined by western blot analysis. Representative results from blots are included in each panel and samples for each mouse in all groups were scanned and plotted in bar graph format. Results are presented as mean ± SEM. Differences between treatment groups was tested for significance by one-way ANOVA followed by Tukey’s multiple comparison test. Symbols (*, p < 0.05; **, p < 0.01; ***, p < 0.001) indicate significant differences relative to the respective controls.
Fig. 9.

Effects of METH on core body temperature in TPH2−/− mice. Wild-type or TPH2−/− mice (N = 5–8/group) were treated as described in the legend to Fig. 7 and core body temperatures were recorded by telemetry at 20 min intervals. Results are presented as group means. SEMs are omitted for the sake of clarity and were always < 10% of the respective mean values. Areas under the curve (AUC) for all treatment groups were calculated using GraphPad Prism 5 with respect to an arbitrary baseline set to 35°C for each group. AUC values for untreated TPH2−/−mice (927) did not differ from wild-type controls (1153) whereas wild-type (1707) and TPH2−/−mice (1731) treated with METH were significantly higher (p < 0.001) than their respective controls. The effects of METH on core temperatures were significantly greater in TPH2−/− mice by comparison to wild-type controls only at the 20, 140 and 380 min time points (p < 0.05; Student’s T-test).
Discussion
The rationale for testing endogenous striatal 5HT for its influence on METH-induced damage to DA nerve endings of the striatum is compelling. Striatal 5HT homeostasis is disrupted by METH in much the same manner as is seen for DA. An essential role for endogenous DA in METH-induced toxicity to the DA system has been established (Albers and Sonsalla 1995, Thomas et al. 2008, Schmidt et al. 1985, Wagner et al. 1983, Thomas et al. 2009) but a similar role for 5HT in this effect has scarcely been studied. The powerful release of 5HT from vesicles into the cytoplasm and the synapse under conditions of heightened oxidative stress caused by METH, would expose the neurotransmitter to a variety of reactants (e.g., superoxide, hydroxyl radical, nitric oxide, peroxynitrite, see Introduction) that could lead to the local production of numerous 5HT-derived neurotoxins (Wrona and Dryhurst 1988, Wrona and Dryhurst 1998, Wrona and Dryhurst 2001, Wrona et al. 1995, Wrona et al. 1997b). 5HT itself can even bind to nitric oxide synthase and increase its production of oxygen-based radicals (Breard and Grillon 2009, Breard et al. 2007). However, despite this strong precedence, the present data do not assign a role for endogenous 5HT in METH-induced toxicity to DA nerve endings of the striatum. Increases in 5HT levels using 5HTP do not modify METH-induced reductions in DA, TH or DAT, and leave METH-induced hyperthermia unchanged. Partial reductions in 5HT using PCPA are likewise without effect on reductions in DA, TH and DAT caused by METH. Mice lacking the gene for TPH2 are devoid of brain 5HT and TPH2 protein, and show the same METH-induced reductions in striatal DA, TH and DAT as seen in wild-type controls.
In light of the fact that METH can increase 5HT release in striatum, several groups have examined if blockade of the SERT or 5HT receptors modulates METH-induced DA neurotoxicity. Results with the SERT blocker fluoxetine show conflicting results with regard to METH-induced toxicity to the DA system. Fluoxetine has been shown to enhance (Ricaurte et al. 1983) or have no effect (Matuszewich and Yamamoto 2004) on METH-induced reductions in striatal DA. The fluoxetine-induced enhancement of toxicity was attributed to increased brain concentrations of METH (Ricaurte et al. 1983). Fluoxetine and other SERT inhibitors provide substantial protection against MDMA- (Schmidt and Taylor 1987, Li et al. 2010, Sanchez et al. 2001, Goni-Allo et al. 2006) and METH-induced (Rau et al. 2006) (Fleckenstein et al. 1997a) damage to the 5HT neuronal system. In contrast, fluoxetine does not protect against MDMA-induced deficits to the DA neuronal system (O'Shea et al. 2001) but it does partially prevent the malonate potentiation of MDMA reductions in DA (Goni-Allo et al. 2006). The possibility that 5HT released into the synapse by METH is contributing to DA neurotoxicity via 5HT receptor mechanisms has been ruled out as well by studies showing that 5HT2 and 5HT3/4 receptor antagonists (Johnson et al. 1994, Johnson et al. 1988) fail to prevent drug-induced reductions in DA neurochemical function. In sum, it does not appear that the amount of 5HT in nerve endings and in the synaptic space exerts any influence on METH-induced toxicity to the striatal DA system.
A number of investigators have shown that reserpine leads to substantial increases in DA neurotoxicity after METH treatment and this effect has generally been attributed to alterations in DA homeostasis (Albers and Sonsalla 1995, Thomas et al. 2008, Thomas et al. 2009, Wagner et al. 1983, Yuan et al. 2001). Similarly, mice lacking the gene for VMAT2 show increased toxicity after METH treatment, effects attributed to reduced storage but increased release of DA (Fumagalli et al. 1999, Guillot et al. 2008). However, reserpine and null mutations in the gene for VMAT2 also disrupt 5HT homeostasis, making it difficult to attribute alterations in METH toxicity solely to DA. Our present results, and particularly those in mice lacking TPH2 and brain 5HT, resolve this interpretational difficulty by showing that TPH2−/− mice are equally susceptible to METH-induced toxicity to the DA system as are wild-type mice. Together, these results indicate that endogenous 5HT does not contribute to or influence METH toxicity to DA nerve endings of the striatum.
A role for endogenous NE in the toxic actions of METH on striatal DA nerve endings has also been indicated and derives from the general neuroprotective and antioxidant properties attributed to this monoamine (Heneka et al. 2002, Heneka et al. 2003, Kalinin et al. 2006). With regard to METH, Fornai and colleagues have shown that conditions associated with reduced striatal NE lead to substantial increases in drug-induced toxicity to DA nerve endings. These conditions include treatment of animals with DSP-4, a selective NE neurotoxin (Fornai et al. 1995, Fornai et al. 1999, Fornai et al. 1996), ablation of the gene for dopamine β-hydroxylase and inhibition of NE synthesis with the dopamine β-hydroxylase inhibitor fusaric acid (Weinshenker et al. 2008). The interpretational problems that arise from the use of reserpine and VMAT2 knockout mice mentioned previously also apply in the case of NE, because disruption of vesicle storage capacity severely depletes all monoamines including NE. The input of NE to the striatum is very small by comparison to DA and 5HT, comprising levels that are from 2–5% of the other monoamines, and dopamine β-hydroxylase levels in striatum are among the lowest in the CNS (Ross and Reis 1974). DSP-4 lesions of the locus coeruleus reduce striatal NE levels by about 30% (Fornai et al. 1999) indicating an extremely powerful influence of NE over the sensitivity of DA nerve endings to METH-induced toxicity. Unfortunately, our present approach cannot resolve the issue of whether NE depletion in reserpinized mice is the mechanism by which METH-toxicity is increased and remains an open question.
Many of the studies that have investigated the role of endogenous 5HT, DA or NE in METH-induced neurotoxicity depended by necessity on the use of drugs or neurotoxins to reduce the levels of the desired neurotransmitter. The non-specific nature of monoamine depletion by reserpine has already been discussed above. PCPA is another such drug and is the classical TPH inhibitor used to lower brain 5HT levels (Koe and Weissman 1966). PCPA is only moderately effective and useful in this regard because larger doses used to cause the greatest losses in 5HT also result in significant reductions in striatal DA [see for example (Dailly et al. 2006)]. PCPA is also a powerful inhibitor of liver phenylalanine hydroxylase (Koe and Weissman 1966) and will result in hyper-phenylalaninemia, a condition that could complicate its specificity as elevated levels of phenylalanine in blood could compete with other large neutral amino acids (e.g., tyrosine, tryptophan) for uptake into brain. The depletion of brain 5HT by PCPA is also relatively short-lived and in the doses used in our experiments (see Fig. 4), 5HT returns to untreated control levels in 1–2 days. The use of mice lacking the gene for TPH2 allows a superior approach to these types of experiments. The TPH2−/− mice are shown presently to be devoid of TPH2 protein, tryptophan hydroxylating activity and 5HT using several complementary approaches. Supplementary results establish further that the expression of SERT in the TPH2−/− mice is the same as wild-type controls, and TPH2−/− mice also express NPT-2, the protein product of the neomycin resistance cassette used in the targeting vector for positive selection of homologous recombination events. These data establish that 5HT neurons are intact in TPH2−/− mice but they totally lack their neurotransmitter content. TPH2−/− mice showed similar psychostimulant responses to METH as wild-type controls (data not shown) and METH-induced toxicity within DA nerve endings of the striatum was the same as controls.
The responses of mice to METH neurotoxicity after dramatic shifts in the status of their brain 5HT content is all the more interesting when considered in light of the role of hyperthermia in amphetamine-induced neuronal damage. It is generally observed that the neurotoxicity caused by METH and MDMA is accentuated by elevated ambient or core temperatures and prevented by low ambient temperature or by treatments that cause hypothermia (Yamamoto et al. 2010, Green et al. 2004). Nonetheless, it seems clear that this relation does not hold in the present experiments. Increases in 5HT with 5HTP injections do not change either METH-induced hyperthermia or neurotoxicity. The effects of 5HT reductions on METH toxicity are quite paradoxical. PCPA provides near-total protection against METH-induced hyperthermia but does not change its neurotoxicity. TPH2−/− mice actually show significantly enhanced hyperthermia after 3 of the 4 METH injections (1°C above core temperatures measured in wild-type mice given METH) yet toxicity within striatal DA nerve endings was not changed. It is known that genetically modified mice lacking 5HT neurons (Hodges et al. 2008) or TPH2 (Alenina et al. 2009) have difficulty maintaining core temperature control and show rapid losses of body heat within 1–2 hours of a cold challenge. The present work suggests further that mice lacking brain 5HT also show exaggerated responses to at least drug-induced hyperthermic challenges.
While development and expansion of our TPH2−/− colony was just underway, several other groups have published accounts of a similar mutant mouse line. Gutknecht et al. (Gutknecht et al. 2008) published a paper on a TPH2 knockout mouse that offered limited phenotype characterization. Savelieva and colleagues created knockout lines for TPH1, TPH2 and a double TPH1/TPH2 knockout, and these mice exhibited a relatively mild phenotype (Savelieva et al. 2008). More recently, Bader and colleagues described a TPH2 null mutant with more significant autonomic dysfunction and growth deficiencies (Alenina et al. 2009). Finally, Yadav et al. reported that brain derived 5HT was important in leptin regulation of bone mass and energy expenditure through the use of TPH2−/− mice (Yadav et al. 2009). The fact that at least 5 independent groups are working with mice lacking TPH2 attests to the high level of interest and value in studying the molecular and neurobiological roles played by brain 5HT. Removal of the TPH2 gene generally results in > 95% reduction in brain 5HT content with varying effects on behavior and survival (Alenina et al. 2009, Yadav et al. 2009, Savelieva et al. 2008). Our TPH2−/− mice were totally devoid of 5HT and tryptophan hydroxylating capability and their overall phenotype agrees most with the work presented by Alenina et al. (Alenina et al. 2009), particularly with regard to survival and growth retardation. In addition, the TPH2−/− mice used presently and in other papers appear to have a fully intact 5HT neuronal system. This sets the TPH2−/− mouse apart from mice with null mutations in the genes for PET1 (Hendricks et al. 2003), Lmx1b and Nkx2.2 (Cheng et al. 2003), all of which show severe reductions in CNS 5HT content because of a near-complete absence of central 5HT neurons. Work with the TPH2−/−mouse has revealed some very interesting results and the present approach represents a new extension of this model organism into the study of psychostimulant and neurotoxic drugs of abuse.
Supplementary Material
Acknowledgments
This work was supported by NIH grants (DA020680 (DMT), DA010756 (DMK), DA017327 (DMK) and by the Department of Veteran’s Affairs (DMT, DMK). We are indebted to Dr. Roxanne A. Vaughn for providing DAT antibodies.
Abbreviations used
- 5HT
serotonin
- 5HTP
5-hydroxytryptophan
- 5HIAA
5-hydroxyindole acetic acid
- AUC
area under the curve
- DA
dopamine
- DAT
dopamine transporter
- HRP
horseradish peroxidase
- METH
methamphetamine
- NPT-2
neomycin phosphotransferase-2
- NE
norepinephrine
- PBS
phosphate-buffered saline
- PCPA
para-chlorophenylalanine
- ROS
reactive oxygen species
- TH
tyrosine hydroxylase
- TPH2
tryptophan hydroxylase 2
- SERT
serotonin transporter
References
- Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective agents. J Pharmacol Exp Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- Alenina N, Kikic D, Todiras M, et al. Growth retardation and altered autonomic control in mice lacking brain serotonin. Proc Natl Acad Sci U S A. 2009;106:10332–10337. doi: 10.1073/pnas.0810793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger UV, Gu XF, Azmitia EC. The substituted amphetamines 3,4-methylenedioxymethamphetamine, methamphetamine, p-chloroamphetamine and fenfluramine induce 5-hydroxytryptamine release via a common mechanism blocked by fluoxetine and cocaine. Eur J Pharmacol. 1992;215:153–160. doi: 10.1016/0014-2999(92)90023-w. [DOI] [PubMed] [Google Scholar]
- Bowyer JF, Robinson B, Ali S, Schmued LC. Neurotoxic-related changes in tyrosine hydroxylase, microglia, myelin, and the blood-brain barrier in the caudate-putamen from acute methamphetamine exposure. Synapse. 2008;62:193–204. doi: 10.1002/syn.20478. [DOI] [PubMed] [Google Scholar]
- Breard M, Grillon C. Serotonin binds to purified neuronal nitric oxide synthase: a possible explanation for ROS production induced by 5HT in the presence of nNOS. Free Radic Res. 2009;43:206–213. doi: 10.1080/10715760802676662. [DOI] [PubMed] [Google Scholar]
- Breard M, Sari MA, Frapart Y, Boucher JL, Ducrocq C, Grillon C. The endogenous neurotransmitter, serotonin, modifies neuronal nitric oxide synthase activities. Free Radic Res. 2007;41:413–423. doi: 10.1080/10715760601105681. [DOI] [PubMed] [Google Scholar]
- Brown JM, Quinton MS, Yamamoto BK. Methamphetamine-induced inhibition of mitochondrial complex II: roles of glutamate and peroxynitrite. J Neurochem. 2005;95:429–436. doi: 10.1111/j.1471-4159.2005.03379.x. [DOI] [PubMed] [Google Scholar]
- Burrows KB, Gudelsky G, Yamamoto BK. Rapid and transient inhibition of mitochondrial function following methamphe tamineor 3, 4-methylenedioxymethamphetamine administration. Eur J Pharmacol. 2000;398:11–18. doi: 10.1016/s0014-2999(00)00264-8. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Jayanthi S, Deng X. Speed kills: cellular and molecular bases of methamphetamine-induced nerve terminal degeneration and neuronal apoptosis. FASEB J. 2003;17:1775–1788. doi: 10.1096/fj.03-0073rev. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Krasnova IN, Jayanthi S, Lyles J. Neurotoxicity of substituted amphetamines: molecular and cellular mechanisms. Neurotox Res. 2007;11:183–202. doi: 10.1007/BF03033567. [DOI] [PubMed] [Google Scholar]
- Cass WA. Attenuation and recovery of evoked overflow of striatal serotonin in rats treated with neurotoxic doses of methamphetamine. J Neurochem. 2000;74:1079–1085. doi: 10.1046/j.1471-4159.2000.0741079.x. [DOI] [PubMed] [Google Scholar]
- Cheng L, Chen CL, Luo P, Tan M, Qiu M, Johnson R, Ma Q. Lmx1b, Pet-1, and Nkx2.2 coordinately specify serotonergic neurotransmitter phenotype. J Neurosci. 2003;23:9961–9967. doi: 10.1523/JNEUROSCI.23-31-09961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailly E, Chenu F, Petit-Demouliere B, Bourin M. Specificity and efficacy of noradrenaline, serotonin depletion in discrete brain areas of Swiss mice by neurotoxins. J Neurosci Methods. 2006;150:111–115. doi: 10.1016/j.jneumeth.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Davidson C, Gow AJ, Lee TH, Ellinwood EH. Methamphetamine neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res Brain Res Rev. 2001;36:1–22. doi: 10.1016/s0165-0173(01)00054-6. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Beyeler ML, Jackson JC, Wilkins DG, Gibb JW, Hanson GR. Methamphetamine-induced decrease in tryptophan hydroxylase activity: role of 5-hydroxytryptaminergic transporters. Eur J Pharmacol. 1997a;324:179–186. doi: 10.1016/s0014-2999(97)00081-2. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Gibb JW, Hanson GR. Differential effects of stimulants on monoaminergic transporters: pharmacological consequences and implications for neurotoxicity. Eur J Pharmacol. 2000;406:1–13. doi: 10.1016/s0014-2999(00)00639-7. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681–698. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Wilkins DG, Gibb JW, Hanson GR. Interaction between hyperthermia and oxygen radical formation in the 5- hydroxytryptaminergic response to a single methamphetamine administration. J Pharmacol Exp Ther. 1997b;283:281–285. [PubMed] [Google Scholar]
- Fornai F, Bassi L, Torracca MT, Scalori V, Corsini GU. Norepinephrine loss exacerbates methamphetamine-induced striatal dopamine depletion in mice. Eur J Pharmacol. 1995;283:99–102. doi: 10.1016/0014-2999(95)00313-a. [DOI] [PubMed] [Google Scholar]
- Fornai F, Giorgi FS, Alessandri MG, Giusiani M, Corsini GU. Effects of pretreatment with N-(2-chloroethyl)-N-ethyl-2- bromobenzylamine (DSP-4) on methamphetamine pharmacokinetics and striatal dopamine losses. J Neurochem. 1999;72:777–784. doi: 10.1046/j.1471-4159.1999.0720777.x. [DOI] [PubMed] [Google Scholar]
- Fornai F, Torracca MT, Bassi L, D'Errigo DA, Scalori V, Corsini GU. Norepinephrine loss selectively enhances chronic nigrostriatal dopamine depletion in mice and rats. Brain Res. 1996;735:349–353. doi: 10.1016/0006-8993(96)00891-8. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J Neurosci. 1999;19:2424–2431. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb JW, Johnson M, Hanson GR. Neurochemical basis of neurotoxicity. Neurotoxicology. 1990;11:317–321. [PubMed] [Google Scholar]
- Goni-Allo B, Ramos M, Herv'as I, Lasheras B, Aguirre N. Studies on striatal neurotoxicity caused by the 3,4-methylenedioxymethamphetamine/ malonate combination: implications for serotonin/dopamine interactions. J Psychopharmacol. 2006;20:245–256. doi: 10.1177/0269881106063264. [DOI] [PubMed] [Google Scholar]
- Green AR, O'Shea E, Colado MI. A review of the mechanisms involved in the acute MDMA (ecstasy)-induced hyperthermic response. Eur J Pharmacol. 2004;500:3–13. doi: 10.1016/j.ejphar.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Guillot TS, Shepherd KR, Richardson JR, Wang MZ, Li Y, Emson PC, Miller GW. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J Neurochem. 2008;106:2205–2217. doi: 10.1111/j.1471-4159.2008.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutknecht L, Waider J, Kraft S, Kriegebaum C, Holtmann B, Reif A, Schmitt A, Lesch KP. Deficiency of brain 5-HT synthesis but serotonergic neuron formation in Tph2 knockout mice. J Neural Transm. 2008;115:1127–1132. doi: 10.1007/s00702-008-0096-6. [DOI] [PubMed] [Google Scholar]
- Hanson GR, Gibb JW, Metzger RR, Kokoshka JM, Fleckenstein AE. Methamphetamine-induced rapid and reversible reduction in the activities of tryptophan hydroxylase and dopamine transporters: oxidative consequences? Ann N Y Acad Sci. 1998;844:103–107. [PubMed] [Google Scholar]
- Hendricks TJ, Fyodorov DV, Wegman LJ, et al. Pet-1 ETS gene plays a critical role in 5-HT neuron development and is required for normal anxiety-like and aggressive behavior. Neuron. 2003;37:233–247. doi: 10.1016/s0896-6273(02)01167-4. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O'Banion MK, Weinberg G, Klockgether T, Feinstein DL. Noradrenergic depletion potentiates beta -amyloid-induced cortical inflammation: implications for Alzheimer's disease. J Neurosci. 2002;22:2434–2442. doi: 10.1523/JNEUROSCI.22-07-02434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Gavrilyuk V, Landreth GE, O'Banion MK, Weinberg G, Feinstein DL. Noradrenergic depletion increases inflammatory responses in brain: effects on IkappaB and HSP70 expression. J Neurochem. 2003;85:387–398. doi: 10.1046/j.1471-4159.2003.01694.x. [DOI] [PubMed] [Google Scholar]
- Hodges MR, Tattersall GJ, Harris MB, McEvoy SD, Richerson DN, Deneris ES, Johnson RL, Chen ZF, Richerson GB. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci. 2008;28:2495–2505. doi: 10.1523/JNEUROSCI.4729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M, Hanson GR, Gibb JW. Effects of dopaminergic and serotonergic receptor blockade on neurochemical changes induced by acute administration of methamphetamine and 3,4-methylenedioxymethamphetamine. Neuropharmacology. 1988;27:1089–1096. doi: 10.1016/0028-3908(88)90002-0. [DOI] [PubMed] [Google Scholar]
- Johnson M, Sonsalla PK, Letter AA, Hanson GR, Gibb JW. Role of the 5-HT2 receptor in the methamphetamine-induced neurochemical alterations. J Pharmacol Exp Ther. 1994;270:97–103. [PubMed] [Google Scholar]
- Kalinin S, Polak PE, Madrigal JL, Gavrilyuk V, Sharp A, Chauhan N, Marien M, Colpaert F, Feinstein DL. Beta-amyloid-dependent expression of NOS2 in neurons: prevention by an alpha2-adrenergic antagonist. Antioxid Redox Signal. 2006;8:873–883. doi: 10.1089/ars.2006.8.873. [DOI] [PubMed] [Google Scholar]
- Koe BK, Weissman A. p-Chlorophenylalanine: a specific depletor of brain serotonin. J Pharmacol Exp Ther. 1966;154:499–516. [PubMed] [Google Scholar]
- Krasnova IN, Cadet JL. Methamphetamine toxicity and messengers of death. Brain Res Rev. 2009;60:379–407. doi: 10.1016/j.brainresrev.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS, Cho AK, Melega W. Hippocampus norepinephrine, caudate dopamine and serotonin, and behavioral responses to the stereoisomers of amphetamine and methamphetamine. J Neurosci. 1995;15:1308–1317. doi: 10.1523/JNEUROSCI.15-02-01308.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn DM, Billingsley ML. Tyrosine hydroxylase: Purification from PC -12 cells, characterization, and production of antibodies. Neurochem Int. 1987;11:463–475. doi: 10.1016/0197-0186(87)90036-2. [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Francescutti-Verbeem DM, Thomas DM. Dopamine quinones activate microglia and induce a neurotoxic gene expression profile: relationship to methamphetamine-induced nerve ending damage. Ann N Y Acad Sci. 2006;1074:31–41. doi: 10.1196/annals.1369.003. [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Sakowski SA, Geddes TJ, Wilkerson C, Haycock JW. Phosphorylation and activation of tryptophan hydroxylase 2: identification of serine-19 as the substrate site for calcium, calmodulin-dependent protein kinase II. J Neurochem. 2007;103:1567–1573. doi: 10.1111/j.1471-4159.2007.04855.x. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Card JP, Hastings TG. Microglial activation precedes dopamine terminal pathology in methamphetamine-induced neurotoxicity. Exp Neurol. 2004;187:47–57. doi: 10.1016/j.expneurol.2004.01.010. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li IH, Huang WS, Shiue CY, et al. Study on the neuroprotective effect of fluoxetine against MDMA-induced neurotoxicity on the serotonin transporter in rat brain using micro-PET. Neuroimage. 2010;49:1259–1270. doi: 10.1016/j.neuroimage.2009.07.072. [DOI] [PubMed] [Google Scholar]
- Matuszewich L, Yamamoto BK. Effects of chronic stress on methamphetamine-induced dopamine depletions in the striatum. Ann N Y Acad Sci. 2004;1032:312–314. doi: 10.1196/annals.1314.049. [DOI] [PubMed] [Google Scholar]
- Michelsen KA, Schmitz C, Steinbusch HW. The dorsal raphe nucleus--from silver stainings to a role in depression. Brain Res Rev. 2007;55:329–342. doi: 10.1016/j.brainresrev.2007.01.002. [DOI] [PubMed] [Google Scholar]
- O'Shea E, Esteban B, Camarero J, Green AR, Colado MI. Effect of GBR 12909 and fluoxetine on the acute and long term changes induced by MDMA ('ecstasy') on the 5-HT and dopamine concentrations in mouse brain. Neuropharmacology. 2001;40:65–74. doi: 10.1016/s0028-3908(00)00106-4. [DOI] [PubMed] [Google Scholar]
- Rau KS, Birdsall E, Volz TJ, et al. Methamphetamine administration reduces hippocampal vesicular monoamine transporter-2 uptake. J Pharmacol Exp Ther. 2006;318:676–682. doi: 10.1124/jpet.105.099200. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Fuller RW, Perry KW, Seiden LS, Schuster CR. Fluoxetine increases long-lasting neostriatal dopamine depletion after administration of d-methamphetamine and d-amphetamine. Neuropharmacology. 1983;22:1165–1169. doi: 10.1016/0028-3908(83)90075-8. [DOI] [PubMed] [Google Scholar]
- Ross RA, Reis DJ. Effects of lesions of locus coeruleus on regional distribution of dopamine-beta-hydroxylase activity in rat brain. Brain Res. 1974;73:161–166. doi: 10.1016/0006-8993(74)91016-6. [DOI] [PubMed] [Google Scholar]
- Sanchez V, Camarero J, Esteban B, Peter MJ, Green AR, Colado MI. The mechanisms involved in the long-lasting neuroprotective effect of fluoxetine against MDMA ('ecstasy')-induced degeneration of 5-HT nerve endings in rat brain. Br J Pharmacol. 2001;134:46–57. doi: 10.1038/sj.bjp.0704230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savelieva KV, Zhao S, Pogorelov VM, Rajan I, Yang Q, Cullinan E, Lanthorn TH. Genetic disruption of both tryptophan hydroxylase genes dramatically reduces serotonin and affects behavior in models sensitive to antidepressants. PLoS ONE. 2008;3:e3301. doi: 10.1371/journal.pone.0003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 1985;233:539–544. [PubMed] [Google Scholar]
- Schmidt CJ, Taylor VL. Depression of rat brain tryptophan hydroxylase activity following the acute administration of methylenedioxymethamphetamine. Biochem Pharmacol. 1987;36:4095–4102. doi: 10.1016/0006-2952(87)90566-1. [DOI] [PubMed] [Google Scholar]
- Steinbusch HW. Distribution of serotonin-immunoreactivity in the central nervous system of the rat-cell bodies and terminals. Neuroscience. 1981;6:557–618. doi: 10.1016/0306-4522(81)90146-9. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Dowgiert J, Geddes TJ, Francescutti-Verbeem D, Liu X, Kuhn DM. Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett. 2004a;367:349–354. doi: 10.1016/j.neulet.2004.06.065. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. The newly synthesized pool of dopamine determines the severity of methamphetamine-induced neurotoxicity. J Neurochem. 2008;105:605–616. doi: 10.1111/j.1471-4159.2007.05155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. Increases in cytoplasmic dopamine compromise the normal resistance of the nucleus accumbens to methamphetamine neurotoxicity. J Neurochem. 2009;109:1745–1755. doi: 10.1111/j.1471-4159.2009.06094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther. 2004b;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- Tian C, Murrin LC, Zheng JC. Mitochondrial fragmentation is involved in methamphetamine-induced cell death in rat hippocampal neural progenitor cells. PLoS One. 2009;4:e5546. doi: 10.1371/journal.pone.0005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz TJ, Fleckenstein AE, Hanson GR. Methamphetamine-induced alterations in monoamine transport: implications for neurotoxicity, neuroprotection and treatment. Addiction. 2007;102(Suppl 1):44–48. doi: 10.1111/j.1360-0443.2007.01771.x. [DOI] [PubMed] [Google Scholar]
- Wagner GC, Lucot JB, Schuster CR, Seiden LS. Alpha-methyltyrosine attenuates and reserpine increases methamphetamine- induced neuronal changes. Brain Res. 1983;270:285–288. doi: 10.1016/0006-8993(83)90602-9. [DOI] [PubMed] [Google Scholar]
- Wall SC, Gu H, Rudnick G. Biogenic amine flux mediated by cloned transporters stably expressed in cultured cell lines: amphetamine specificity for inhibition and efflux. Mol Pharmacol. 1995;47:544–550. [PubMed] [Google Scholar]
- Weinshenker D, Ferrucci M, Busceti CL, et al. Genetic or pharmacological blockade of noradrenaline synthesis enhances the neurochemical, behavioral, and neurotoxic effects of methamphetamine. J Neurochem. 2008;105:471–483. doi: 10.1111/j.1471-4159.2007.05145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf WA, Ziaja E, Arthur RA, Jr, Anastasiadis PZ, Levine RA, Kuhn DM. Effect of tetrahydrobiopterin on serotonin synthesis, release, and metabolism in superfused hippocampal slices. J Neurochem. 1991;57:1191–1197. doi: 10.1111/j.1471-4159.1991.tb08279.x. [DOI] [PubMed] [Google Scholar]
- Wrona MZ, Dryhurst G. Further insights into the oxidation chemistry of 5-hydroxytryptamine. J Pharm Sci. 1988;77:911–917. doi: 10.1002/jps.2600771102. [DOI] [PubMed] [Google Scholar]
- Wrona MZ, Dryhurst G. Oxidation of serotonin by superoxide radical: implications to neurodegenerative brain disorders. Chem Res Toxicol. 1998;11:639–650. doi: 10.1021/tx970185w. [DOI] [PubMed] [Google Scholar]
- Wrona MZ, Dryhurst G. A putative metabolite of serotonin, tryptamine-4,5-dione, is an irreversible inhibitor of tryptophan hydroxylase: possible relevance to the serotonergic neurotoxicity of methamphetamine. Chem Res Toxicol. 2001;14:1184–1192. doi: 10.1021/tx010037c. [DOI] [PubMed] [Google Scholar]
- Wrona MZ, Lemordant D, Lin L, Blank CL, Dryhurst G. Oxidation of 5-hydroxytryptamine and 5,7-dihydroxytryptamine. A new oxidation pathway and formation of a novel neurotoxin. J Med Chem. 1986;29:499–505. doi: 10.1021/jm00154a013. [DOI] [PubMed] [Google Scholar]
- Wrona MZ, Waskiewicz J, Han QP, Han J, Li H, Dryhurst G. Putative oxidative metabolites of 1-methyl-6-hydroxy-1,2,3,4-tetrahydro-beta-carboline of potential relevance to the addictive and neurodegenerative consequences of ethanol abuse. Alcohol. 1997a;14:213–223. doi: 10.1016/s0741-8329(96)00144-9. [DOI] [PubMed] [Google Scholar]
- Wrona MZ, Yang Z, McAdams M, O'Connor-Coates S, Dryhurst G. Hydroxyl radical-mediated oxidation of serotonin: potential insights into the neurotoxicity of methamphetamine. J Neurochem. 1995;64:1390–1400. doi: 10.1046/j.1471-4159.1995.64031390.x. [DOI] [PubMed] [Google Scholar]
- Wrona MZ, Yang Z, Zhang F, Dryhurst G. Potential new insights into the molecular mechanisms of methamphetamine-induced neurodegeneration. NIDA Res Monogr. 1997b;173:146–174. [PubMed] [Google Scholar]
- Yadav VK, Oury F, Suda N, et al. A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell. 2009;138:976–989. doi: 10.1016/j.cell.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto BK, Bankson MG. Amphetamine neurotoxicity: cause and consequence of oxidative stress. Crit Rev Neurobiol. 2005;17:87–117. doi: 10.1615/critrevneurobiol.v17.i2.30. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Moszczynska A, Gudelsky GA. Amphetamine toxicities: classical and emerging mechanisms. Ann N Y Acad Sci. 2010;1187:101–121. doi: 10.1111/j.1749-6632.2009.05141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wrona MZ, Dryhurst G. 5-hydroxy-3-ethylamino-2-oxindole is not formed in rat brain following a neurotoxic dose of methamphetamine: evidence that methamphetamine does not induce the hydroxyl radical-mediated oxidation of serotonin. J Neurochem. 1997;68:1929–1941. doi: 10.1046/j.1471-4159.1997.68051929.x. [DOI] [PubMed] [Google Scholar]
- Yuan J, Callahan BT, McCann UD, Ricaurte GA. Evidence against an essential role of endogenous brain dopamine in methamphetamine-induced dopaminergic neurotoxicity. J Neurochem. 2001;77:1338–1347. doi: 10.1046/j.1471-4159.2001.00339.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.