

Abstract
The partial unfolding of human lysozyme underlies its conversion from the soluble state into amyloid fibrils observed in a fatal hereditary form of systemic amyloidosis. To understand the molecular origins of the disease, it is critical to characterize the structural and physicochemical properties of the amyloidogenic states of the protein. Here we provide a high-resolution view of the unfolding process at low pH for three different lysozyme variants, the wild-type protein and the mutants I56T and I59T, which show variable stabilities and propensities to aggregate in vitro. Using a range of biophysical techniques that includes differential scanning calorimetry and nuclear magnetic resonance spectroscopy, we demonstrate that thermal unfolding under amyloidogenic solution conditions involves a cooperative loss of native tertiary structure, followed by progressive unfolding of a compact, molten globule-like denatured state ensemble as the temperature is increased. The width of the temperature window over which the denatured ensemble progressively unfolds correlates with the relative amyloidogenicity and stability of these variants, and the region of lysozyme that unfolds first maps to that which forms the core of the amyloid fibrils formed under similar conditions. Together, these results present a coherent picture at atomic resolution of the initial events underlying amyloid formation by a globular protein.
Introduction
Many debilitating and often fatal disorders, including Alzheimer’s and Parkinson’s diseases, the transmissible spongiform encephalopathies, type II diabetes, and a range of systemic amyloidoses, are associated with the deposition of normally soluble proteins as insoluble aggregates in various types of tissue.(1) Although the sequences and native structures of the proteins involved in the different conditions vary greatly, the aggregates found in patients exhibit very similar biophysical properties. These include their fibrillar nature and cross-β structure, where β-strands are oriented perpendicularly to the axis of the fibril.(1) In many cases the formation of amyloid fibrils requires the monomeric precursor protein to undergo a series of substantial conformational rearrangements. A detailed characterization of the different species involved in these conformational transitions is crucial for developing an understanding of the mechanism behind misfolding diseases.2−6 Such species are also key therapeutic targets for developing drugs to treat or prevent amyloid-related diseases.7,8 Partially folded states, denatured states, disordered protein ensembles, and molten globules are possible precursors to amyloid fibrils or other aggregates and have therefore been the object of extensive research.9−16
Human lysozyme is a small, well-characterized globular protein with a native structure that can be divided into two domains: the α domain (residues 1 to 38, and 86 to 130) and the β domain (residues 39 to 85) that primarily contain α-helical and β-sheet secondary structure, respectively(18) (see Figure 1a,b). In 1993, Pepys and co-workers reported that two variants of human lysozyme are responsible for a hereditary non-neuropathic systemic amyloidosis.(20) Given the significant body of data on the structure, dynamics, and folding of both hen and human lysozymes, the link to human disease renders this protein an ideal system to probe the relationships between protein folding and aggregation. The first two single-point mutations found to be linked to lysozyme amyloidosis were I56T and D67H, and other amyloidogenic and non-amyloidogenic mutations have also been identified more recently (for a review, see Dumoulin et al.(21)). The observation that only certain mutational variants form fibrils in vivo raises the question of which factors regulate the amyloidogenicity of this protein. A comparative thermal denaturation study has shown that the two amyloidogenic variants I56T and D67H are less stable than the wild-type protein (WT)1 , suggesting that a destabilization of the native state is, at least in part, responsible for the process of fibril formation. Indeed at pH 5.0 the midpoints of thermal unfolding of the I56T and D67H variants were reported to be 12 ± 2 °C lower than that of the WT protein.22−25 However, the naturally occurring and non-amyloidogenic T70N variant also shows a decrease in the unfolding temperature in vitro of approximately 4 °C relative to the WT but is not observed to form detectable quantities of fibrils or to give rise to disease in individuals carrying this mutation.24,26 In addition, under in vitro conditions, T70N does not form fibrils at rates comparable to the amyloidogenic variants. It therefore appears that the disease-associated mutational variants need to be destabilized enough to allow detectable amyloid fibril formation to take place under physiological conditions but not so destabilized as to trigger clearance by the cell.
Figure 1.

Structure of human lysozyme indicating the location of destabilizing mutations. (a) Representation of the secondary structure of the native state of WT human lysozyme, indicating glycine residues (using DSSP(17) on pdb entry 1LZ1(18)). Strands are represented by arrows, α-helices (A to D) by large boxes, and 310 helices (h1 to h3) by small boxes. The disulfide bridges C6−C128, C30−C116, C65−C81, and C77−C95 are shown in purple. The α and β domains are shown in blue and yellow, respectively. (b) Tertiary structure of WT human lysozyme with the same color coding as in panel a. All structures are drawn using vmd(19) with the secondary structure defined by DSSP.(17) Vmd only represents strands comprising three residues and more; therefore, strand 3 (residues 59−60) is not shown. The colored spheres represent residues 56 (blue) and 59 (red), sites of the mutations I56T and I59T studied herein. (c) Normalized signal of the near-UV circular dichroism thermal unfolding traces obtained for the WT (black), I59T (red) and I56T (blue) variants, at pH 1.2.
Hydrogen/deuterium (H/D) exchange experiments on the amyloidogenic variants I56T and D67H have revealed the presence of a partially folded intermediate state that is transiently populated under native conditions and characterized by the unfolding of the β-domain and one of the helices (helix C of the α-domain).22,27,28 In addition, thermal unfolding experiments carried out in vitro under equilibrium conditions in the presence of a fluorescent dye sensitive to hydrophobic patches (1-anilinonaphthalene-8-sulfonic acid or ‘ANS’) indicate the presence of partially unfolded species near to the midpoint of thermal denaturation.22,23,28 On the basis of these results, a reduction in the global cooperativity of the variants of lysozyme found in disease has emerged as a key determinant for their amyloidogenicity: the non-cooperative unfolding behavior leads to the formation of partially folded species that are able to associate to form highly organized fibrils, the deposition of which is characteristic of amyloid diseases. Despite the importance of partially folded states in this mechanism, their transient nature and low populations at equilibrium under solution conditions commonly used in in vitro studies (pH > 5) have precluded a direct characterization of these species at high resolution.
In this paper, we use low pH conditions to provide such high-resolution structural evidence on the nature of the equilibrium thermal unfolding behavior of human lysozyme. Using NMR spectroscopy in combination with a wide range of biophysical techniques, we studied three variants of human lysozyme that have variable stabilities and propensities to aggregate in vitro, namely the WT protein and the I56T and I59T mutants (see Figure 1).(29) As stated above, the I56T variant is linked to familial lysozyme systemic amyloidosis, and the I59T mutant has been designed to have properties that are intermediate between those of I56T and the WT lysozymes.20,25 Our results reveal that under the acidic destabilizing conditions commonly used to form amyloid fibrils in vitro, human lysozyme is in a dynamic equilibrium between its native conformation and a broad ensemble of non-native conformations: the members of this non-native ‘denatured’ ensemble contain both molten globular and unfolded regions. Molten globular regions, in which secondary structure elements are present but tertiary interactions are disorganized to varying degrees, are predominant at lower temperatures and unfold gradually, and with local cooperativity, as they become increasingly disordered at higher temperatures.
Results
Amyloidogenic Species Are Populated Near the Denaturation Temperatures
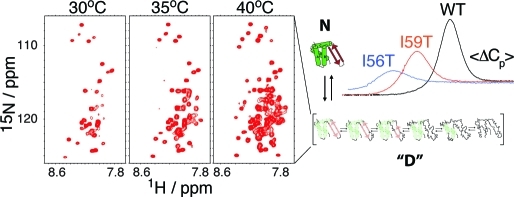
The strategy used in this study exploits low pH conditions to destabilize the native state and to observe partially unfolded states of lysozyme under equilibrium conditions.(30) Lowering the pH from 5.0 to 1.2 results in a substantial decrease in the stability of the native state of all three variants, thereby enabling a comparison of the progressive denaturation of the WT protein and the I59T and I56T variants at temperatures below 50 °C (see Figure 1c). 1H−15N backbone HSQC spectra recorded at 20 °C are typical of a folded protein, confirming that the native structure is preserved at the lower temperatures in all three variants at pH 1.2 (see Figure 2). The temperature at which the tertiary structure is lost differs significantly between the three variants, showing that the I56T and I59T single-point mutations have a large and differential effect on the stability of the native state: 48.5 ± 0.3 °C for WT, 35.4 ± 0.5 °C for I59T, and 22.6 ± 0.3 °C for I56T lysozyme (see Figure 1c, and Table S1 in the Supporting Information). The proteins are stable for days at this pH at low temperature (<35 °C), and for several hours at the higher temperatures, enabling detailed NMR analysis to be carried out on the chemically intact protein, and allowing the equilibrium unfolding of all three variants to be compared in detail without the addition of denaturant.
Figure 2.

1H−15N HSQC spectra of I59T at pH 1.2 recorded at different temperatures. At 20 °C, the protein is fully folded (a), whereas at 50 °C, the protein is fully unfolded (c). Near the midpoint of the unfolding transition, at 35 °C, the protein shows resonances typical of both folded and unfolded states (b). The region boxed in panel (c) indicates the peaks corresponding to glycine residues in the unfolded state.
Incubation of 1 mM solutions of WT lysozyme at temperatures near the midpoint of unfolding (∼47.5 °C) and with stirring leads to the essentially complete conversion of the soluble forms of the protein into amyloid fibrils within 4−5 days. In order to accelerate the aggregation process and avoid prolonged incubation times at elevated temperatures and low pH, which can cause hydrolysis of the protein,31,32 aliquots (2% v/v) of preformed fibril seeds were added to the solutions, resulting in more than 95% of the monomeric protein becoming incorporated in fibrils within one day of incubation. The fibrils obtained contain non-hydrolyzed, full length monomeric protein as confirmed by SDS-PAGE electrophoresis and have all the morphologic characteristics of amyloid structures: transmission electron microscopy images show that they are long and unbranched, they bind thioflavin-T, an amyloid-specific dye, and they present a high degree of β-sheet structure, as indicated by CD (see Figure S1 in the Supporting Information). The observation that amyloidogenic precursors are populated near the temperatures where tertiary structure is lost prompted us to study the nature and relative abundance of such species.
Cooperative Denaturation Is Followed by Non-cooperative Progressive Unfolding
Monitoring the unfolding transition of all three lysozyme variants by means of circular dichroism spectroscopy (CD) at different wavelengths reveals that the tertiary structure (near-UV region, 270 nm) is disrupted more readily, i.e. at lower temperatures, than the secondary structure (far-UV region, 222 nm) (see Figure 3a and Table S1, Figure S3, and Figure S4a in the Supporting Information). The difference in melting temperatures detected by near- and far-UV CD increases as the stability of the native state decreases; hence, the temperature difference between the two midpoints is largest for I56T and smallest for the WT protein. These data show that a simple two-state model fails to describe the complexity of the unfolding process. In addition to the native and unfolded states, at least one more state exists with a secondary structure content similar to that of the native state but with a very significant loss of tertiary structure. Interestingly, the thermal unfolding of the secondary structure is essentially identical for the I56T and I59T variants, whereas the tertiary structure is more strongly destabilized for the I56T than for the I59T variant (see Figure S3 in the Supporting Information).
Figure 3.

Thermal unfolding of human lysozyme monitored using CD spectroscopy and DSC: a pseudo-two-state model. (a) Equilibrium thermal unfolding of WT, I59T, and I56T lysozyme, followed by near-UV (270 nm, red) and far-UV (222 nm, black) CD at pH 1.2. Thermal melts were fitted to a two-state model (fits in solid lines). For more details, see Experimental Section and the Supporting Information. (b) Evolution of excess heat capacity with temperature shown for WT (black), I59T (red), and I56T (blue) at pH 1.2. Solid lines represent the experimental data, while dashed lines show curves simulated using the thermodynamic parameters obtained from the fitting the CD data to a three-state model (see the Supporting Information). The reversibility of the thermal denaturation monitored by DSC is shown in Figure S2 of the Supporting Information. (c) Proposed model: scheme of the native state N and the denatured ensemble “D”. The populations of the native state (dashed blue lines) and denatured ensemble “D” (dashed green lines) are derived by fitting the near-UV CD first-order transition to a pseudo-two-state model.
A three-state model defining native, intermediate, and unfolded states accounts essentially perfectly for the far- and near-UV CD data,(30) and allows the apparent population of each state to be determined for all three variants (see Figure S4a,b in the Supporting Information). In addition, knowledge of the populations of the three states enabled us to deconvolute the far- and near-UV CD data to extract separate spectra of the native, intermediate, and unfolded states (see Figure S4c in the Supporting Information). As expected, the intermediate lacks a well-defined tertiary structure, as indicated by the loss of the native near-UV CD signal, yet preserves an essentially native-like secondary structure signature in the far-UV CD region, indicative of molten globular character (see Figure S4c in the Supporting Information). The population of the intermediate state and the temperature window where it is significantly populated both increase as the stability of the native state decreases.
In order to investigate further the nature of the unfolding transition, we performed differential scanning calorimetry (DSC) experiments on all three variants. DSC allows the identification of merged, multiple transitions that may not be well-resolved by spectroscopic techniques, provided that these transitions induce a detectable change in both heat capacity and enthalpy.(33) In the present case, the unique heat absorption peak observed by DSC is characteristic of a two-state transition, where the temperature midpoint of the unfolding transition is identical within error to that seen by near-UV CD (see Figure 3a,b, Experimental Section, and Supporting Information Table S1); that is, the observed transition corresponds to the loss of tertiary structure. Furthermore, simulated DSC thermograms, based on the thermodynamic parameters obtained from the three-state fit to the far- and near-UV CD data, show profiles that clearly differ from the experimental ones (compare plain and dashed lines in Figure 3b). We therefore conclude that only one conformational transition is detected by DSC that, according to the three-state model, corresponds to the cooperative conversion of the native state into the intermediate state. By contrast, the unfolding of the intermediate does not follow a cooperative first-order transition, but is instead best described as a continuous (second-order) ‘phase transition’ with a progressive loss of structure as the temperature is increased. Therefore, the DSC data question the validity of a three-state model invoking two first-order transitions.
The three-state model defined by the CD data is still useful in an operational sense but needs to be improved with the intermediate and unfolded states replaced by an ensemble of denatured states separated by marginal energy barriers: in this improved model the ‘denatured ensemble’ consists of a collection of non-native states possessing different degrees of unfolding. The composition of this ensemble changes with temperature, being on average more molten globule-like at low temperatures, and more unfolded as the temperature is increased (see Figure 3c). To obtain populations for the native state and the ‘denatured ensemble’, we have used the near-UV CD unfolding curve only and fitted it to this pseudo-two-state model (dashed lines in Figures 3 and 4). The model that we propose here is, therefore, a significant improvement on the three-state model used until now for human lysozyme. This situation is reminiscent of the thermal unfolding of the molten globule of the structurally homologous protein α-lactalbumin whose unfolding does not give rise to any noticeable heat absorption peak and for which a similar ‘denatured ensemble’ has been proposed.15,16
Figure 4.

NMR evidence for non-cooperative unfolding. Black dots connected by lines represent the temperature dependence of individual 1H−15N HSQC cross-peak volumes of the native state (left), normalized relative to the volume of each peak at 5 °C, and the denatured ensemble (right), normalized relative to the volume of each peak at 55 °C (see comment for WT in the Experimental Section). All non-overlapping NMR cross-peaks are shown for the decay of the native state (left), whereas the increases in cross-peak volumes of glycine residues only are shown for the denatured ensemble (right). Glycine residues have been selected because they have all been assigned in the denatured state of all three variants (see Figure 5b). Increases in cross-peak volumes for 46 assigned residues of the denatured ensemble of I56T are shown in Figure S5 in the Supporting Information and show similar results, therefore justifying the fact that only glycines are considered here. The decay of the integrated signal of selected native side chain methyl groups is shown in light blue circles in the left column (see raw data in Figure S6 in the Supporting Information). Full lines represent populations derived by simultaneously fitting far- and near-UV CD data to the three-state model: the populations of the native, intermediate, and unfolded states are indicated by the blue, red, and green lines, respectively (the population of the native state of the I56T variant is normalized to its value at 5 °C). Dashed lines show the results obtained by fitting the near-UV CD data to the pseudo-two-state model: the populations of the native state and the ‘denatured ensemble’ are shown in blue and green, respectively.
NMR Spectroscopy Provides Direct Evidence of Progressive Unfolding
To characterize the unfolding process at the level of individual residues, we recorded 1H−15N backbone HSQC spectra of all three variants over a wide range of temperatures, acquiring spectra at every 2.5 °C between 5 and 55 °C. At low temperature, the 1H−15N HSQC spectrum of each variant is characteristic of a folded protein with a large number of well resolved and dispersed cross-peaks (see Figure 2a). By studying the decrease in volume of non-overlapping backbone cross-peaks as a function of temperature, we observe that the resonances of residues distributed throughout the protein disappear in a concerted fashion, confirming that the unfolding of the native state is highly and globally cooperative (see Figure 4, left column). NMR resonances shifted upfield of 0.5 ppm in 1D NMR spectra are characteristic of buried methyl groups in folded proteins; we observe that these resonances decrease in intensity with increasing temperature similarly to those of backbone amides, showing at the level of individual residues that the secondary and tertiary structures are lost simultaneously in the unfolding of the native state, in agreement with our previous work (see light blue circles on the left column of Figure 4, Experimental Section, and raw data in Figure S6 in the Supporting Information).(30) The relative decreases in the volume of the NMR signals corresponding to the native state agree very well with the population of this state derived from near-UV CD, showing that both techniques probe the disappearance of the highly cooperative native state (see Figure 4, left column).
At higher temperatures, 1H−15N HSQC cross-peaks collapse into the central region of the 1H spectrum with little chemical shift dispersion as expected for a denatured protein (see Figure 2c). Two sets of NMR signals, corresponding to folded and denatured species, are observed at temperatures near the denaturation midpoints identified by CD (see Figure 2b). However, only a subset of the resonances from the denatured state is present in the NMR spectra recorded under these conditions, but additional resonances appear gradually as the temperature is increased further (see Figures 4, right column, and Figure 5). This behavior is exemplified by I56T (see Figure 5a): at 30 °C we observe only 26 backbone amide cross-peaks out of 130 residues. As discussed further below, this temperature dependence is similar to that observed for the low pH molten globular state of α-lactalbumin, where cross-peaks appear as the temperature is increased.(34)
Figure 5.

Unfolding of the denatured ensemble of human lysozyme monitored by NMR spectroscopy. (a) 1H−15N HSQC spectra of the I56T mutant recorded at different temperatures show the gradual appearance of the peaks from different residues. The lowest contour level was adjusted so as not to display the weak signals of the native state at 30 °C, which has a population of ca. 20% at this temperature. The lowest contour level, the number of contours as well as the increments between them are constant across all temperatures. Changing the lowest contour level displayed does not affect the results qualitatively and only slightly affects the temperature at which a peak first appears; moreover, it affects all residues in a similar way, enabling robust comparisons to be made. (b) Region of the 1H−15N HSQC spectra (boxed in panel a) containing all resonances from glycine residues in the denatured ensemble. Resonances from the locally folded and unfolded residues are shown in black and red, respectively. The temperatures of disappearance of resonances from the native state, as well as appearance of resonances from the denatured ensemble, are summarized for all three variants in Figure S7 in the Supporting Information.
The build-up of cross-peak intensity from the denatured state is markedly non-concerted, especially in the case of the I56T and I59T variants (see Figure 4, right column). Moreover, the range of temperatures over which NMR cross-peaks from the denatured state appear is closely similar to the range defined by the population profiles of denatured conformations without native tertiary interactions (dashed line) and without secondary structure (full line) determined by CD spectroscopy. This result indicates that NMR is able to distinguish between classes of residues with different unfolding behaviors, ranging from those that are fully unfolded at low temperatures where native tertiary structure is initially lost, to those that become fully unfolded only upon complete denaturation of the secondary structure elements.
Unfolding of the Denatured Ensemble Is Locally Cooperative
Residue specific assignments of the denatured state provide structural insights into the locally cooperative unfolding process (see Experimental Section). To make direct comparisons between the unfolding behavior of the different variants, we focus on a well-resolved region of the spectrum that has been completely assigned in both the folded and denatured states for all three variants. Human lysozyme has eleven glycine residues and, as they are distributed throughout the native fold, these residues act as very valuable probes of the structural properties of different regions of sequence (see Figure 1). Glycine resonances cluster in an isolated and well-dispersed region of the 1H−15N HSQC spectrum of non-native lysozyme (see squared region in Figure 5a) thus enabling us to determine accurately the relative increases in cross-peak volumes of the denatured ensemble as the temperature is increased. As shown in Figure 4, the relative volumes of the glycine cross-peaks of non-native lysozyme vary significantly over a wide range of temperatures, showing that the unfolding of non-native states follows a non-cooperative process. Calculation of the expected water exchange rates of amide protons of the unfolded form of human lysozyme (http://hx2.med.upenn.edu/) reveals that the exchange process is very slow and that the magnitudes of the rates do not correlate with the temperature of the appearance of the corresponding peaks in the NMR spectrum, therefore ruling out the possibility that line broadening is due to exchange with water (see Figure S8 in the Supporting Information).(35) Expanded plots of the glycine region of the HSQC spectra show the gradual appearance of resonances from the non-native states of the protein as the temperature is increased (see Figure 5b). Importantly, the temperature range over which glycine cross-peaks of non-native states appear is observed to differ between WT, I56T, and I59T; the range increases as the stability of the native state decreases. As can be seen from Figure 5b, this range is around 5 °C for WT, 10 °C for I59T, and 27.5 °C for I56T, directly reflecting the increasing degree of global non-cooperativity in the phase transition that accompanies the thermal unfolding of the three variants of lysozyme (see Figure 4).
The partial assignment obtained for I56T enables us to identify which parts of lysozyme unfold first. Figure 6 maps the differential and gradual appearance of resonances of locally unfolded states onto the native protein structure. The results clearly show that the β-hairpin is the first structural element to unfold, followed by the rest of the β-domain, helix C, both 310 helices 1 and 2, and some residues at the chain termini. The rest of the protein, which comprises helices A, B, D and 310 helix 3, unfolds at higher temperatures, although residues located in loops between secondary structure elements tend to unfold at lower temperatures than the elements themselves. For all three variants, the glycine residues that unfold at the lowest temperatures are G48, G55, G72, G68, and G105, all of which are located in the β-domain or in 310 helix 2 (see Figure 5b) The results suggest that the unfolding process of the denatured ensemble is locally cooperative and structurally similar for all three variants, with the β-domain, helix C, both 310 helices 1 and 2, and some residues of the N- and C-termini unfolding first. However, the temperature window over which unfolding takes place, which indicates the degree of global non-cooperativity of the unfolding of the denatured ensemble, is modulated by the single-point mutations of the variants.
Figure 6.

Residue specific view of the locally cooperative unfolding of human lysozyme. (a) Structural location of individual residues assigned in the 1H−15N HSQC spectrum of denatured lysozyme. The color code indicates the temperature of the first appearance of the corresponding cross-peaks in the 1H−15N HSQC spectra of the denatured state of I56T, according to Figure 5. (b) As in panel a, but highlighting glycine residues.
Characterization of the Denatured Ensemble Using the Temperature Evolution of 1HN and 15N Chemical Shifts
The temperature evolution of chemical shifts, as provided by the temperature series of HSQCs, provides additional information on the denatured ensemble (see Figure 7a). The temperature coefficients of amide protons, defined as the slope of a linear fit of 1HN amide chemical shift versus temperature, vary across glycine residues but do not correlate with the temperature at which they appear nor with the protein sequence (see Figure 7b). Values more positive than −4.5 ppb/K have been attributed to the existence of intramolecular hydrogen bonds.36−38 This is the case for three glycines (G16, G37, and G105), located in the α-domain of the protein, whereas the remaining glycines, located in both the α- and β-domains, have values that are more negative than −4.5 ppb/K. These results suggest that part of the α-domain could possess some residual structure at high temperature. However, the interpretation of amide temperature coefficients is not straightforward, especially in the case of chemically exchanging systems like lysozyme at low pH.(39)
Figure 7.

Temperature evolution of 1HN and 15N chemical shifts of the denatured ensemble. (a) The glycine-rich region of the HSQC spectrum of the denatured ensembles of human lysozyme variants. In each case the top spectrum, in red, corresponds to a temperature of 50 °C; spectra were recorded every 2.5 °C and are all shown with the same contour levels as in Figure 5. (b) Temperature coefficients of 1HN amide chemical shifts of glycine residues of the denatured ensemble. The temperature coefficients are determined as the slope of a linear fit of chemical shifts versus temperature. Results are shown for the WT (black), the I59T (red), and the I56T (green) variants. (c) Plot of residuals of 1HN and 15N chemical shifts, after subtraction of the best first-order fit. The P-value shown in the inset of each graph relates to the comparison of fits to first- and second-order polynomial functions. Only residues for which one chemical shift (1HN or 15N) has P < 0.05 are shown.
The temperature evolution of glycine 1HN and 15N chemical shifts were also analyzed in terms of their deviation from linearity, which reports on possible conformational heterogeneity.40−42 For all three variants, we used a P-value analysis to compare the fits to first- and second-order polynomial functions: we observe that nearly all glycine residues show a linear evolution of their 1HN and 15N chemical shifts with temperature, i.e., do not undergo any important structural changes in the denatured ensemble after becoming visible in the HSQC spectrum (see residuals in Figure 7c). The exceptions are G48 in the I56T and I59T variants, and G72 in the I56T variant, where both residues are located in the β-domain of the protein: the P-values for G72 in I56T, and G48 in I59T, are significantly larger than those for G48 in I56T, showing that the extent of conformational exchange is most significant for G48 in I56T. Moreover, deviation from linearity for G48 is larger in I56T than in I59T, and non-existent in the WT protein, suggesting that the extent of conformational heterogeneity experienced by G48 within the denatured state correlates with the decrease in native state stability (a similar conclusion holds for G72 for which deviation from linearity is only observed for I56T). We should note here that the deviation from linearity is minimal and only becomes observable when performing a statistical test: therefore, although the conformational heterogeneity probed by this analysis is statistically significant, it is of rather small magnitude. We attribute this conformational heterogeneity to slight differences in chemical shift, and therefore structure, of G48 and G72 between the denatured states at lower and higher temperature and is most pronounced for G48 in the I56T variant, as this residue unfolds at lower temperatures. Overall, this analysis shows that, once resonances corresponding to the denatured ensemble appear in the NMR spectrum, their chemical shift temperature evolution is not indicative of major conformational rearrangements.
Discussion
In this report, we have provided a detailed characterization of the energy landscape of monomeric human lysozyme at low pH with the aim of providing key structural insights into the nature of the amyloidogenic species populated under equilibrium conditions. We present here a comparative study of the complete thermal unfolding transition of the three variants I56T, I59T, and WT under relatively modest temperatures and in the absence of denaturants. Using low pH conditions has allowed us to manipulate the folding energy landscape by greatly destabilizing the native state and, for the first time for human lysozyme, to observe directly non-native states that are populated in amyloidogenic conditions. The present investigation is therefore distinct from previous studies based on H/D exchange techniques that indirectly detect partially folded species that are transiently populated as a result of fluctuations from the native state.22,27,28
Using a combination of DSC, CD, and NMR spectroscopy, we show that human lysozyme unfolds in a relatively complex process that involves a cooperative transition from the native structure into a denatured ensemble that unfolds progressively with increasing temperature; the cooperative and first-order transition detected by near-UV CD and DSC corresponds essentially exclusively to the loss of tertiary structure. In the denatured ensemble, tertiary contacts are disrupted and secondary structure elements unfold independently, but in a locally cooperative manner. At lower temperatures, the properties of the denatured ensemble are characteristic of a molten globule: it does not give rise to detectable NMR resonances for most of its residues, lacks tertiary contacts as shown by near-UV CD, and retains most of the secondary structure of the native state as seen by far-UV CD. In addition, we observe that the NMR resonances characteristic of fully unfolded conformations do not appear simultaneously at a given temperature but rather become evident gradually (see Figures 4 and 5). These observations are consistent with a model in which unfolding proceeds through a continuum of states that are relatively close in energy and separated by marginal barriers, such that they interconvert relatively rapidly (see Figure 3c). In agreement with this model, the increase in cross-peak volumes of denatured resonances takes place at higher temperatures than the near-UV CD transition (green dashed lines in Figure 4). This behavior is consistent with the observation that no heat absorption peak is detected by DSC; hence, unfolding of the molten globule is not associated with any ‘latent heat’.
Our observations are in agreement with previous reports on the thermal unfolding of hen egg white lysozyme(43) as well as with numerous previous calorimetric and NMR studies of α-lactalbumins, proteins that are homologous to lysozymes;15,16,34 however, whereas α-lactalbumin populates essentially 100% of a molten-globular species at 20 °C and low pH, the energy landscape of human lysozyme at low pH is significantly more complex, as its native state is more stable and is therefore highly populated at low temperatures. Therefore, α-lactalbumins seem to behave at low pH as extremely destabilized mutants of human lysozyme, a phenomenon that can be attributed to the loss of a stabilizing Ca2+ ion in the former group of proteins at low pH.(44)
A major advance in our study has been to record NMR spectra describing the whole of the unfolding transition, yielding, in particular, a structural description of the states populated in the course of the locally cooperative unfolding of the denatured ensemble. Residue-specific information from NMR spectroscopy has enabled us to map the unfolding process of the denatured ensemble onto the native structure, thereby revealing which parts of human lysozyme unfold first and are therefore of lower conformational stability. Within the denatured state, each secondary structure element unfolds individually in a continuous transition, resulting in the formation of a wide ensemble of partially folded conformations. This unfolding process is structurally similar for all three variants studied here, with the chain termini, the β-domain, α helix C, and 310 helix 2 from the α-domain unfolding at temperatures lower than the rest of the protein (comprising α helices A, B, and D and 310 helix 3). Within the β-domain, the I56T variant shows that the β-hairpin is the first structural element to unfold. The regions of the protein that unfold first in the denatured ensemble are the ones that are transiently unfolded under native conditions at higher pH (pH 5.0, 37 °C), as detected indirectly by H/D exchange techniques: both observations are therefore in agreement and suggest that these regions are of intrinsically lower stability than the remainder of the protein structure.22,27,28 The temperature evolution of amide chemical shifts of the denatured ensemble suggests the presence of some intramolecular hydrogen bonding, and therefore a modest degree of residual structure in the α-domain. Once structural elements of the denatured ensemble unfold, however, there is little conformational rearrangement within this locally unfolded conformation, as indicated by the fact that the chemical shift temperature evolution of most glycine residues is essentially linear. Interestingly, both the β-domain and helix C have been found to form the core of human lysozyme fibrils prepared at pH 2.0.(31) As we demonstrate here that the β-domain and helix C have marginal individual stability, being the first regions of the protein to unfold once the tertiary structure is disrupted, this unfolding process is likely to be at the origin of the formation of amyloid fibrils at low pH.
The single-point mutations I59T and I56T not only affect the stability of the native state of the protein, as assessed by near-UV CD, therefore influencing the temperature at which the denatured ensemble starts to unfold, but also determine the extent of non-cooperativity in the unfolding of the denatured ensemble, as measured by the temperature window over which structural elements of the denatured ensemble progressively unfold. The more destabilized the native state, the lower the temperature at which the denatured ensemble starts to be populated, and the more pronounced its non-cooperative unfolding. Interestingly, both the native state destabilization and the extent to which cooperativity is lost within the denatured ensemble correlate with the amyloidogenicity of the variant in other solution conditions (pH 5.0), as well as with the expression levels of lysozyme in Pichia pastoris.(29) Although the results of this study do not directly report on the structural properties of the amyloidogenic state of lysozyme under physiological conditions, they strongly indicate that the propensity of human lysozyme to aggregate is closely related to the shape of the energy landscape of the monomeric protein,(25) that has been probed in the present study.
Experimental Section
The I56T and I59T variants and WT human lysozyme were expressed in Aspergillus niger (I56T) or Pichia pastoris (I59T, WT) and purified as described previously.24,45 All experiments were carried out with the protein samples in 50 mM phosphate buffer adjusted to pH 1.2 with HCl. Buffers for NMR experiments included 10% D2O.
Circular Dichroism
Circular dichroism experiments were carried out using a Jasco J-810 spectropolarimeter equipped with a Peltier holder. Spectra were recorded using cells of 0.1 and 1 cm path length for the far- and near-UV, respectively, with a protein concentration of ca. 20 μmol/L. Heat-induced unfolding transitions were monitored from 5 to 90 °C at a rate of 0.5 °C/min or 1 °C/min (the data obtained were independent of the heating rate). Data points were acquired every 0.5 °C with a response time of 2 s and a bandwidth of 1 nm. Fitting to a two-state model was carried out after normalization of the unfolding transition; the three mutants were fitted simultaneously, with common baselines for the native and the denatured states (see equations in the Supporting Information). Fitting to a three-state model was carried out for each mutant separately, after normalization of both near- and far-UV transitions (see equations in the Supporting Information).(33) The ΔCp of the first transition was taken as 1.6 kcal/mol·K for I59T and WT, and 1.3 kcal/mol.K for I56T, according to previous studies.(46) Because the theoretical ΔCp calculated for the unfolding of the native state to the completely unfolded state is 1.6 kcal/mol·K, and therefore very close to the measured ΔCp of the first transition, we assume that the ΔCp for the second transition is much smaller than this value and arbitrarily set it at 0.2 kcal/mol·K for all three variants.(47) Distributing the ΔCp equally over the two transitions (for example with ΔCp = 0.8 kcal/mol.K for both transitions) had very little effect on the fitting, validating our approach to the data analysis.
Differential Scanning Calorimetry
Heat capacities were measured as a function of temperature using a VP-DSC microcalorimeter (Microcal LLC, Northampton, MA), with protein concentrations of ca. 40 μmol/L and a scanning rate of 1 °C/min, from 10 to 70 °C. The baseline obtained by heating a buffer solution was subtracted from the protein unfolding thermogram, and the excess heat capacity (taking the native state as reference) was obtained by subtracting a progressive baseline between the native and unfolded states. The reversibility of the transition was assessed by overlaying the first and second thermal unfolding curves, the latter being obtained by reheating the sample after cooling inside the calorimetric cell. The reversibility was greater than 90% for all variants (see Figure S1 in the Supporting Information). Van’t Hoff to calorimetric enthalpy ratios were close to unity for the I59T and WT variants (it was not possible to estimate it for I56T because of the lack of native state baseline at low temperatures), and data could only be fitted to a two-state model (see equations in the Supporting Information).
Nuclear Magnetic Resonance Spectroscopy
15N ammonium sulfate and 13C methanol were used to label the protein produced in Pichia pastoris with 15N and 13C isotopes, respectively. Fast HSQC schemes for 1H−15N 2D spectra were used for monitoring the unfolding process, and spectra were acquired from 5 to 55 °C, every 2.5 °C.(48) The backbone resonances of the folded state of the I59T variant were assigned at 25 °C using HNCA, HNCACB, and CBCA(CO)NH experiments.(49) Conventional NMR characterization of the non-native ensemble of lysozyme, that requires several days of measurements at high temperature and low pH (pH 1.2), has previously been limited by the degradation of the sample. Using a procedure based on a 3D version of the 15N zz-exchange experiments, however, enabled data collection to be achieved in 1−2 days: the denatured state of the I59T variant was partially assigned (ca. 82%), by observation of the transfer of magnetization at 35 °C between cross-peaks arising from native and non-native states.50,51 Some resonances of the I56T variant could be correlated with those of the I59T variant by overlaying the spectra of the denatured states, resulting in the assignment of ca. 48% of resonances of I56T. Spectra processing was done with nmrPipe(52) and spectra visualization with ‘Sparky’ (http://www.cgl.ucsf.edu/home/sparky/). Peak volumes of HSQC cross-peaks were determined assuming Gaussian lineshapes, and overlapping peaks were discarded. In Figure 4, the traces of the increase in peak volumes for the WT are slightly to the left of the green dashed line, because normalization was carried out relative to a temperature of 55 °C, where the far-UV for WT has achieved only 70% of its transition. 1D spectra were processed using TOPSPIN (http://www.bruker-biospin.com/topspin.html).
Acknowledgments
A.D. is grateful for support from Boerhinger Ingelheim Fonds through a Ph.D. scholarship and from Murray Edwards College, Cambridge, through a Junior Research Fellowship. N.C. is a Human Frontiers Science Program (HFSP) Long-term Fellow (LT000795/2009). J.R.K. thanks BBSRC (U.K.) for support. S.T.D.H. is a recipient of a HFSP long-term Fellowship (LT0798/2005) and is supported by the Postdoctoral Research Abroad Program of National Science Council of the Republic of China, Taiwan (NSC97-2917-1-564-102). M.F.M. acknowledges support from IRB Barcelona. M.D. is a Research Associate of the Belgian F.R.S.-F.N.R.S and acknowledges support from the Belgian Government (IAP P6-19 to C.M.D and M.D.) D.N. and the NMR Facility of the Department of Biochemistry, Cambridge, U.K., are supported by the BBSRC, Cancer Research UK, and the Wellcome Trust. M.A. is supported by the Swedish Research Council, the Göran Gustafsson Foundation for Research in Natural Sciences and Medicine, and the Knut and Alice Wallenberg Foundation. X.S. acknowledges support from the Leverhulme Trust, the Wellcome Trust, ICREA, IRB Barcelona, and MICINN (CTQ2009-08850). C.M.D. acknowledges support from BBSRC (BB/E019927/1), the Wellcome Trust, the European Commission (project LSHM-CT-2006-037525), and the Leverhulme Trust.
Supporting Information Available
Thermodynamic equations used to fit the spectroscopic data (CD and DSC), table of thermodynamic parameters resulting from fitting CD and DSC data, and figures showing the reversibility of the unfolding transition assessed by DSC, the characterization of amyloid fibrils of human lysozyme, the normalized CD signal, the thermal unfolding analyzed using a three-state model, the temperatures of disappearance and appearance of NMR resonances in HSQC spectra, the raw 1D NMR data, the predicted water exchange rates of amide protons, and the temperature increase in peak volume of NMR resonances from the denatured state of the I56T variant. This material is available free of charge via the Internet at http://pubs.acs.org.
Footnotes
Abbreviations: WT, wild-type; NMR, nuclear magnetic resonance; HSQC, heteronuclear single quantum coherence; UV, ultraviolet; CD, circular dichroism; DSC, differential scanning calorimetry.
Supplementary Material
References
- Chiti F.; Dobson C. M. Annu. Rev. Biochem. 2006, 75, 333–366. [DOI] [PubMed] [Google Scholar]
- Teilum K.; Smith M. H.; Schulz E.; Christensen L. C.; Solomentsev G.; Oliveberg M.; Akke M. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 18273–18278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. Nat. Chem. Biol. 2009, 5, 15–22. [DOI] [PubMed] [Google Scholar]
- Calloni G.; Lendel C.; Campioni S.; Giannini S.; Gliozzi A.; Relini A.; Vendruscolo M.; Dobson C. M.; Salvatella X.; Chiti F. J. Am. Chem. Soc. 2008, 130, 13040–13050. [DOI] [PubMed] [Google Scholar]
- Campioni S.; Mossuto M. F.; Torrassa S.; Calloni G.; de Laureto P. P.; Relini A.; Fontana A.; Chiti F. J. Mol. Biol. 2008, 379, 554–567. [DOI] [PubMed] [Google Scholar]
- Soldi G.; Bemporad F.; Chiti F. J. Am. Chem. Soc. 2008, 130, 4295–4302. [DOI] [PubMed] [Google Scholar]
- Sacchettini J. C.; Kelly J. W. Nat. Rev. Drug Discovery 2002, 1, 267–275. [DOI] [PubMed] [Google Scholar]
- Cohen F. E.; Kelly J. W. Nature 2003, 426, 905–909. [DOI] [PubMed] [Google Scholar]
- Mittag T.; Forman-Kay J. D. Curr. Opin. Struct. Biol. 2007, 17, 3–14. [DOI] [PubMed] [Google Scholar]
- Gerum C.; Wirmer-Bartoschek J.; Schwalbe H. Angew. Chem., Int. Ed. 2009, 48, 9452–9456. [DOI] [PubMed] [Google Scholar]
- Klein-Seetharaman J.; Oikawa M.; Grimshaw S. B.; Wirmer J.; Durchardt E.; Ueda T.; Imoto T.; Smith L. J.; Dobson C. M.; Schwalbe H. Science 2002, 295, 1719–1722. [DOI] [PubMed] [Google Scholar]
- Radford S. E.; Dobson C. M.; Evans P. A. Nature 1992, 358, 302–307. [DOI] [PubMed] [Google Scholar]
- Miranker A.; Radford S. E.; Karplus M.; Dobson C. M. Nature 1991, 349, 633–636. [DOI] [PubMed] [Google Scholar]
- Arai M.; Kuwajima K. Adv. Protein Chem. 2000, 53, 209. [DOI] [PubMed] [Google Scholar]
- Griko Y. V.; Freire E.; Privalov P. L. Biochemistry 1994, 33, 1889–1899. [DOI] [PubMed] [Google Scholar]
- Griko Y. V. J. Protein Chem. 1999, 18, 361–369. [DOI] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. Biopolymers 1983, 22, 2577–637. [DOI] [PubMed] [Google Scholar]
- Artymiuk P. J.; Blake C. C. F. J. Mol. Biol. 1981, 152, 737–762. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics Modell. 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Pepys M. B.; Hawkins P. N.; Booth D. R.; Vigushin D. M.; Tennent G. A.; Soutar A. K.; Totty N.; Nguyan O.; Blake C. C. F.; Terry C. J.; Feest T. G.; Zalin A. M.; Hsuan J. J. Nature 1993, 362, 553–557. [DOI] [PubMed] [Google Scholar]
- Dumoulin M.; Kumita J. R.; Dobson C. M. Acc. Chem. Res. 2006, 39, 603–610. [DOI] [PubMed] [Google Scholar]
- Booth D. R.; Sunde M.; Bellotti V.; Robinson C. V.; Hutchinson W. L.; Fraser P. E.; Hawkins P. N.; Dobson C. M.; Radford S. E.; Blake C. C. F.; Pepys M. B. Nature 1997, 385, 787–793. [DOI] [PubMed] [Google Scholar]
- Dumoulin M.; Last A. M.; Desmyter A.; Decanniere K.; Canet D.; Larsson G.; Spencer A.; Archer D. B.; Sasse J.; Muyldermans S.; Wyns L.; Redfield C.; Matagne A.; Robinson C. V.; Dobson C. M. Nature 2003, 424, 783–788. [DOI] [PubMed] [Google Scholar]
- Johnson R. J. K.; Christodoulou J.; Dumoulin M.; Caddy G. L.; Alcocer M. J. C.; Murtagh G. J.; Kumita J. R.; Larsson G.; Robinson C. V.; Archer D. B.; Luisi B.; Dobson C. M. J. Mol. Biol. 2005, 352, 823–836. [DOI] [PubMed] [Google Scholar]
- Hagan C.; Johnson R. J. K.; Dhulesia A.; Dumoulin M.; Dumont J.; Genst E. E.; Christodoulou J.; Robinson C. V.; Dobson C. M.; Kumita J. R. Protein Eng. Des. Sel. 2010, 23, 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G.; Garcia J.; Mangione P.; Giorgetti S.; Corazza A.; Viglino P.; Chiti F.; Andreola A.; Dumy P.; Booth D.; Hawkins P. N.; Bellotti V. J. Biol. Chem. 2003, 278, 25910–25918. [DOI] [PubMed] [Google Scholar]
- Canet D.; Last A. M.; Tito P.; Sunde M.; Spencer A.; Archer D. B.; Redfield C.; Robinson C. V.; Dobson C. M. Nat. Struct. Biol. 2002, 9, 308–315. [DOI] [PubMed] [Google Scholar]
- Dumoulin M.; Canet D.; Last A. M.; Pardon E.; Archer D. B.; Muyldermans S.; Wyns L.; Matagne A.; Robinson C. V.; Redfield C.; Dobson C. M. J. Mol. Biol. 2005, 346, 773–788. [DOI] [PubMed] [Google Scholar]
- Kumita J. R.; Johnson R. J. K.; Alcocer M. J. C.; Dumoulin M.; Holmqvist F.; MacCammon M. G.; Robinson C. V.; Archer D. B.; Dobson C. M. FEBS J. 2006, 273, 711–720. [DOI] [PubMed] [Google Scholar]
- Haezebrouck P.; Joniau M.; Dael H. V.; Hooke S. D.; Woodruff N. D.; Dobson C. M. J. Mol. Biol. 1995, 246, 382–387. [DOI] [PubMed] [Google Scholar]
- Frare E.; Mossuto M. F.; de Laureto P. P.; Dumoulin M.; Dobson C. M.; Fontana A. J. Mol. Biol. 2006, 361, 551–561. [DOI] [PubMed] [Google Scholar]
- Mossuto M. F.; Dhulesia A.; Devlin G.; Frare E.; Kumita J. R.; de Laureto P. P.; Dumoulin M.; Fontana A.; Dobson C. M.; Salvatella X.. J. Mol. Biol. 2010, 402, 783−796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremades N.; Velazquez-Campoy A.; Freire E.; Sancho J. Biochemistry 2007, 47, 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramboarina S.; Redfield C. J. Mol. Biol. 2003, 330, 1177–1188. [DOI] [PubMed] [Google Scholar]
- Bai Y.; Milne J. S.; Mayne L.; Englander S. W. Proteins: Struct., Funct., Bioinf. 1993, 17, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalicky J. J.; Selsted M. E.; Pardi A. Proteins: Struct., Funct., Bioinf. 1994, 20. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Rance M.; Houghten R. A.; Lerner R. A.; Wright P. E. J. Mol. Biol. 1988, 201, 161–200. [DOI] [PubMed] [Google Scholar]
- Baxter N. J.; Williamson M. P. J. Biomol. NMR 1997, 9, 359–369. [DOI] [PubMed] [Google Scholar]
- Andersen N. H.; Neidigh J. W.; Harris S. M.; Lee G. M.; Liu Z.; Tong H. J. Am. Chem. Soc. 1997, 119, 8547–8561. [Google Scholar]
- Baxter N. J.; Hosszu L. L. P.; Waltho J. P.; Williamson M. P. J. Mol. Biol. 1998, 284, 1625–1639. [DOI] [PubMed] [Google Scholar]
- Williamson M. P. Proteins: Struct., Funct., Bioinf. 2003, 53, 731–739. [DOI] [PubMed] [Google Scholar]
- Teilum K.; Poulsen F. M.; Akke M. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 6877–6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai S.; Hirai M. Biophys. J. 1999, 76, 2192–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima K. FASEB J. 1996, 10, 102–109. [DOI] [PubMed] [Google Scholar]
- Spencer A.; Morozova-Roche L.; Noppe W.; Mackenzie D. A.; Jeenes D. J.; Joniau M.; Dobson C. M.; Archer D. B. Protein Expression Purif. 1999, 16, 171–180. [DOI] [PubMed] [Google Scholar]
- Funahashi J.; Takano K.; Yamagata Y.; Yutani K. Protein Eng. Des. Sel. 1999, 12, 841–850. [DOI] [PubMed] [Google Scholar]
- Gómez J.; Hilser V.; Xie D.; Freire E. Proteins 1995, 22, 404–12. [DOI] [PubMed] [Google Scholar]
- Mori S.; Abeygunawardana C.; Johnson M. O.; van Zijl P. C. M. J. Magn. Reson. 1995, 108, 94–98. [DOI] [PubMed] [Google Scholar]
- Sattler M.; Schleucher J.; Griesinger C. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 93–158. [Google Scholar]
- Farrow N. A.; Zhang O.; Forman-Kay J. D.; Kay L. J. Biomol. NMR 1994, 4, 727–734. [DOI] [PubMed] [Google Scholar]
- Palmer A. G.; Kroenke C. D.; Loria J. P. Methods Enzymol. 2001, 339, 204–238. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. J. Biomol. NMR 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.