

Abstract
Objective:
While the hallmark of amyotrophic lateral sclerosis (ALS) is corticospinal tract in combination with lower motor neuron degeneration, the clinical involvement of both compartments is characteristically variable and the site of onset debated. We sought to establish whether there is a consistent signature of cerebral white matter abnormalities in heterogeneous ALS cases.
Methods:
In this observational study, diffusion tensor imaging was applied in a whole-brain analysis of 24 heterogeneous patients with ALS and well-matched healthy controls. Tract-based spatial statistics were used, with optimized voxel-based morphometry of T1 images to determine any associated gray matter involvement.
Results:
A consistent reduction in fractional anisotropy was demonstrated in the corpus callosum of the ALS group, extending rostrally and bilaterally to the region of the primary motor cortices, independent of the degree of clinical upper motor neuron involvement. Matched regional radial diffusivity increase supported the concept of anterograde degeneration of callosal fibers observed pathologically. Gray matter reductions were observed bilaterally in primary motor and supplementary motor regions, and also in the anterior cingulate and temporal lobe regions. A post hoc group comparison model incorporating significant values for fractional anisotropy, radial diffusivity, and gray matter was 92% sensitive, 88% specific, with an accuracy of 90%.
Conclusion:
Callosal involvement is a consistent feature of ALS, independent of clinical upper motor neuron involvement, and may reflect independent bilateral cortical involvement or interhemispheric spread of pathology. The predominantly rostral corticospinal tract involvement further supports the concept of independent cortical degeneration even in those patients with ALS with predominantly lower motor neuron involvement clinically.
GLOSSARY
- ALS
= amyotrophic lateral sclerosis;
- ALSFRS-R
= revised Amyotrophic Lateral Sclerosis Functional Rating Scale;
- CC
= corpus callosum;
- CST
= corticospinal tract;
- DD
= disease duration;
- DTI
= diffusion tensor imaging;
- FA
= fractional anisotropy;
- FTD
= frontotemporal dementia;
- GM
= gray matter;
- LMN
= lower motor neuron;
- MD
= mean diffusivity;
- PLS
= primary lateral sclerosis;
- PMA
= progressive muscular atrophy;
- RD
= radial diffusivity;
- UMN
= upper motor neuron;
- WM
= white matter.
A major issue in amyotrophic lateral sclerosis (ALS) is phenotypic heterogeneity. While ALS is characterized by simultaneous upper motor neuron (UMN) and lower motor neuron (LMN) degeneration, phenotypes are recognized in which degeneration in one or more compartments appears dominant, termed progressive muscular atrophy (PMA) where there is LMN-only involvement clinically and primary lateral sclerosis (PLS) where involvement is UMN only. The nature of this observed spectrum of compartmentalization of motor neuron pathology is not understood, and extremes can present a diagnostic challenge early in the disease course.
The disappointing progress in therapeutic trials in ALS, despite advances in the understanding of pathogenesis,1 has been partly attributed to a lack of biomarkers, although there are emerging candidates.2 A particular challenge for any biomarker is that it must hold true across a range of phenotypes, and, in addition to diagnostic and therapeutic monitoring value, resolving issues about the onset and spread of pathology in ALS may also be important in identifying the at-risk population.
Diffusion tensor imaging (DTI) is now established as a robust noninvasive MRI tool to perform in vivo neuropathologic study of white matter (WM) neuronal tracts.3 We studied a group of heterogeneous patients with ALS of variable UMN involvement clinically using DTI at 3 Tesla to achieve high spatial and angular resolution. Analysis of the entire brain was used to detect common regions of WM damage that might inform concepts of focality and spread. Associated gray matter (GM) volumetric changes were explored using an optimized voxel-based morphometric protocol.
METHODS
Participants.
Consecutively consenting patients with sporadic ALS were recruited from the Oxford Motor Neuron Disease Care & Research Centre as part of the Oxford Study for Biomarkers in Motor Neuron Disease (BioMOx, www.biomox.net). All patients were initially diagnosed by 1 of 2 experienced ALS neurologists (K.T., M.R.T.) according to revised El Escorial criteria. Two patients with PLS and PMA (i.e., no detectable LMN or UMN signs, respectively, at the time of scanning), were excluded from the current analysis presented as there were insufficient numbers to permit meaningful direct comparison with the larger ALS group, given that their inclusion within the same spectrum is still debated.
Twenty-four closely age- and gender-matched healthy controls were recruited for comparison.
ALS clinical and functional measures.
All patients underwent clinical examination and El Escorial classification on the day of study (M.R.T.). Patients' functional status was measured using the revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R). A quantitative assessment of clinical UMN involvement was based on a scale used in a previous ALS neuroimaging study.4 This UMN score was based upon the number of pathologic reflexes (recorded by M.R.T. in advance of the scan), elicited from 15 body sites: glabellum, orbicularis oris, masseter (jaw jerk), biceps, triceps and finger jerks bilaterally, and knee, ankle, and Babinksi responses bilaterally. Disease duration (DD) was calculated from symptom onset to scan date in months. In view of the central hypothesis concerning a focal cortical onset to the disease, prior to analysis patients with ALS were classified by the presumed dominant hemisphere for disease onset, defined as the contralateral hemisphere to the initial laterality of limb symptoms reported.
Standard protocol approvals, registrations, and patient consents.
Ethical approval for all procedures was obtained prior to study (08/H0605/85 and 07/K1604/43). Written informed consent was obtained from all participants.
Image acquisition.
Scans were performed at the Oxford Centre for Clinical Magnetic Resonance Research using a 3-T Siemens Trio scanner (Siemens AG, Erlangen, Germany) with a 12-channel head coil. Whole-brain diffusion-weighted imaging was performed using a spin echo sequence (repetition time/echo time = 9,300/94 msec, field of view 192 mm, 2 mm isotropic resolution, b value = 1,000 s/mm2, 60 isotropically distributed gradients). High-resolution 3-dimensional T1-weighted MRI scans were acquired using a magnetization-prepared rapid gradient echo sequence (repetition time/echo time = 2,040/4.7 msec, flip angle 8°, field of view 192 mm, 1 mm isotropic resolution).
Image analysis and statistics.
Detailed methods are provided online (e-Methods on the Neurology® Web site at www.neurology.org). In summary, whole-brain analysis of fractional anisotropy (FA), mean diffusivity (MD), radial diffusivity (RD), and axial diffusivity maps was carried out using tract-based spatial statistics. Group comparison and correlation with UMN scores, ALSFRS-R, and DD within the patient group were carried out using permutation-based nonparametric inference within the framework of the general linear model. An optimized voxel-based morphometry approach was used to identify any group-related differences in GM using permutation-based nonparametric inference. Correlation analyses between diffusion and GM values extracted for each subject, using a mask of the significant group-related differences, were carried out with Spearman rank correlation. Finally, we explored the sensitivity, specificity, and accuracy values to distinguish heterogeneous ALS from controls, using a model comprising significant overlapping group-related differences for all diffusion indices and significant GM results.
RESULTS
Participants.
Twenty-four consecutively enrolled patients with ALS underwent MRI with closely age- and gender-matched healthy control subjects (table 1).
Table 1 Participant characteristics
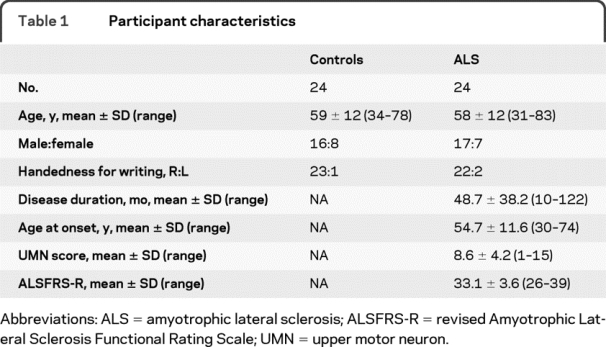
The breakdown of patients by revised El Escorial criteria clinically was definite (n = 7), probable/probable laboratory-supported (n = 9), and possible (n = 8). First limb of involvement was right-sided in 14 and left-sided in 9, plus 1 patient with progressive bulbar palsy without clinical limb involvement to date. Analysis of the data without image rotation according to laterality did not materially alter the results reported, neither did the exploratory inclusion of the 2 excluded patients with PLS and PMA, respectively, though the latter will be the subject of future dedicated group comparison.
Whole-brain group differences in FA replicated earlier postmortem study.
FA was reduced in the ALS patient group compared to healthy controls within the corpus callosum (CC) and bilaterally in WM tracts known to extend from the central CC to primary motor and premotor cortex, including parts of the rostral corticospinal tract (CST) (p < 0.05 corrected). Additional, weaker regional differences were observed caudally in the CST, including brainstem (p < 0.05 uncorrected). All changes, notably including the CC, were strikingly similar to those documented in a previously published postmortem pathologic study5 (figure 1). Whole-brain FA (and GM) reductions were not influenced by the inclusion of DD as a nuisance covariate in the analysis (figure e-1).

Figure 1 Regional fractional anisotropy (FA) reductions in amyotrophic lateral sclerosis group whole-brain comparison with healthy controls, alongside published postmortem observations
Consistent corpus callosum (CC) and rostral corticospinal tract (CST) tract involvement was seen despite the inclusion of a large number of patients with few clinical upper motor neuron signs. Similar white matter tract degeneration sections alongside those taken from an historical pathologic study5 (left-side images of A–D) confirmed prominent involvement of the CC and rostral CST (A and B, thick black lines indicating primary motor cortex), with caudal CST changes seen in uncorrected FA results (A, C, and D). Images shown were corrected (red–yellow scale) and uncorrected (red-only scale) for multiple comparisons (p < 0.05; radiologic convention used for display in all images).
Whole-brain FA correlated with UMN score in the CST but not CC in patients with ALS.
Within the patient group, significant voxel-wise correlations between FA and clinical measures were observed. For reduced FA vs increased UMN score, correlation was seen bilaterally in the CST (p < 0.05 corrected), but not in the CC. For reduced FA vs lower ALSFRS-R, a weaker correlation (p < 0.05 uncorrected) was found in several regions, including parts of the CC. Correlation between increased FA and longer DD also localized bilaterally to the CST (p < 0.05 corrected; figure e-2).
Whole-brain group differences in RD overlapped with FA differences.
Increased RD values were observed in patients with ALS compared with controls in the CC and bilaterally in the WM connecting primary and premotor cortex (p < 0.05 corrected). These regions colocalized with FA reductions (figure 2). No significant group differences were found in mean or axial diffusivity.

Figure 2 Regional fractional anisotropy (FA) reductions and radial diffusivity (RD) increases in amyotrophic lateral sclerosis (ALS) group whole-brain comparison with healthy controls
The close overlap between FA reductions (top panel, red–yellow) and RD (lower panel, blue) findings suggests that involvement of the corpus callosum and rostral corticospinal tract in ALS reflects a secondary demyelinating process due to an anterograde Wallerian degeneration. Both measures were corrected for multiple comparisons (p < 0.05; radiologic convention used for display in all cases with sagittal sections specifically marked for side).
Whole-brain GM volumetric group differences showed spatial correspondence to FA change.
Analysis of GM volumetric differences between patients with ALS and controls revealed widespread areas of reduction including primary, premotor, and supplementary motor cortices, cingulate cortex, and temporal lobe regions. There was good anatomic correspondence between motor-related GM atrophy and DTI-derived (FA, RD) changes (figure 3). Regional volume changes were bilateral despite our standardization for laterality of limb onset, and in the uncorrected data (not shown) there was bilateral temporal lobe involvement, suggesting that apparent lateralization in this area was a thresholding issue.

Figure 3 Gray matter (GM) and fractional anisotropy (FA) reductions in amyotrophic lateral sclerosis (ALS) group whole-brain comparison with healthy controls
Widespread GM reductions (green) in the primary and supplementary motor cortices, anterior cingulate gyrus, and temporal lobes were found, many with close relation to the regional white matter changes revealed by FA reductions (red–yellow). Motor region–related GM involvement was bilateral despite correction for laterality of limb onset, with the possibility that the corpus callosum involvement reflects independent bilateral cortical processes, or interhemispheric spread of pathology in ALS. Images were corrected for multiple comparisons (p < 0.05; radiologic convention used for display in all cases with sagittal sections specifically marked for side).
Combining measures.
Further post hoc investigation showed that both group-related differences in FA and RD were highly correlated with GM differences across subjects (FA and GM: ρ = 0.64, p < 10−4; RD and GM: ρ = −0.61, p < 10−4; figure 4).

Figure 4 Scatterplot of mean fractional anisotropy (FA), radial diffusivity (RD), and gray matter (GM) values in amyotrophic lateral sclerosis (ALS) and healthy control groups
For each subject, the mean values for FA and RD (expressed in mm2.s−1), obtained in those regions found to be significantly different between the 2 groups using tract-based spatial statistics, were plotted separately against GM values obtained in those regions found to be volumetrically significantly different using voxel-based morphometry. Correlations were seen in FA vs GM (ρ = 0.64, p < 10−4) and RD vs GM (ρ = −0.61, p < 10−4) for both control (blue) and ALS (red) patient groups, with reasonable separation of the two.
Discriminant analysis using the combination of all 3 measures improved the classification of the subjects into 2 groups with 92% sensitivity, 88% specificity, and 90% accuracy, performing better than discriminant analyses based on each one of these measures considered separately (table e-1).
DISCUSSION
This study demonstrated a consistent involvement of the CC and rostral CST across a heterogeneous group of patients with ALS, including those with little clinical UMN involvement, supporting the concept of an independent cerebral pathogenic process in ALS. While CC involvement might relate to interhemispheric spread, it might equally reflect secondary damage due to independent bilateral cortical processes. Multimodal MRI has the potential to discriminate heterogeneous ALS from controls, and can generate noninvasive biomarkers.
DTI is sensitive to the motion of water molecules. Tightly confined water movement in intact neuronal pathways will be anisotropic (i.e., strongly directional, along the main direction of the fibers), whereas damaged pathways will result in less restricted movement of water in multiple directions leading to a reduction in anisotropy. The application of DTI to ALS has been refined over a decade from the initial experience,6 with CST and more widespread WM changes reported to varying degrees.2 Such studies have generally involved more homogeneous patients with ALS with UMN signs in 2 or more body territories.
Our assessment of UMN involvement was based on reflexes. The localization of FA and UMN score correlations to the CSTs supports our view that pathologic reflexes at the bedside accurately reflect pathology in this neuronal compartment in a quantitative way. Several targeted DTI studies in ALS have also reported a relationship between clinical UMN involvement and CST FA,7–9 or brainstem FA10 reductions.
A debate over spinal anterior horn retrograde11 vs anterograde corticomotoneuronal degeneration12 continues, though the 2 concepts are not mutually exclusive. Clinicopathologic studies support a cortical as well as spinal focus.13 The whole-brain FA reductions found in our study were most marked in the rostral CST, despite a broad range of UMN involvement clinically in our participants, including nearly one-third of patients categorized as only possible ALS as a result of few UMN signs. This rostral emphasis has been observed before14 and supports an active cortical process, rather than UMN involvement occurring solely due to dying back from a focal onset of pathology in the anterior horn of the cord.
A within-group positive correlation was found between DD and CST FA. A potential confound was the negative correlation noted between DD and UMN scores in our patient group (ρ = −0.61, p = 0.002). More speculatively the CST may be more resistant to disease-related damage in some patients with ALS or such individuals have a higher baseline FA, either way then reflected in a longer disease course. Given that post hoc analysis showed that the effect in FA was largely due to a reduction in RD, we speculate that longevity might reflect more myelinated or higher packing density of axons in this pathway. Longitudinal DTI studies in ALS are needed to confirm the true nature of these observations, but have been scarce due to the practical challenge of MRI in those for whom there is rapid progression and disability. One study has demonstrated progressive FA decreases in the CST.15
Postmortem study specifically noted CC degeneration to be prominent in ALS,5 and there was a striking similarity in the distribution of the degenerating fibers when compared with our study (see figure 1). Several DTI studies have also reported variable degrees of FA change within the CC as part of a wider array of cerebral WM changes in ALS.14,16,17 Changes in MD reported within parts of the CC14,18 were not detected in our study.
Diffusion tensor tractography can been used to parcellate regions according to their wider cortical connectivity.19 This confirms that the fibers of the mid-body of the CC, where we found the strongest group-related differences in FA, appear largely to link to the motor and premotor cortices. This central portion of the CC was also noted to be most markedly involved in postmortem pathologic study.5 In a previous ALS study employing tract-based spatial statistics, the authors concluded that the central CC FA reductions observed were able to discriminate PLS from ALS.20 DTI studies of patients with hereditary spastic paraparesis have also demonstrated prominent CC involvement, notably not seen in the LMN-only slowly progressive disorder X-linked spinobulbar muscular atrophy.21 Therefore CC appears to be a common pathway in UMN disorders, with the reported PLS findings likely confirming this phenotype as an extreme end of a continuum with ALS.
Indirect clinical evidence of central CC involvement in ALS comes from the observation of mirror movements in patients, reflecting impaired interhemispheric inhibition.22 In a combined study of mirror movements in patients with ALS using DTI targeted to specific areas of the pyramidal tract and the central CC (where our most significant FA reductions were also located), it was demonstrated that a ratio of the FA values from these regions correlated strongly with the observed abnormal co-movements and also, like our study, with ALSFRS-R.23 Our MRI protocol required patients to lie flat and still for least 30 minutes and this is often impossible in the more disabled stages of ALS, compounded by orthopnea due to simultaneous diaphragmatic weakness. For this reason our study contained a rather narrow range of ALSFRS-R scores, which may have limited the power to demonstrate a stronger relationship of CC involvement to disability.
Transcallosal inhibition measured by transcranial magnetic stimulation has demonstrated reduced inhibition in ALS in the earliest stages of the disease, and importantly (like our findings) in individuals without clinical UMN involvement.24 Studies in presymptomatic patients with mutations of the superoxide dismutase-1 gene suggest that cortical excitability,25 as well as LMN loss,26 is an early event associated with the development of symptoms.
Others have noted cerebral MD increases in relation to DD in ALS.10,27,28 Our finding of RD changes closely matched to the regions of FA reduction, in a U-shape linking the rostral CSTs and CC, is noteworthy. Studies of individual DTI parameter changes in relation to surgical callosotomy for epilepsy29 suggest that our observed changes may represent a secondary demyelination in response to cortical Wallerian degeneration. Relative preservation of axonal architecture (seen also postmortem5) might also then explain the lack of MD and axial diffusivity changes.
A synthesis of neurophysiologic and our DTI findings supports a view that cortical involvement early in ALS pathogenesis may be followed quickly by callosal involvement, either through independent bilateral cortical pathology or interhemispheric spread. Hypotheses about focality of onset and spread of disease in ALS have indirectly implicated the CC (see figure in reference 13), and others have speculated that the CC might be a “conduit” for pathologic spread within a corticomotoneuronal model of ALS.30 Although asymmetry of symptom onset is poorly understood, the observation of concordance for handedness and laterality of upper limb onset in ALS may provide further indirect support for an independent cortical vulnerability, when considered alongside studies that have noted greater cortical excitability and altered cerebral activation patterns in the dominant vs nondominant hand.31
It is clear also that ALS pathology extends beyond the CST, sharing pathologic as well as clinical features with some forms of frontotemporal dementia (FTD). It is noteworthy therefore that CC without CST involvement was reported in a DTI study of FTD,32 and that the anterior part of the CC projects to the dorsolateral prefrontal cortex,19 thought to be involved in ALS.4 We speculate that more anterior CC involvement might reflect frontotemporal pathology in ALS. Quantitative measurement of cortical neuronal loss in postmortem ALS brains is challenging and may be unrevealing.33 Beyond the minority of ALS cases with frank FTD, significant atrophy of either GM or WM is not typical. MRI is potentially more sensitive to such changes, and several studies have demonstrated widespread GM changes including regional overlap with our study18,27,34–36 (reviewed in reference 37). While the involvement of the temporal lobes we noted is in keeping with the known spectrum of cognitive and pathologic overlap with FTD, none of our subjects had overt cognitive impairment, and we do not draw any firm conclusions in the absence of formal neuropsychological assessment.
Despite our observations of consistent callosal involvement, the sine qua non of ALS is LMN degeneration at the anterior horns, for which any unifying model of pathogenesis must account. Assuming a LMN process proceeds in parallel, a common link (if not direct connection) may be a shared vulnerability through interneuronal inhibitory influences.38 The lack of knowledge of the at-risk population and uncertainty over where the clinical horizon lies in relation to the start of the pathologic cascade currently hampers MRI study of the very earliest changes beyond rare familial cases.39 Early peridiagnostic as well as longitudinal studies will be important for validation of the present findings.
Notwithstanding a need for dedicated studies in cases of pure PMA as well as in the earliest stages of typical ALS, we speculate, like others,30 that ALS may be fundamentally a cortical neurodegenerative disorder. Rapid increase in human lifespan may have exposed a vulnerability of the aging brain for the specialized motor functions that have evolved on the basis of much shorter survival. An important challenge is to understand why the rate of spread of pathology is so variable across the wider spectrum of motor neuron disease. Whether LMN involvement is driven through direct connectivity with a cortically based process or indirectly, perhaps through a shared interneuronopathy for example, will be the subject of further research.
ACKNOWLEDGMENT
Ther authors thank Care Centre Research Assistant Melanie Lord and Centre Coordinator/Specialist Nurse Rachael Marsden and the ALS patients and their carers.
DISCLOSURE
Dr. Filippini receives research support from the Gordon Small Charitable Trust. Dr. Douaud receives license fee and royalty payments from the University of Oxford for FSL software and receives research support from the Engineering and Physical Sciences Research Council (EPSRC). Dr. Mackay and S. Knight report no disclosures. Dr. Talbot serves on a scientific advisory board for Agenzia di Ricerca per la Sclerosi Laterale Amiotrofica (AriSLA); serves on the editorial board of Neuropathology and Applied Neurobiology; receives royalties from the publication of Medicine at a Glance (Blackwell Science, 2002), Motor Neuron Disease: The Facts (Oxford University Press, 2009), and Motor Neuron Disease: A Practical Manual (Oxford University Press, 2010); and receives research support from the Motor Neurone Disease Association and SMA Trust. Dr. Turner receives royalties from the publication of The Brain: A Beginner's Guide (Oneworld, 2008) and Motor Neuron Disease: A Practical Manual (Oxford University Press, 2010); serves as a consultant for Evalueserve, IMS Hospital Group Ltd., Smartanalyst Inc., Scisive, and Guidepoint Global; and receives research support from the Medical Research Council, the Motor Neurone Disease Association Lady Edith Wolfson Fellowship.
Supplementary Material
Address correspondence and reprint requests to Dr. Martin Turner, Department of Clinical Neurology, West Wing Level 3, John Radcliffe Hospital, Oxford, OX3 9DU, UK martin.turner@clneuro.ox.ac.uk
Supplemental data at www.neurology.org
*These authors contributed equally to this work.
Study funding: The Oxford Motor Neuron Disease Care & Research Centre receives funding from the Motor Neurone Disease Association UK Care Centre Program.
Disclosure: Author disclosures are provided at the end of the article.
Received April 9, 2010. Accepted in final form June 21, 2010.
REFERENCES
- 1.Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol 2009;65(suppl 1):S3–S9. [DOI] [PubMed] [Google Scholar]
- 2.Turner MR, Kiernan MC, Leigh PN, Talbot K. Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol 2009;8:94–109. [DOI] [PubMed] [Google Scholar]
- 3.Johansen-Berg H, Behrens TE. Diffusion MRI: From Quantitative Measurement to in-vivo Neuroanatomy. Academic Press; 2009. [Google Scholar]
- 4.Turner MR, Cagnin A, Turkheimer FE, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [(11)C](R)-PK11195 positron emission tomography study. Neurobiol Dis 2004;15:601–609. [DOI] [PubMed] [Google Scholar]
- 5.Smith MC. Nerve fibre degeneration in the brain in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 1960;23:269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis CM, Simmons A, Jones DK, et al. Diffusion tensor MRI assesses corticospinal tract damage in ALS. Neurology 1999;53:1051–1058. [DOI] [PubMed] [Google Scholar]
- 7.Wong JC, Concha L, Beaulieu C, Johnston W, Allen PS, Kalra S. Spatial profiling of the corticospinal tract in amyotrophic lateral sclerosis using diffusion tensor imaging. J Neuroimaging 2007;17:234–240. [DOI] [PubMed] [Google Scholar]
- 8.Roccatagliata L, Bonzano L, Mancardi G, Canepa C, Caponnetto C. Detection of motor cortex thinning and corticospinal tract involvement by quantitative MRI in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2009;10:47–52. [DOI] [PubMed] [Google Scholar]
- 9.Iwata NK, Aoki S, Okabe S, et al. Evaluation of corticospinal tracts in ALS with diffusion tensor MRI and brainstem stimulation. Neurology 2008;70:528–532. [DOI] [PubMed] [Google Scholar]
- 10.Hong YH, Lee KW, Sung JJ, Chang KH, Song IC. Diffusion tensor MRI as a diagnostic tool of upper motor neuron involvement in amyotrophic lateral sclerosis. J Neurol Sci 2004;227:73–78. [DOI] [PubMed] [Google Scholar]
- 11.Chou SM, Norris FH. Amyotrophic lateral sclerosis: lower motor neuron disease spreading to upper motor neurons. Muscle Nerve 1993;16:864–869. [DOI] [PubMed] [Google Scholar]
- 12.Eisen A, Kim S, Pant B. Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron? Muscle Nerve 1992;15:219–224. [DOI] [PubMed] [Google Scholar]
- 13.Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 2009;73:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sage CA, Van Hecke W, Peeters R, et al. Quantitative diffusion tensor imaging in amyotrophic lateral sclerosis: revisited. Hum Brain Mapp 2009;30:3657–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sage CA, Peeters RR, Gorner A, Robberecht W, Sunaert S. Quantitative diffusion tensor imaging in amyotrophic lateral sclerosis. Neuroimage 2007;34:486–499. [DOI] [PubMed] [Google Scholar]
- 16.Sach M, Winkler G, Glauche V, et al. Diffusion tensor MRI of early upper motor neuron involvement in amyotrophic lateral sclerosis. Brain 2004;127:340–350. [DOI] [PubMed] [Google Scholar]
- 17.Senda J, Ito M, Watanabe H, et al. Correlation between pyramidal tract degeneration and widespread white matter involvement in amyotrophic lateral sclerosis: a study with tractography and diffusion-tensor imaging. Amyotroph Lateral Scler 2009;10:288–294. [DOI] [PubMed] [Google Scholar]
- 18.Agosta F, Pagani E, Rocca MA, et al. Voxel-based morphometry study of brain volumetry and diffusivity in amyotrophic lateral sclerosis patients with mild disability. Hum Brain Mapp 2007;28:1430–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chao YP, Cho KH, Yeh CH, Chou KH, Chen JH, Lin CP. Probabilistic topography of human corpus callosum using cytoarchitectural parcellation and high angular resolution diffusion imaging tractography. Hum Brain Mapp 2009;30:3172–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciccarelli O, Behrens TE, Johansen-Berg H, et al. Investigation of white matter pathology in ALS and PLS using tract-based spatial statistics. Hum Brain Mapp 2009;30:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Unrath A, Muller HP, Riecker A, Ludolph AC, Sperfeld AD, Kassubek J. Whole brain-based analysis of regional white matter tract alterations in rare motor neuron diseases by diffusion tensor imaging. Hum Brain Mapp 2010 (in press). [DOI] [PMC free article] [PubMed]
- 22.Karandreas N, Papadopoulou M, Kokotis P, Papapostolou A, Tsivgoulis G, Zambelis T. Impaired interhemispheric inhibition in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2007;8:112–118. [DOI] [PubMed] [Google Scholar]
- 23.Bartels C, Mertens N, Hofer S, et al. Callosal dysfunction in amyotrophic lateral sclerosis correlates with diffusion tensor imaging of the central motor system. Neuromuscul Disord 2008;18:398–407. [DOI] [PubMed] [Google Scholar]
- 24.Wittstock M, Wolters A, Benecke R. Transcallosal inhibition in amyotrophic lateral sclerosis. Clin Neurophysiol 2007;118:301–307. [DOI] [PubMed] [Google Scholar]
- 25.Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008;131:1540–1550. [DOI] [PubMed] [Google Scholar]
- 26.Aggarwal A, Nicholson G. Detection of preclinical motor neurone loss in SOD1 mutation carriers using motor unit number estimation. J Neurol Neurosurg Psychiatry 2002;73:199–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellis CM, Suckling J, Amaro E Jr, et al. Volumetric analysis reveals corticospinal tract degeneration and extramotor involvement in ALS. Neurology 2001;57:1571–1578. [DOI] [PubMed] [Google Scholar]
- 28.Cosottini M, Giannelli M, Siciliano G, et al. Diffusion-tensor MR imaging of corticospinal tract in amyotrophic lateral sclerosis and progressive muscular atrophy. Radiology 2005;237:258–264. [DOI] [PubMed] [Google Scholar]
- 29.Concha L, Gross DW, Wheatley BM, Beaulieu C. Diffusion tensor imaging of time-dependent axonal and myelin degradation after corpus callosotomy in epilepsy patients. Neuroimage 2006;32:1090–1099. [DOI] [PubMed] [Google Scholar]
- 30.Eisen A. Amyotrophic lateral sclerosis: evolutionary and other perspectives. Muscle Nerve 2009;40:297–304. [DOI] [PubMed] [Google Scholar]
- 31.Turner MR, Wicks P, Brownstein CA, et al. Concordance between site of onset and limb dominance in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2010 (in press). [DOI] [PubMed]
- 32.Matsuo K, Mizuno T, Yamada K, et al. Cerebral white matter damage in frontotemporal dementia assessed by diffusion tensor tractography. Neuroradiology 2008;50:605–611. [DOI] [PubMed] [Google Scholar]
- 33.Gredal O, Pakkenberg H, Karlsborg M, Pakkenberg B. Unchanged total number of neurons in motor cortex and neocortex in amyotrophic lateral sclerosis: a stereological study. J Neurosci Methods 2000;95:171–176. [DOI] [PubMed] [Google Scholar]
- 34.Kassubek J, Unrath A, Huppertz HJ, et al. Global brain atrophy and corticospinal tract alterations in ALS, as investigated by voxel-based morphometry of 3-D MRI. Amyotroph Lateral Scler Other Motor Neuron Disord 2005;6:213–220. [DOI] [PubMed] [Google Scholar]
- 35.Chang JL, Lomen-Hoerth C, Murphy J, et al. A voxel-based morphometry study of patterns of brain atrophy in ALS and ALS/FTLD. Neurology 2005;65:75–80. [DOI] [PubMed] [Google Scholar]
- 36.Grosskreutz J, Kaufmann J, Fradrich J, Dengler R, Heinze HJ, Peschel T. Widespread sensorimotor and frontal cortical atrophy in amyotrophic lateral sclerosis. BMC Neurol 2006;6:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grosskreutz J, Peschel T, Unrath A, Dengler R, Ludolph AC, Kassubek J. Whole brain-based computerized neuroimaging in ALS and other motor neuron disorders. Amyotroph Lateral Scler 2008;9:238–248. [DOI] [PubMed] [Google Scholar]
- 38.Turner MR, Hammers A, Al Chalabi A, et al. Distinct cerebral lesions in sporadic and ‘D90A’ SOD1 ALS: studies with [11C]flumazenil PET. Brain 2005;128:1323–1329. [DOI] [PubMed] [Google Scholar]
- 39.Ng MC, Ho JT, Ho SL, et al. Abnormal diffusion tensor in nonsymptomatic familial amyotrophic lateral sclerosis with a causative superoxide dismutase 1 mutation. J Magn Reson Imaging 2008;27:8–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.