

Abstract
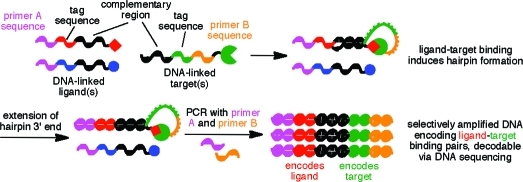
Interaction-dependent PCR (IDPCR) is a solution-phase method to identify binding partners from combined libraries of small-molecule ligands and targets in a single experiment. Binding between DNA-linked targets and DNA-linked ligands induces formation of an extendable duplex. Extension links codes that identify the ligand and target into one selectively amplifiable DNA molecule. In a model selection, IDPCR resulted in the enrichment of DNA encoding all five known protein−ligand pairs out of 67 599 possible sequences.
Recent advances in genome and proteome research have led to a dramatic increase in the number of macromolecular targets of interest to the life sciences. The rapid identification of ligands to this expanding number of targets is a major scientific and technological challenge. To this end, a variety of target-oriented high-throughput screening methods have been developed. Two fundamental limitations to target-oriented screening methods are (i) the requirement that each target of interest must successively be assayed against libraries of potential ligands and (ii) the general reliance on immobilized targets or ligands.(1) The first constraint limits assay throughput significantly when researchers are interested in multiple targets or in ligand specificity. The second limitation adds immobilization, washing, and/or elution steps to the screening process and is a source of artifacts that arise from matrix binding, multivalent binding, or loss of native target structure.(2) A solution-phase method to simultaneously reveal all ligand−target binding pairs from a single solution containing libraries of ligands and libraries of targets could in principle overcome both limitations and significantly increase the efficiency and effectiveness of target-oriented screening efforts. Here we report the development of such a method, interaction-dependent PCR (IDPCR).
IDPCR is based on the melting temperature (Tm) difference between duplex DNA formed intramolecularly versus intermolecularly, a difference that we recently exploited to couple covalent bond formation with PCR amplification.(3) We hypothesized that binding of a target to its ligand would increase the effective molarity of single-stranded DNA (ssDNA) oligonucleotides linked to the target and ligand, promoting duplex formation between complementary regions on each strand that are otherwise too short to hybridize (Figure 1a). The resulting hairpin could serve as a starting point for primer extension. Crucially, only the newly extended hairpin contains in a single DNA strand two primer (or primer-binding) sequences that enable subsequent PCR amplification. IDPCR therefore results in the selective amplification of those DNA sequences previously attached to, and therefore encoding, ligand−target pairs (Figure 1a). In contrast to traditional target-based selections, which rely on the physical separation of active molecules from inactive ones, IDPCR selectively amplifies DNA encoding active library members.
Figure 1.

(a) Overview of IDPCR. (b) IDPCR with streptavidin as the target (1a-SA) and biotin (2a-biotin, Kd = 40 pM) or desthiobiotin (2b-desthiobiotin, Kd = 2 nM) as ligands was analyzed by qPCR and PAGE (21 cycles of PCR). IDPCR reports the interaction of (c) trypsin and antipain (Ki = 100 nM); (d) carbonic anhydrase and carboxy benzene sulfonamide (CBS, Ki = 3.2 μM) or Gly-Leu-CBS (GLCBS, Ki = 9 nM); and (e) a DNA aptamer and daunomycin (Dn) (Dn, Kd = 272 nM) or doxorubicin (Dx). PAGE gels in (c), (d), and (e) show DNA after 20, 24, and 23 cycles of PCR, respectively.
In order for hairpin extension to report target−ligand binding, it must occur under conditions that both allow binding to take place and enable selective extension of intramolecular duplexes over intermolecular duplexes. Studies on DNA polymerase-mediated extension at 37 °C suggested that a 6-nt complementary region was optimal for enabling intramolecular but not intermolecular duplexes to be extended (Supporting Information Figure S1). We investigated whether binding between a small molecule and a protein could replace a covalent linkage in a DNA hairpin and support extension and PCR. Biotin and streptavidin (SA) (Kd = 40 pM)(4) were chosen as an initial ligand−target pair. We reacted SA with NHS ester-linked ssDNA 1a (the target strand) to generate 1a-SA and also synthesized an oligonucleotide (the ligand strand) conjugated at its 3′ end with biotin to provide 2a-biotin. The target and ligand strands shared a 6-nt complementary region. Negative control ligand−DNA conjugates lacking biotin (2) or incapable of hybridizing to 1 (3a-biotin) were also prepared. Each ligand−DNA conjugate (2, 2a-biotin, or 3a-biotin) was individually incubated under identical conditions with 1a-SA and Klenow fragment DNA polymerase at 37 °C and then subjected to qPCR to determine the threshold cycle (CT) value.
Consistent with our hypothesis, the sample containing 1a-SA and 2a-biotin underwent far more efficient PCR amplification than all of the negative controls, resulting in CT values ≥ 5 cycles lower (corresponding to ≥32-fold more extension product) than those of the 1a-SA + 2 control, the 1a-SA + 3a-biotin control, or a sample containing 1a-SA + 2a-biotin but lacking Klenow (Figure 1b). A positive control, containing a 10-nt complementary region that hybridizes to 2 independent of target−ligand binding, exhibited a comparable CT value to that of 1a-SA + 2a-biotin sample. Importantly, the addition of excess free biotin abrogated the IDPCR of 2a-biotin with 1a-SA (Figure 1b). IDPCR was surprisingly tolerant of ligand−DNA linker lengths between ∼28 and 123 atoms (Supporting Information Figure S2). Together, these results indicate that specific ligand−target binding can promote DNA extension and trigger the selective PCR amplification of DNA sequences linked to ligand−target pairs.
Next we tested the ability of IDPCR to report ligand−target interactions of much lower affinity (Kd = ∼2 nM to ∼3 μM; protein target affinities of DNA-linked ligands other than biotin were measured and found to be within 5-fold of the reported affinities for the free small molecules (Supporting Information Figure S3)). Ligand strand 2 was conjugated to a lower affinity SA ligand, desthiobiotin (Kd = 2 nM).5,6 When 2b-desthiobiotin was combined with 1a-SA and subjected to Klenow extension and qPCR, 2b-desthiobiotin was amplified with an efficiency comparable to that of 2a-biotin (Figure 1b). Similarly, trypsin and antipain (Ki = 100 nM)(7) were conjugated to DNA to form 1b-trypsin and 2c-antipain. The 1b-trypsin + 2c-antipain pair resulted in a 7-cycle CT advantage relative to negative controls lacking ligand, containing 2 conjugated to unrelated ligands, or containing an excess of free antipain (Figure 1c and Supporting Information Figures S2 and S3). Likewise, DNA encoding carbonic anhydrase II (CA) (1c-CA) and CA ligands 4-carboxy benzene sulfonamide (CBS) (Kd = 3.2 μM)(8) (2d-CBS) and Gly-Leu-CBS (Kd = 9 nM)(9) (2e-GLCBS) was amplified far more efficiently (6- to 7-cycle ΔCT) than control reactions lacking ligand (2), containing a mismatched complementary region (3c-CBS), or containing an excess of free GLCBS (Figure 1d and Supporting Information Figures S2 and S3). Collectively, these results suggest that IDPCR can serve as a general method to detect a wide variety of small molecule−protein interactions of varying affinities.
In principle, any class of intermolecular interaction can be detected by IDPCR. Next we tested the ability of IDPCR to selectively amplify DNA sequences encoding nucleic acid aptamer−ligand pairs.(10) A 68-nt DNA aptamer that binds daunomycin (Kd = 272 nM)(11) and doxorubicin was synthesized at the 5′ end of the target strand to generate 1d-aptamer. Daunomycin or doxorubicin was conjugated to the ligand strand to afford 2g-Dn or 2h-Dx, respectively. Consistent with the above results for protein targets, IDPCR reactions containing both 1d-aptamer and either 2g-Dn or 2h-Dx were amplified more efficiently than samples with 2f in place of 2g-Dn (ΔCT = 8 cycles) or samples containing free doxorubicin (ΔCT = 4 cycles) (Figure 1e). These results indicate that IDPCR can be used to selectively amplify DNA linked to small molecule−aptamer pairs.
We wondered if the covalent bond between the target and the target oligonucleotide could be replaced by a noncovalent interaction, resulting in IDPCR of a ternary complex between two DNA-linked ligands and a multivalent target.5,12,13 To test this possibility, we conjugated biotin to the target strand to generate 1e-biotin, which can hybridize with 2a-biotin. We hypothesized that in the presence of SA a ternary complex of SA and two DNA-linked biotin ligands would form, enabling DNA hybridization, extension, and amplification. Indeed, IDPCR in this “sandwich” mode detected as little as 2 × 10−19 mol (200 zmol) of SA (Supporting Information Figure S4). These results suggest the potential of IDPCR for the sensitive detection of multivalent analytes in sandwich assays.
Since applications of IDPCR include library screening, we performed a series of model selections to test the ability of IDPCR to selectively enrich DNA encoding authentic ligands in the presence of an excess of nonbinding small molecule−DNA conjugates (Figure 2a). A 1:10, 1:100, or 1:1000 ratio of 2i-biotin/2k-GLCBS was combined with 1a-SA and subjected to Klenow extension and PCR. The same mixtures of 2i-biotin/2k-GLCBS were also subjected to IDPCR in the presence of 1 (without SA) as a control. When IDPCR was performed with 1a-SA, the biotin-encoding sequence 2i was strongly enriched among the resulting PCR products (Figure 2b). In contrast, IDPCR with 1 resulted in no enrichment of the biotin-linked strand. Similarly strong enrichment was observed for CA-GLCBS binding and for DNA aptamer−daunomycin binding in the presence of large excesses of nonbinding conjugates (Figure 2c−e). These findings demonstrate that IDPCR can enrich DNA encoding a ligand ∼100-fold over DNA encoding small molecules without target affinity for a variety of protein and nucleic acid targets.
Figure 2.

(a) IDPCR with a single target in the presence of mock ligand library. (b) Mixtures of 2i-biotin and excess 2k-GLCBS were subjected to IDPCR with 1a-SA or 1. (c) Mixtures of 2k-GLCBS and excess 2i-biotin were subjected to IDPCR with 1c-CA or 1. (d) Mixtures of 2n-Dn and excess 2l were subjected to IDPCR against 1f-aptamer or 1h. (e) Mixtures of 1g-aptamer and excess unstructured DNA (1h) were subjected to IDPCR with 2g-Dn or 2f. The DNA in (b), (c), (d), and (e) was digested with EcoRI, HindIII, NsiI, or NsiI, respectively.
Finally, we tested the ability of IDPCR to simultaneously evaluate all possible combinations of multiple ligands and multiple targets in a single solution. We performed a model selection in which five small-molecule ligands (biotin, desthiobiotin, GLCBS, CBS, and antipain) and three targets (SA, CA, and trypsin), each conjugated to unique sequence tags, were present in one solution containing a 250-fold excess of DNA-linked ligand (hexylamine) and a 250-fold excess of a DNA-linked target (glutathione S-transferase) not known to interact with the other ligands or targets. The negative control ligand and target were conjugated to libraries of 256 different sequence tags. The resulting solution therefore contained equimolar quantities of each of 261 ligand sequences and each of 259 target sequences, collectively representing 67 599 possible ligand−target sequence combinations. A control sample was prepared identically except using DNA lacking any protein targets. Both samples were subjected to IDPCR followed by high-throughput DNA sequencing.
For each of the three different proteins in the library, the most highly enriched sequences relative to the control sample correspond to their known ligands (Figure 3), despite the large excess of nonbinding ligands and the fact that ligand−target affinities span 5 orders of magnitude. The mean enrichment factor across all 67 599 possibilities was 1.4, while the enrichment factors corresponding to the five known ligand−target pairs ranged from 75 to 3000. Only three enrichment factors above 75 were observed among presumed nonbinding pairs out of 67 594 possibilities, representing a low false positive rate (Supporting Information Tables S1 and S2). These results establish the ability of IDPCR to evaluate a small-molecule library for affinity to a protein target library in a single experiment and suggest that IDPCR can identify ligand−target pairs across a wide range of affinities in a highly multiplexed format.
Figure 3.

(a) A model library of DNA-encoded ligands mixed with a model library of DNA-encoded targets allows multiplexed detection of binding pairs. (b) IDPCR was used to perform a model selection on an equimolar 261-member DNA−ligand library and an equimolar 259-member DNA−target library containing five known protein−ligand pairs out of 67 599 possible combinations. For each protein target, the most highly enriched sequences (blue bars) relative to a control lacking proteins corresponded to the known protein−ligand pairs, labeled A−E in the plot. A: biotin + SA; B: desthiobiotin + SA; C: GLCBS + CA; D: CBS + CA; E: trypsin + antipain.
In conclusion, we have developed IDPCR as a general method for selectively amplifying DNA sequences encoding ligand−target pairs. IDPCR can be applied to a wide variety of targets and potential ligands and to our knowledge is one of the first methods that can identify ligand−target pairs from libraries of small molecules and libraries of targets in a single solution.(14) From a practical perspective, IDPCR requires DNA-linked ligands (which can be prepared by any of several methods in current use by a variety of laboratories)12,15 and DNA-linked targets (prepared here by simple nonspecific conjugation) but is highly sensitive, takes place entirely in the solution phase, and can be performed in a few hours using routine equipment. We anticipate that IDPCR will significantly enhance efforts to discover new ligands and targets, to reveal target-binding specificities of small molecules, and to detect low-abundance analytes.
Acknowledgments
This work was supported by NIH GM065865, the Kavli Foundation, and HHMI. L.M.M. is an NSF Graduate Research fellow. D.J.G. is an American Cancer Society-Canary Foundation fellow. C.E.D. acknowledges fellowships from the Swiss National Science Foundation and the Novartis Foundation. We are grateful to Matt McGregor for programming assistance.
Supporting Information Available
Experimental methods, supporting experimental data, and complete ref (15a). This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Inglese J.; Johnson R. L.; Simeonov A.; Xia M.; Zheng W.; Austin C. P.; Auld D. S. Nat. Chem. Biol. 2007, 3, 466. [DOI] [PubMed] [Google Scholar]; b Zhu Z.; Cuozzo J. J. Biomol. Screening 2009, 14, 1157. [DOI] [PubMed] [Google Scholar]
- Vijayendran R. A.; Leckband D. E. Anal. Chem. 2001, 73, 471. [DOI] [PubMed] [Google Scholar]
- Gorin D. J.; Kamlet A. S.; Liu D. R. J. Am. Chem. Soc. 2009, 131, 9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green N. M. Methods Enzymol. 1990, 184, 62. [DOI] [PubMed] [Google Scholar]
- Dumelin C. E.; Scheuermann J.; Melkko S.; Neri D. Bioconjugate Chem. 2006, 17, 366. [DOI] [PubMed] [Google Scholar]
- Torreggiani A.; Fini G. Biospectroscopy 1998, 4, 197. [DOI] [PubMed] [Google Scholar]
- Otto H. H.; Schirmeister T. Chem. Rev. 1997, 97, 133. [DOI] [PubMed] [Google Scholar]
- West G. M.; Tang L.; Fitzgerald M. C. Anal. Chem. 2008, 80, 4175. [DOI] [PubMed] [Google Scholar]
- Mincione F.; Starnotti M.; Menabuoni L.; Scozzafava A.; Casini A.; Supuran C. T. Bioorg. Med. Chem. Lett. 2001, 11, 1787. [DOI] [PubMed] [Google Scholar]
- a Ellington A. D.; Szostak J. W. Nature 1990, 346, 818. [DOI] [PubMed] [Google Scholar]; b Robertson D. L.; Joyce G. F. Nature 1990, 344, 467. [DOI] [PubMed] [Google Scholar]; c Tuerk C.; Gold L. Science 1990, 249, 505. [DOI] [PubMed] [Google Scholar]; For a general review see:; d Wilson D. S.; Szostak J. W. Annu. Rev. Biochem. 1999, 68, 611. [DOI] [PubMed] [Google Scholar]
- Wochner A.; Menger M.; Orgel D.; Cech B.; Rimmele M.; Erdmann V. A.; Gloekler J. Anal. Biochem. 2008, 373, 34. [DOI] [PubMed] [Google Scholar]
- Melkko S.; Scheuermann J.; Dumelin C. E.; Neri D. Nat. Biotechnol. 2004, 22, 568. [DOI] [PubMed] [Google Scholar]
- Sprinz K. I.; Tagore D. M.; Hamilton A. D. Bioorg. Med. Chem. Lett. 2005, 15, 3908. [DOI] [PubMed] [Google Scholar]
- a Bowley D. R.; Jones T. M.; Burton D. R.; Lerner R. A. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fredriksson S.; Gullberg M.; Jarvius J.; Olsson C.; Pietras K.; Gustafsdottir S. M.; Ostman A.; Landegren U. Nat. Biotechnol. 2002, 20, 473. [DOI] [PubMed] [Google Scholar]; c Hofstadler S. A.; Griffey R. H. Chem. Rev. 2001, 101, 377. [DOI] [PubMed] [Google Scholar]
- a Clark M. A.; et al. Nat. Chem. Biol. 2009, 5, 647. [DOI] [PubMed] [Google Scholar]; b Mannocci L.; Zhang Y.; Scheuermann J.; Leimbacher M.; De Bellis G.; Rizzi E.; Dumelin C. E.; Melkko S.; Neri D. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 17670. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gorska K.; Huang K.-T.; Chaloin O.; Winssinger N. Angew. Chem., Int. Ed. 2009, 48, 7695. [DOI] [PubMed] [Google Scholar]; d Halpin D. R.; Harbury P. B. PLoS Biol. 2004, 2, 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hansen M. H.; Blakskjaer P.; Petersen L. K.; Hansen T. H.; Hojfeldt J. W.; Gothelf K. V.; Hansen N. J. V. J. Am. Chem. Soc. 2009, 131, 1322. [DOI] [PubMed] [Google Scholar]; f Kanan M. W.; Rozenman M. M.; Sakurai K.; Snyder T. M.; Liu D. R. Nature 2004, 431, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kleiner R. E.; Dumelin C. E.; Tiu G. C.; Sakurai K.; Liu D. R. J. Am. Chem. Soc. 2010, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Tse B. N.; Snyder T. M.; Shen Y.; Liu D. R. J. Am. Chem. Soc. 2008, 130, 15611. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Doyon J. B.; Snyder T. M.; Liu D. R. J. Am. Chem. Soc. 2003, 125, 12372. [DOI] [PubMed] [Google Scholar]; j Gartner Z. J.; Liu D. R. J. Am. Chem. Soc. 2001, 123, 6961. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Gartner Z. J.; Tse B. N.; Grubina R.; Doyon J. B.; Snyder T. M.; Liu D. R. Science 2004, 305, 1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.