

Abstract
Proteases have an important role in many signalling pathways, and represent potential drug targets for diseases ranging from cardiovascular disorders to cancer, as well as for combating many parasites and viruses. Although inhibitors of well-established protease targets such as angiotensin-converting enzyme and HIV protease have shown substantial therapeutic success, developing drugs for new protease targets has proved challenging in recent years. This in part could be due to issues such as the difficulty of achieving selectivity when targeting protease active sites. This Perspective discusses the general principles in protease-based drug discovery, highlighting the lessons learned and the emerging strategies, such as targeting allosteric sites, which could help harness the therapeutic potential of new protease targets.
Proteins are among the most stable biological polymers. Peptide bonds can withstand hours in boiling concentrated acid, yet they last no more than microseconds in the presence of a specific protease. The study of proteolysis goes back at least to the nineteenth century with the description of pepsin by Schwann in 1836 and of trypsin by Corvisart in 1856. Since then, proteases have been identified in almost every organism, have been discovered to play a part in most biological pathways1 and have been implicated in almost every disease.
Historically, much of the focus has been on the role of proteases in coagulopathies, inflammation, infectious diseases, cancer and degenerative diseases, and some protease inhibitors have been developed into highly successful drugs. For example, inhibitors of the human protease angiotensin-converting enzyme (ACE), such as captopril, have been used in the treatment of cardiovascular disorders, primarily hypertension and congestive heart failure, for several decades2. In addition, inhibitors of the HIV protease, such as ritonavir, atazananvir and tipranavir (Aptivus; Pfizer/Boehringer Ingelheim), have had a key role in transforming the treatment of HIV infection since their introduction in the mid-1990s3 (see TABLE 1 for examples). Inhibitors of the proteases thrombin and factor Xa together have current global sales of US$1 billion, which is anticipated to rise to $3.5 billion by 2014 (REF. 4), whereas antihypertensive drugs that act on the proteases in the renin–angiotensin system currently have over $6 billion global sales1. Indeed, at present, we estimate that 5–10% of all pharmaceutical targets being pursued for drug development are proteases.
Table 1.
Examples of successful strategies applied for the discovery of protease inhibitors
| Disease | Protease (class) |
Drug name (trade name; company) |
Drug type |
Drug structure | Hits by | Date on market |
Refs |
|---|---|---|---|---|---|---|---|
| Hypertension | ACE (metallo) |
Captopril (Capoten; Brystol-Myers Squibb) |
Peptidic |
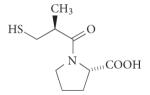
|
De novo (structure- based drug screening |
1981 | 2 |
| Hypertension | Renin (aspartic) |
Aliskiren (Tekturna/Rasilez; Novartis/Speedel) |
Non- peptidic |
|
De novo (structure- based drug screening) |
2007 | 108 |
| HIV/AIDS | HIV protease (aspartic) |
Tipranavir (Aptivus; Pfizer/ Boehringer Ingelheim) |
Non- peptidic |
|
Derived from a non-peptidic screening programme |
2005 | 3 |
| Cancer | Proteasome (threonine) |
Bortezomib (Velcade; Millennium) |
Peptidic |
|
De novo (systematic optimization studies) |
2003 | 27 |
| Diabetes | DPP4 (serine) |
Sitagliptin (Januvia; Merck) |
Non- peptidic |
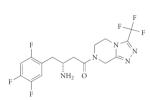
|
Optimization of a class of β-amino acids |
2006 | 35 |
| Coagulation | Thrombin (serine) |
Desirudin (Revasc/Iprivask; Novartis) |
Peptidic | Polypeptide (65 amino acid residues) | Isolated from the medicinal leech Hirudo medicinalis |
1998 | 129 |
| Coagulation | Factor Xa (serine) |
Rivaroxaban (Xarelto; Bayer) |
Non- peptidic |
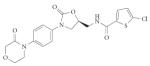
|
Product from HTS and subsequent de novo systematic optimization studies |
2008 | 130 |
| Coagulation | Thrombin (serine) |
Dabigatran (Rendix/Pradaxa; Boehringer Ingelheim) |
Non- peptidic |
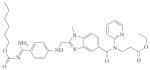
|
De novo (systematic optimization studies) |
2008 | 131 |
| Coagulation | Thrombin (serine) |
Argatroban (Acova; GlaxoSmithKline) |
Non- peptidic |
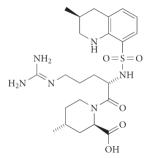
|
De novo (systematic optimization studies) |
2000 | 132 |
ACE, angiotensin-converting enzyme; DPP, dipeptidyl peptidase; HTS, high-throughput screening.
The general strategy for therapeutically targeting proteases is to identify a specific inhibitor — generally a small molecule — that blocks the active site. As discussed below, discovery efforts for new inhibitors have typically been based on the structure of known protease substrates, presenting a substantial challenge for the development of peptidomimetic compounds that have the pharmacokinetic characteristics needed to be suitable as a drug. Furthermore, proteolytic pathways generally consist of close homologues that have an identical catalytic mechanism and similar substrate specificity profiles. Consequently, relatively large peptidic or peptidomimetic inhibitors are often required to achieve potent and selective active-site inhibition, adding to the challenge of identifying compounds with the appropriate drug-like properties.
So the two key questions in protease-based drug discovery are: how specific should an inhibitor be to deliver a therapeutic outcome and what are the best ways to achieve the specificity needed? Despite the successes of active-site targeted inhibitors in helping to gain fundamental scientific information and the successes in the design of some highly valuable drugs, the limited number of new protease inhibitors introduced during the past decade (about six), as well as several high-profile failures, indicates a need to explore alternative approaches. TABLE 1, which samples currently approved protease inhibitors based on protease types, is notable for its brevity and the absence of new targets that have not already been known for decades (see REF. 1 for more complete tables). This highlights two additional important questions for protease-based drug discovery: are there common reasons for the apparent lack of recent success, and if so, are alternative strategies available to enhance the likelihood of success in the future?
Here, we discuss principles for targeting proteases, with a focus on drug discovery. After providing an introduction to general protease biology and their role in disease, as well as the requirements for the design of a protease inhibitor, we examine the current strategies being pursued for protease inhibitor development, and discuss key issues such as the development of reversible inhibitors or irreversible inhibitors. We then explore the new insights into protease biology and structure that are shaping emerging therapeutic approaches, such as the development of allosteric protease modulators and of biological protease inhibitors.
Proteolytic pathways
Proteases and their inhibitors are popular experimental models for structural biologists, for protein engineers and in studies of protein folding. There have been huge strides in the elucidation of their mechanisms of action, their biology and their structure over the past 10 years. The number of known proteases has also increased substantially and the MEROPS protease database now lists 679 known and putative human peptidases, and an additional 423 non-catalytic homologues in humans alone (as of August 2010).
These proteases fall into five main mechanistic classes: the metalloproteinases (~200), serine proteases (~180), cysteine proteases (~160), threonine proteases (~30) and aspartic acid proteases (~25), with the remaining proteases belonging to groups with an unknown or unclassified catalytic mechanism5. Proteases in established classes cleave peptide bonds through several different mechanisms. Serine proteases, cysteine proteases and amino-terminal nucleophile hydrolases (threonine proteases) possess an enzyme side chain in the active cleft that participates in catalysis (so-called covalent catalysis). By contrast, aspartic acid proteases and metalloproteinases carry out peptide-bond hydrolysis without covalent participation, primarily by generating a highly reactive water molecule (FIG. 1).
Figure 1. Tetrahedral intermediates formed during peptide cleavage by proteases.

All proteases accelerate the formation of a transition state in peptide-bond hydrolysis. This state, termed the tetrahedral intermediate, is a prerequisite for peptide-bond scission in all protease mechanistic classes. In the case of serine (Ser), cysteine (Cys) and threonine (Thr) proteases, the tetrahedral intermediate involves a stable covalent bond to the enzyme’s catalytic nucleophile (Nuc), whereas metalloproteinases and aspartic acid (Asp) proteases use a non-covalent acid–base mechanism. The tetrahedral intermediate is difficult to simulate; protein engineers and chemists have yet to invent or evolve an independent chemical structure or fold that can efficiently catalyse peptide-bond cleavage. There are examples in which catalytic antibodies, for example, have been evolved to efficiently hydrolyse activated esters, but these ester bonds are extremely different from the resonance-stabilized peptide bond124. This is a surprising gap in our knowledge and remains fertile ground for future work.
From the biological and the drug design point of view, the most important feature of a protease is its ability to recognize and to process its putative substrate or substrates. The intrinsic specificity of a protease is defined by the optimal peptide sequence or motif that binds to the specificity determinants or specificity pockets of the active site cleft, and can be greatly enhanced by exosites and allosteric sites (FIG. 2). Proteases were previously thought to bind their substrates with the peptide chain running through the catalytic cleft in an extended conformation, although this concept may now need to be adjusted to explain the ability of proteases to cleave in apparently well-structured α-helices6.
Figure 2. Active site, exosite and allosteric site.

Three proteases of known structure have been chosen to illustrate the components identified in many (but not all) proteases that define their specificity and their control of activity. Surfaces of sentrin-specific protease 2 (SENP2) (Protein Data Bank code 1TGZ125) (a) and thrombin (Protein Data Bank code 1EB1 (REF. 126)) (b) with substrate or inhibitor residues contacting the active site cleft (shown in red sticks) and with substrate or inhibitor residues contacting the exosite (shown in green sticks) are shown. In SENP2, the exosite is composed of a large disperse region that occupies the bulk of the small ubiquitin-like modifier (SUMO) substrate, which is required for directing specificity and enhancing activity76. By contrast, the thrombin exosite consists of a relatively small cationic patch that is required for efficient catalysis of fibrinogen and for binding to the inhibitor hirudin21. The surface of Vibrio cholerae RtxA toxin (Protein Data Bank code 3GCD127) (c) centres on the allosteric natural small-molecule activator inositol hexakisphosphate (shown in white), with the active site bound peptide (shown in red) underlying a loop in the translucent surface. In the case of SENP2, the exosite is potentially quite large, complicating small-molecule control. However, in thrombin and RtxA the exosite or the allosteric site is composed of surfaces compatible with small-molecule targeting.
The concept of proteases acting in proteolytic cascades was developed by Davie, Ratnoff and MacFarland during their pioneering work on blood coagulation around 40 years ago7-10. Since that time, several other proteolytic cascades have been postulated, for example, in fibrinolysis, complement fixation, apoptosis and gastrulation (BOX 1). Blood coagulation is viewed as the model proteolytic cascade in which information is passed through a pathway involving sequential activations of protease zymogens, with a minimal pathway requiring the proteases factor VII, factor X and prothrombin (BOX 1). Besides signal amplification, this cascade provides multiple regulation points, which are presumed to allow fine-tuning. Indeed, most proteolytic cascades have endogenous inhibitors that target the activated proteases, which can act either as ‘buffers’ or ‘thresholds’ to hold in check a pathway that has become inappropriately activated11. However, this is extremely difficult to verify in vivo, so the multiple-regulation-point concept awaits experimental validation.
Box 1. Examples of proteolytic cascades.
Blood clotting
The classical model for thrombus generation involves a cascade of activating events, involving the sequential activation of a series of zymogens of serine proteases to produce thrombin. Thrombin converts fibrinogen to fibrin, which forms the blood clot8. The rapid activation of thrombin can be visualized by in vivo substrate imaging97. A clot takes a minute or two to form, but the terminal clotting protease thrombin is activated within seconds after tissue injury in vivo97.
The cascade is initiated through two pathways. In the intrinsic pathway all zymogens and cofactors are constituents of the blood. By contrast, in the extrinsic pathway, tissue factor, which is expressed on the surface of all non-vascular cells, comes into contact with flowing blood to initiate blood coagulation. The concept of these two distinct pathways has sufficed for many years, but recent observations have blurred the distinctions. For example, deficiencies of proteins in the contact phase of the intrinsic pathway do not lead to haemorrhagic disorders97, and thrombin can activate factor XI, a zymogen of the intrinsic pathway99. Significantly, it has also been realized that in vitro coagulation is not an accurate reflection of the in vivo process, and some putative coagulation proteases have dropped from our understanding of blood coagulation in vivo. For example, a physiological role for Hageman factor (factor XII) and prekallikrein in the coagulation pathway is unlikely because individuals lacking this activity have no bleeding complications, even though both proteases accelerate blood coagulation in vitro and in mouse models of pathological coagulopathies100.
Fibrinolysis
To prevent excessive fibrin accumulation, fibrinolysis promotes the local dissolution of thrombi and promotes wound healing by re-establishing blood flow. Fibrinolysis is carried out by the plasminogen activation system, consisting of serine proteases that convert the inactive zymogen plasminogen to the active serine protease plasmin22. There are two plasminogen activators: urokinase-type plasminogen activator and tissue-type plasminogen activator. Plasmin, in turn, activates localized extracellular proteolytic activity, which catalyses the relatively nonspecific degradation of extracellular proteins.
Complement fixation
The complement system, comprising a group of more than 34 serum proteins, is activated by antigen-bound immunoglobulin or by membrane components on Gram-negative bacteria or fungi. This stimulates inflammation, antigen phagocytosis, and in some cases, direct lysis of cells. Complement fixation is activated through three protease cascade pathways: the classical, the alternative and the mannan-binding lectin pathway101. All pathways involve the cleavage of complement component C3, the most important activation step in complement fixation, and have a common final pathway called the terminal pathway or the membrane attack complex.
Apoptosis
Apoptosis, the most intensively studied form of programmed cell death, is regulated in mammalian cells by the cysteine proteases known as caspases. Like the blood coagulation pathway, caspases exist as zymogens awaiting an appropriate activation signal102. Again, similar to blood coagulation, two cascades are known: the extrinsic pathway and the intrinsic pathway. The extrinsic pathway is initiated by a signal from a so-called death receptor, such as the surface receptor Fas, which results in the activation of caspase 8 or caspase 10 (REF. 103). The intrinsic pathway is thought to be triggered by translocation into the mitochondria of a pro-apoptotic BCL2 family member; for example, BAX, which allows the release of pro-apoptotic factors, such as cytochrome c, from the mitochondria. Cytochrome c then triggers the formation of a supramolecular structure called an apoptosome, which results in the activation of caspase 9 (REF. 104). Activated caspase 8, caspase 9 or caspase 10 then generate the catalytically active forms of caspase 3 and caspase 7, which constitute the main apoptotic effectors.
Gastrulation
During Drosophila embryogenesis, cell fates along the dorsoventral axis are determined by a gradient of the extracellular morphogen Spätzle, which activates the receptor Toll. Spätzle carries the signal needed for ventral and lateral development, and genetic evidence suggests it is activated by four serine proteases — Nudel, Gastrulation defective, Snake and Easter — sequentially in a proteolytic cascade similar to blood clotting105.
For some other postulated proteolytic cascades, the term ‘cascade’ warrants some revision12. For example, in proteolytic matrix remodelling, the existence of a cascade, or even of a proteolytic pathway, is questioned by considerable biochemical and genetic evidence13. Perhaps the concept of a redundant network of interactions is more appropriate in this case14. It is now considered that many of the metalloproteinases once thought to degrade the matrix might in fact be limited processing enzymes, the substrates of which are cytokines and growth factors13. It is also unclear whether the plasminogen activator–plasmin–metalloproteinase pathway exists anywhere but in vitro, and the 30-year-old concept that the matrix must be broken down to allow cell migration (in normal or in metastatic instances) is under close scrutiny and revision14-16. Indeed, the possibility that the proteolytic degradation of the extracellular matrix might not be required for migration at all has been investigated17.
There are other instances in which a series of required proteolytic steps does not necessarily denote a pathway or a cascade. For example, chromatid separation during anaphase requires the protease separase. Separase is under control of securin, which must be degraded by the proteasome18. The timing of securin degradation is controlled by the anaphase-promoting complex — a ubiquitin E3 ligase19. Ubiquitin itself must be generated by the carboxy-terminal proteolytic processing of polymeric precursors or from the recycling of conjugates, which are all dependent on deubiquitylating enzymes (DUBs)20. Therefore, in principle, we have a sequence of proteases: DUB–proteasome–separase; however, it is debatable whether this could be called a pathway or a cascade.
Roles of proteases in disease
As proteases have a role in almost all biological pathways and networks, their misregulation is implicated in a broad range of diseases. Some of the best known include thrombin and plasmin in coagulopathies and in bleeding disorders21,22; matrix metalloproteinases (MMPs) in cancer and in inflammation14,23; renin and ACE in hypertension24; staphylococcal and streptococcal proteases in necrotic skin infections and in the destruction of haemostasis25,26; and cysteine cathepsins, caspases and the proteasome in cancer and in neurodegeneration27-29.
Throughout most of the pharmaceutical literature and the practice related to proteases, the prevailing view is that active proteases have detrimental roles in disease, although there might not be much formal proof of this. In cancer and in inflammation, for example, many different proteases are upregulated and/or activated and the common approach is to target them for inhibition. Whereas proteases are frequently prognosticators of poor outcome (prostate-specific antigen is the classic example), they also have a role in apoptosis induction. Turning apoptosis-inducing proteases such as caspases ‘back on’ can reinstate the normal death mechanisms that have been overcome in cancer cells30. However, getting the balance right is crucial — too much apoptosis results in the inappropriate death of neuronal cells (neurodegeneration)31, whereas too little apoptosis can give rise to cancer28. MMPs and cathepsins are also frequently upregulated in cancer29,32,33; however, their exact role remains controversial and must be investigated on a case-by-case basis34. Previous drug trials with MMP inhibitors in cancer showed that the drugs had serious side effects and illustrate the importance of careful investigation of the function of proteases in disease (BOX 2).
Box 2. Clinical studies of matrix metalloproteinase (MMP) inhibitors.
Almost two decades ago, several pharmaceutical companies (including Agouron, Bayer, British Biotech and Roche) moved MMP inhibitors into human trials based on the assumption that they would be effective in controlling connective tissue diseases. Many of these trials turned out to be major failures. Owing to serious complications, the trials were terminated early, seeding deep apprehension of MMP-based therapy106.
The severe off-target effects were discovered to be based on two principal misconceptions: first, that the drugs were going to be selective for specific MMPs, and second, that these MMPs had an exclusive role in matrix turnover. The assumptions turned out to be wrong and it is now clear that MMPs are also vital for immune regulation, which may be a principal reason for their failure in cancer and inflammation trials107. Moreover, the goals were set too high and the drug companies added an additional goal of oral bioavailability23. Thus, at least in these contexts, some MMPs and their close relatives should be thought of as the ‘good guys’ in disease.
To overcome the danger of off-target effects, it is essential to either thoroughly characterize the biological roles or understand the degree of crossreactivity of inhibitors to closely related members of an enzyme family, before embarking on drug campaigns. As exemplified by the Novartis Expertise Platform in Proteases, and probably hastened by the failure to take this into account in the early development of MMP inhibitors, considerable effort is now made to counterscreen all members of a protease family (paralogues)106.
Protease inhibitor drug discovery
Having decided that a particular protease has an important role in a given pathology, the general therapeutic strategy taken is to identify a specific inhibitor — generally a small molecule — that could block the action of the protease. There are a limited number of strategies available to pharmaceutical chemists in search of this goal, which usually target the active site, initially by mimicking the structure of a peptide substrate. This is exemplified by the campaign of the novartis and Speedel collaboration that resulted in the drug aliskiren (Tekturna/Rasilez; novartis) to inhibit renin, which has a key role in the regulation of blood pressure (BOX 3; TABLE 1). ACE inhibitors such as captopril, HIV protease inhibitors such as ritonavir (norvir; Abbott laboratories) and dipeptidyl peptidase 4 inhibitors such as sitagliptin (Januvia; Merck), are also paradigms for the development of protease inhibitors that target the active site in an intrinsic subsite occupancy mode (TABLE 1).
Box 3. The generation of renin inhibitors — a classic protease inhibitor campaign.
Renin cleaves angiotensinogen to form angiotensin I and is viewed as an excellent target for blood pressure regulation, especially as another protease (angiotensin-converting enzyme) acting in the same biochemical pathway is the target of extremely successful protease inhibitor drugs108.
Inhibitor development timeline
First generation
These were based on the substrate specificity of renin, an example of which is H-142 (half-maximal inhibitory concentration (IC50) = 10 nM). However, owing to the peptide origin of these molecules their metabolic stability was not satisfactory108,109.
Second generation
These were peptidomimetics in the form of dipeptide transition-state analogues, an example of which is remikiren (IC50 = 0.7 nM). These compounds were much more efficient in the inactivation of the enzyme and also possessed better metabolic stability. However, they had extremely low oral bioavailability and particularly high doses were needed in humans to achieve the desired drug to target ratio110.
Third generation
These were based on non-peptide-like compounds discovered using classical lead to drug methods with the application of X-ray crystallography. Currently the best known and effective representative of this group is aliskiren (Tekturna/Rasilez; Novartis) (IC50 = 0.6 nM), which was co-developed by Speedel and Novartis, and received marketing approval in the United States and in the European Union in 2007. Aliskiren possesses good metabolic stability, and its oral bioavailability is approximately 2.5% with a half-life of around 24 hours111.

The active site of proteases has a high propensity to accommodate organic molecules. In a study that mapped the binding surface of porcine pancreatic elastase using multiple solvent crystal structures35, it was shown that organic molecules bind almost exclusively in the active centre, whereas the remaining surface of the elastase protein had almost no binding affinity towards them. A more detailed analysis showed that these molecules bind in the pockets of the protease that are known to be targeted by previously developed inhibitors. Interestingly, the binding of the organic molecules also overlapped with the binding hot spots that were determined using a protein–ligand interaction database analysis.
Knowledge of the intrinsic subsite occupancy — the inherent preference of a protease to bind specific side chains in its specificity pockets (FIG. 2) — is vital for guiding active-site directed protease drug discovery. In principle, a good substrate can be converted to a good inhibitor by replacement of the part of a substrate that directly reacts with the active site of the protease (covalently or non-covalently) with a chemical ‘warhead’ targeting the catalytic mechanism. The importance of this methodology to drug discovery is exemplified by the identification of sitagliptin by Merck36-38 (BOX 4).
Box 4. Case study: the development of sitagliptin.
The stabilization in blood of glucagon-like peptide 1, a key glucoregulatory hormone, by the inhibition of its inactivating protease dipeptidyl peptidase 4 (DPP4) is a validated approach for the treatment of type 2 diabetes. Development of a DPP4 inhibitor at Merck began in 1999 after the company in-licensed threo-isoleucyl thiazolidide and allo-isoleucyl thiazolidide leads that were based on substrate specificity studies with peptidomimetics that simulated substrate cleavage sequences attached to a reactive electrophilic warhead112. Off-target inhibition of related DPPs led to refocusing, through structure–activity relationship studies, on screening leads that identified the highly selective β-amino acid piperazine series, and ultimately the pharmacologically more potent and selective triazolopiperazine series113. Optimization led to the development of sitagliptin (Januvia; Merck), a highly selective DPP4 inhibitor for the treatment of type 2 diabetes, which was approved by the US Food and Drug Administration in 2006.

Identification of subsite preference
The traditional procedures for determining protease subsite preference are positional scanning approaches to define the best fit into the protease active site cleft39. This can be done by using combinatorial substrate libraries or combinatorial inhibitor libraries, which generally consist of natural amino acids (peptides) linked to a fluorogenic reporter, or more recently to peptidic derivatives of drug-like fragments40, which explore the S1–S4 specificity pockets of the protease catalytic cleft40-42 (FIG. 3). Most positional scanning techniques are restricted to the unprimed side of the catalytic cleft, and exploration of the entire cleft requires methods that are chemically less tractable. This is generally accomplished using fluorogenic libraries that contain internal fluorophores equipped with an appropriate quencher and designed to span the length of the cleft43.
Figure 3. Probing protease active sites.

In standard protease notation, substrates are cleaved between the P1 (amino-terminal) and P1′ (carboxy-terminal) residues, with P1,2,3 … n and P’1,2,3 … n residues increasing towards the N terminus of the protein and towards the C terminus respectively. The corresponding pockets on the protease that accept the substrate side chain are designated S or S’ accordingly128. The schematic representation of the active site cleft of proteases shows the prime S1′-S3′ pockets accommodating specific amino acid side chains identified on the C-terminal side of the scissile bond and unprimed sites S1–S4 identified on the N-terminal side of the scissile bond (indicated by scissors). Shown also are the types of chemical moiety that can be used to investigate protease specificity either as individual substrates or as combinatorial libraries. The application of positional scanning substrate libraries allows exploration of the individual pockets of the protease by synthesis of a set of sublibraries. These generally consist of natural amino acids linked to a fluorogenic reporter, or, for a wider exploration of the catalytic cleft, of peptides that contain fluorophores and a quencher. The cleavage preference for each sublibrary is determined from fluorescence yields generated when the fluorophore is separated from the quencher by cleavage of the peptide bond.
So far, these methods have determined the intrinsic subsite preferences of more than 150 individual proteases44-48. They have led to the identification of active substrates and the subsequent optimization of inhibitors for cruzaine (also known as cruzipain)49, a cysteine protease of Trypanosoma cruzi that is the causative agent of Chagas disease. Also, inhibitors of cathepsin S50, which is implicated in autoimmune diseases, and inhibitors of the hepatitis C virus protease51 have been identified. Positional scanning methods and complete platforms for inhibitor screening were also developed for screening MMPs. Such screening resulted in peptide inhibitors for MMPs with in vivo protective effects against endotoxic shock52-54.
An orthogonal method of substrate screening using mixture-based oriented peptide libraries allows separate identification of the unprimed and primed subsite preferences of the catalytic cleft without the need for fluorogenic or colourimetric reporters55. This has delivered specificity information on several MMPs and on anthrax lethal factor56,57.
An interesting reverse method related to the classic substrate libraries was recently developed by Sanofi–Aventis, in which the reactive electrophilic group of previously developed drug candidates for cathepsin K and cathepsin S was replaced with a cleavable peptide bond attached to a fluorophore–quencher pair. In this way a relatively selective substrate for cathepsin K was developed, which may become useful for cell-based screening of the protease58. Substrate phage techniques, in which a randomized peptide sequence is assessed for cleavage by a protease, also remain popular for determining the substrate cleft subsite preferences, although validation of the cleavage site can be laborious59-61.
It is important to note that these methods have yet to deliver reliable information on natural protease substrates1. There are now hundreds of structures that show how inhibitors, both natural and synthetic, bind to protease active sites, but there are still surprisingly few structures of proteases with their natural substrate bound. Like other enzyme families, such as kinases, one cannot predict natural substrates from intrinsic subsite occupancy, and only in some cases has this been successfully proved (caspases being the best example)62. Consequently, the only way to determine the identity of natural substrates is by directly determining the products of proteolysis ex vivo.
Finally, a recent burst of proteomic methods has added technological possibilities to the discovery of natural substrates and extended subsite preferences, related in scope to substrate phage techniques, as reviewed in REF. 63. Currently, these proteomic methods are only capable of delivering small subsets of the breadth of natural protease substrates and of exploring a limited amount of the combinatorial complexity that constitutes a protease cleft’s subsites, but we anticipate substantial advances in the next few years.
The discovery of inhibitors, informed by the elucidation of intrinsic subsite preferences, is often supplemented by methods that make no assumption and require no knowledge of protease substrate specificity, such as high-throughput screening (HTS) or fragment-based screening. These methods may even have an advantage because such screens are not restricted to identifying compounds that target the active site.
Screening methods that require no knowledge of substrate specificity
HTS allows the identification of protease inhibitors by screening compound libraries ranging from hundreds to millions of compounds in size. The main challenge associated with HTS is obtaining hits with high selectivity towards the target and with desired bioavailability and safety profiles. Thus, traditional lead optimization must accompany HTS methods to address specificity issues (see BOX 5 for case study). notwithstanding the classic problem that the number of compounds usually used in HTS (1 × 105 to 1 × 107) cannot cover even a small part of chemical space, which is estimated at 1 × 1040 to 1 × 10128 for all possible small-molecule structure variations, successes in HTS for protease inhibitors have been reported. An example is the identification of specific inhibitors of the Plasmodium falciparum methionine aminopeptidase 1b from a library of 175,000 compounds64.
Box 5. Case study: deubiquitylating enzymes.
About 100 deubiquitylating enzymes (DUBs) have been identified in humans, comprising around 20% of proteases in the proteome. These enzymes remove ubiquitin from specific targets and their target recognition profiles overlap20. The DUB ubiquitin carboxy-terminal hydrolase isozyme L1 (UCHL1) is involved in neuropathic pain, Parkinson’s disease and the progression of non-neuronal tumours. High-throughput screening of 42,000 drug-like compounds for their ability to inhibit this protease led to the identification of several hits, including eight promising isatin oximes114. Further optimization of these isatin oximes led to the discovery of the O-acyl oxime reversible competitive inhibitors for UCHL1 with higher specificity over its close homologue UCHL3. Investigation of lung tumour cell lines with these compounds suggested that UCHL1 has antiproliferative activity and that its expression is a result of the response to tumour growth115. The figure depicts the inhibitors and the half-maximal inhibitory concentration (IC50) for each UCHL1 and UCHL3, and the value for the relative potency of inhibition for the homologues.

Fragment-based screening aims to identify low molecular mass fragments that interact — typically weakly — with the target and then guide their optimization into more potent inhibitors, usually by harnessing structural information. This methodology for lead discovery, which uses nuclear magnetic resonance spectroscopy, mass spectrometry or X-ray crystallography, has been developed since the 1990s and has recently attracted considerable interest resulting from the growing number of compounds identified as viable leads65.
The basic concept of fragment-based lead discovery using nuclear magnetic resonance spectroscopy is to screen libraries of small molecules (fragments of 100–300 daltons) and identify at least two binding sites on the target that bind fragments that can be linked together to generate a lead66. Ideally, each fragment will bind to the target protein with dissociation constant (Kd) values in the high micromolar to millimolar range, and the molecule assembled by linking the fragments will have affinity for the target of substantially less than 1 μM. By focusing on the interactions of fragments with several hot spots on the surface of the enzyme, this method leads to the identification of molecules with binding characteristics similar to natural inhibitors that operate by a two-site-binding paradigm, resulting in selective and tight binding. For example, the natural protease inhibitor XIAP uses the two-site-binding paradigm to generate tight and specific inhibitory domains for caspase 3, caspase 7 and caspase 9 (REF. 67), and the medicinal leech protein hirudin uses the strategy to specifically and tightly inhibit thrombin68.
Several protease inhibitor candidates have been developed using this technology. For example, compound ABT-518 (Abbott laboratories) was developed as an inhibitor of MMPs and is currently in Phase I clinical trials69. Other promising leads identified by this technology include inhibitors of methionine aminopeptidase 2 (Abbott laboratories)70, anthrax lethal factor (Burnham Institute)71 and caspase 3 (Sunesis Pharmaceuticals)72.
Reversible versus irreversible inhibitors
Drug leads identified on the basis of subsite specificity or from HTS or fragment-based screens often act as irreversible inhibitors, as they covalently bind to their targets. For aspartyl and metalloproteinases, which do not form stable covalent bonds during scissile peptide bond processing, the compounds of choice are reversible inhibitors possessing strong ligand properties that are able to tightly interact with the active site. However, it has proved difficult to engineer inhibitors of cysteine, serine or threonine proteases that do not contain an irreversible mechanism-based electrophile. This could be a matter of trial and error, as irreversible inhibitors are much more effective and easier to make.
In the pharmaceutical industry, the view is that drug binding should be reversible, owing to concerns about the antigenicity of covalently modified proteins, and the consequences of suboptimal specificity, especially over periods of extended therapy. Leads with irreversible binding mechanisms are usually not optimized as a result of concerns about putative side effects1,38,73. However, it should be noted that not all covalent binders are irreversible. For example, bortezomib (Velcade; Millennium Pharmaceuticals), which is used in the treatment of multiple myeloma27, covalently binds to the catalytic threonine site of the 26S proteasome with high specificity and affinity, but despite the formation of a covalent adduct the bond is kinetically reversible.
There are also cases in which an irreversible inhibitor can be used therapeutically. For example, one may imagine that irreversible inhibitors of the T. cruzi protease cruzaine would show severe side effects because they also target host cysteine proteases. However, these inhibitors show efficacy and low toxicity because they become concentrated within the parasite, and because antiparasite therapy is in principle a short-term treatment74. Thus, an irreversible inhibitor can be extremely effective when used in an appropriate therapeutic context.
Allosteric and exosite inhibitors — next-generation protease inhibitors?
Failures to develop highly selective active-site targeted inhibitors for some groups of proteases due to overlapping activity or lack of specificity has begun to shift interest to exosite and allosteric interactions. The significance of these regions can be readily appreciated for deubiquitylating and desumoylating proteases, which process their natural substrates with kinetic rates — catalytic constant (kcat) or Michaelis constant (Km) — in the range of 1 × 105 to 1 × 106 M−1 s−1, whereas substrates based only on the linear epitope recognized in the active centre are cleaved with rates that are 5–6 orders of magnitude less efficient75-78. The importance of exosites is exemplified by the thrombin inhibitor desirudin (TABLE 1; FIG. 2), and by the discovery of a ligand binding pocket distinct from the catalytic centre in the aspartic acid protease β-site of amyloid precursor protein cleaving enzyme (BACE) (involved in Alzheimer’s disease). Specific peptides, initially identified from combinatorial phage peptide libraries, bind to this exosite even in the presence of saturating concentrations of active-site directed inhibitors. Binding of peptides to the BACE exosite leads to a concentration-dependent inhibition of proteolysis for amyloid precursor protein-related, protein-based substrates of BACE79.
The impact of allosteric mechanisms in delivering tight and specific inhibition is also exemplified by approaches using dimerization disruptors to inhibit the protease of human Kaposi’s sarcoma-associated herpesvirus80, and designed ankyrin repeats (DARpins) as selective caspase 2 inhibitors81. Caspases, especially those involved in apoptosis, have notoriously overlapping substrate specificity and therefore the development of specific inhibitors based on common strategies has been difficult, if not impossible82. However, screening with protein-based recombinant DARpin libraries produced an extremely selective and potent inhibitor of caspase 2. The caspase 2-specific DARpin was discovered to target a region that connected with the active site and influenced the catalytic conformation of the protease81. Although it would be difficult to render a small molecule from the DARpin, this showed that specificity can be achieved with allosteric inhibitors. One could envisage using this strategy for the development of specific inhibitors for other classes of proteases that have tightly overlapping substrate specificities such as the cathepsins, MMPs, DUBs or small ubiquitin-like modifier proteases.
Biologicals — natural inhibitors and antibodies
Another approach that does not rely on active-site directed small molecules is the use of biological (protein-derived) agents. Although it may be difficult to develop biological agents for intracellular proteases, there is a clear opportunity for secreted or cell surface proteases. After all, the proteases of the coagulation, complement and inflammatory pathways are naturally regulated by proteins of the serpin family83. Although abandoned in the past due to delivery problems, the serpin family member antitrypsin is regaining popularity as a therapeutic inhibitor of its target protease elastase in the control of cystic fibrosis84.
However, natural inhibitors of proteases are often characterized by broad selectivity and they can inhibit multiple related proteases. Protein engineering efforts using natural inhibitors as a starting point have yielded selective inhibitors. As an example, the classic thrombin inhibitor hirudin, initially isolated from the medicinal leech, led the way to selective thrombin antagonists. Another example is ecotin, a natural Escherichia coli protein that can inhibit multiple trypsin-fold proteases and has been engineered, using a library display approach, to create a potent and selective inhibitor of plasma kallikrein85. The Kunitz domain, present in many natural protease inhibitors, has also been a focus of protein engineering efforts to generate specific protease inhibitors86. In fact, a Kunitz domain-based inhibitor of plasma kallikrein, ecallantide (Kalbitor; Dyax) has recently been approved for the treatment of hereditary angioedema87. However, as is the case for many scaffold proteins of non-human origin, there are concerns about their immunogenicity.
An alternative approach to engineering natural macromolecular protease inhibitors is the generation of antibody-based protease inhibitors. Several highly potent and selective protease inhibitors have been generated through classic hybridoma-based methods88 as well as screening of antibody-display libraries89,90. The exquisite specificity of antibodies in principle fulfils the search for inhibitors capable of distinguishing between closely related protease family members. Furthermore, the antibody approach offers a unique promise of being able to probe regions of the protease that are not restricted to the natural inhibitor binding site (that is, the active-site region) and thereby may facilitate the generation of allosteric or exosite inhibitors, as shown for coagulation factor VII91.
The macromolecule approach of developing protease inhibitors has the distinct advantage over the small-molecule approach in that the former can target larger surfaces on the protease and may yield more selective inhibitors. However, these two approaches must not necessarily be at odds with one another. Structural studies of a protein-based inhibitor in complex with its specific protease could provide insights for the rational design of small-molecule inhibitors that mimic the binding interactions in terms of overall chemical architecture.
Conclusions and future directions
As described above, conventional drug discovery strategies targeting proteases have proved useful, at least for the limited number of successful protease inhibitors currently on the market. We have discussed reasons for the limited success, based primarily on conventional approaches that frequently limit the achievable specificity required for successful therapy. A simple formula to decide whether a drug lead is worth pursuing would be desirable; for example, a selectivity ratio in which a paralogue is inhibited at less than 1% compared with the main target. However, in reality this may be difficult to achieve, with the result that decisions are made empirically (BOX 6). Related to this consideration is the realization that protease orthologues in animal models do not always have the same specificity as the human enzyme, thus questioning the validity of such animal models. The need to develop approaches for understanding human proteases in their natural context is a priority that will probably dominate protease inhibitor research, if not most drug research, in the future.
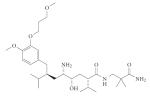
Box 6. Aminopeptidases: targets for antimalarials.
An interesting challenge is posed by drug development for proteases with overlapping substrate specificity, originating from two different organisms, with the aim of inhibiting only one. Such scenarios are encountered in parasite or pathogen infections in humans. An example for this is provided by the aminopeptidases (APNs). The malaria parasite Plasmodium falciparum produces eight different APNs — four methionine APNs, two neutral and one prolyl and one aspartyl — some of which are orthologues of human leucine APN (LAP, class M17) and alanine APN (CD13, class M1)115. These parasite APNs generate a pool of free amino acids through digestion of the peptides formed by other proteases from human haemoglobin in the digestive vacuole of the malaria parasite.
Some substrate specificity studies using fluorogenic substrates or substrate-based inhibitors (phosphonates) suggest substantial specificity overlap of both human and malarial APNs116,117. Phosphinate inhibitors of mammalian porcine kidney LAP have a similar inhibitory potency towards the P. falciparum orthologue (inhibition constant (Ki) = 13.2 nM and Ki = 66.0 nM towards malaria LAP (PfLAP) and mammalian LAP (pkLAP) respectively). But in a Plasmodium chabaudi murine malaria model, these inhibitors reduced the parasite burden by 92% without any serious toxicity118. These data show that in such circumstances, the degree of specificity and selectivity is not as crucial. The key factor seems to be the delivery mechanism responsible for drug distribution in the target cells118. The chemical structures represent the most potent phosphinate dipeptide analogue inhibitors directed against the P. falciparum M17 leucine APN as reported in REF. 118.

With regard to the broad range of technologies that are now available for protease inhibitor design, the question arises as to which delivers the best hits. Targeting the active site using a combination of specificity-based knowledge and structure-based design is a tried and proven strategy. However, the burst in technologies using combinatorial libraries, HTS or fragment-based screening began only in the early 1990s. Although only a minor fraction of approved drugs have been developed with these methods, this is not surprising considering the time necessary for the development of the technology and the subsequent application, and so the question of which strategy might be best remains open.
In the quest to optimize the specificity of protease inhibitors, alternative targets to the active site should also be considered. So far, no allosteric protease inhibitors or inhibitors that target the exosite have reached advanced development. Highly successful protease inhibitors all target the active site, so why risk pursuing allosteric regulators? One important point to consider is that active-site directed inhibitors are inherently competitive with endogenous substrates. Most proteases in vivo are thought to operate in an environment in which they are substrate saturated, and this will slow down and/or weaken competitive inhibitors by the factor of 1 + S/ΣKm, in which ΣKm is the sum of all values for all the substrates in a cell. In addition to the perennial problems of tissue penetration and pharmacokinetic considerations, this concept — backed up by a recent publication on HIV protease inhibitors that operate allosterically92 — could explain why the inhibitory concentration of a protease inhibitor drug in vivo is always several orders larger than in a purified system. By contrast, allosteric or exosite inhibitors need not be affected by substrate concentration.
There is also concern that an allosteric or exosite inhibitor connected to an active-site directed moiety may generate highly specific binding, but will result in an extremely large molecule that will not be viable for commercial inhibitor development. However, as pointed out in FIG. 2, not all exosites or allosteric sites require large binding surfaces, and the proof-of-principle already exists in the form of the thrombin inhibitors based on the exosite binder desirudin. A proof-of-concept application of a small-molecule allosteric inhibitor strategy was reported by Sunesis Pharmaceuticals, who developed low molecular mass inactivators of caspase 3 and caspase 7 using a fragment-based screening technology. Among a selection of molecules were ones that targeted not the active site, but a region at the interface of the caspase dimer, essentially freezing the caspases in their catalytically inactive zymogen conformation72. For technical reasons based on the low affinity covalent tethering method required for the identification of hits, it is challenging to develop these particular compounds directly into drugs. nevertheless, the message is clear: allosteric regulation by small molecules is now a high priority approach to protease inhibition. The evidence that currently available protease inhibitor drugs are highly specific is quite limited, and specificity is a major issue in active-site directed protease development programmes. Therefore, in our opinion, to achieve high specificity and selectivity one must avoid the active site. However, development has been slow in this area, and given the thermodynamic hot spot that defines protease active sites, unconventional approaches are required to target away from the active site93.
Importantly, these considerations also apply to several other potential drug targets that can be regulated by allosteric interactions. Indeed, the hugely successful kinase inhibitor imatinib (Gleevec; novartis) has been shown to operate by a predominantly allosteric mechanism because it does not directly bind to the ATP binding site94, and allosteric modulators of G protein-coupled receptors are emergent leads for compounds that provide high selectivity for this group of therapeutically tractable proteins95.
Given the wealth of mechanistic and structural data on proteases, as well as new fragment screening approaches, we anticipate that strategies to target the allosteric sites of proteases will soon emerge and that research and development in this area is likely to be extensive and profitable. Readers interested in learning more about allosteric inhibitors are directed to an excellent review in REF. 96. Naturally, an equally important part of protease inhibitor therapy, common to all drugs, is to achieve adequate absorption, distribution, metabolism, excretion and toxicology parameters, and only a skilful combination of these parameters with inhibitor selectivity will provide strong drug candidates.
Finally, although small molecules are still the most popular approach to protease inhibitor therapy, the opportunity of using biologicals is increasingly being realized. Future biotechnology drugs in protease therapy may include allosteric-site interactors, biologicals based on antibodies and highly selective natural protease inhibitors, and even proteases themselves (BOX 7).
Box 7. Proteases as therapeutics.
The concept of using proteases as therapeutic agents is not new. Proteases are used in wound debridement (collagenase); for defibrinating wounds in hospitals; for dissolving membranes in diphtheria; or for swelling, for fever and for adhesions reduction after surgery (papain); to reduce ruptured spinal discs (chymopapain); in clot dissolving; and recently for the management of acute ischaemic stroke in adults (recombinant tissue plasminogen activator, Activase; Genentech/Boehringer Ingelheim)119; and in clotting factor replacement (recombinant factor VIIa, NN-1731; Novo Nordisk)120. However, there are currently highly interesting programmes in academic institutions and in biotechnology companies to engineer proteases for specific therapeutic targets. Pioneered by the concept of making inhibitor-resistant versions of tissue plasminogen activator and tissue factor-independent versions of coagulation factor VIIa, the concept of evolving proteases for specific functions is increasingly popular. Evolved proteases promise at least one major advantage over other biologicals such as antibodies. They are catalytic and can therefore be used in smaller amounts, resulting in potentially high efficacy.
Nascent steps in this direction have been taken in two studies in which the substrate specificity and the catalytic efficiency of proteases was altered using Escherichia coli surface endopeptidase OmpT as a scaffold121,122. In these studies, a panel of proteases with new substrate selectivities in the P1 and P1′ positions was generated by screening a library of mutated proteases against specific substrates. An analogous method with similar aims in generating proteases of novel specificity for the treatment of inflammatory and oncogenic diseases is being undertaken by Catalyst Biosciences. Here, the protease platform is oriented towards evolving the protease substrate specificity of a protease scaffold (Alterase) towards the degradation of specific proteins that promote diseases123.
Acknowledgements
The Salvesen laboratory is supported by National Institutes of Health grants CA69381, RR20843 and NS61758, and by the Human Frontier Science Program grant RGP0024. The Drag laboratory is supported by the Foundation for Polish Science and the State for Scientific Research Grant N N401 042838 in Poland. The authors would like to thank E. Madison, B. Turk and the members of their laboratories for helpful discussions.
Glossary
- Active site
Region in the enzyme in which the primary substrate binding site is localized and catalysis occurs. Usually the active site is on the surface of the enzyme and is made up of pockets defined by protein surfaces, which are responsible for the specific binding of the substrate amino acid residues next to the scissile bond.
- Allosteric site
Region of the enzyme that does not participate directly in substrate recognition and processing. Through the ability to interact with specific modulators (proteins or small molecules) the allosteric site enhances or inhibits substrate to product transition through conformational changes in the enzyme.
- Exosite
Region of the enzyme distant from the active site that is responsible for specific substrate–enzyme interactions. Exosite interactions influence the rates of catalysis, and sometimes of substrate specificity, of a given protease. Classic examples are the recognition of fibrinogen by thrombin, ubiquitin recognition by ubiquitin-specific peptidases (deubiquitylating enzymes or DUBs), and small ubiquitin-like modifier (SUMO) recognition by SUMO-specific proteases (SENPs).
Footnotes
Competing interests statement The authors declare no competing financial interests.
DATABASES MEROPS: http://merops.sanger.ac.uk/
UniProtKB: http://www.uniprot.org
FURTHER INFORMATION salvesen laboratory homepage: http://www.sanfordburnham.org/labs/Salvesen/index.html
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Contributor Information
Marcin Drag, Program in Apoptosis and Cell Death Research, Burnham Institute for Medical Research, La Jolla, California 92037, USA.; Division of Medicinal Chemistry and Microbiology, Faculty of Chemistry, Wroclaw University of Technology, Wybrzeze Wyspianskiego 27, 50-370 Wroclaw, Poland..
Guy S. Salvesen, Program in Apoptosis and Cell Death Research, Burnham Institute for Medical Research, La Jolla, California 92037, USA.
References
- 1.Turk B. Targeting proteases: successes, failures and future prospects. Nature Rev. Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 2.Smith CG, Vane JR. The discovery of captopril. FASEB J. 2003;17:788–789. doi: 10.1096/fj.03-0093life. [DOI] [PubMed] [Google Scholar]
- 3.Flexner C, Bate G, Kirkpatrick P. Tipranavir. Nature Rev. Drug Discov. 2005;4:955–956. doi: 10.1038/nrd1907. [DOI] [PubMed] [Google Scholar]
- 4.Melnikova I. The anticoagulants market. Nature Rev. Drug Discov. 2009;8:353–354. doi: 10.1038/nrd2851. [DOI] [PubMed] [Google Scholar]
- 5.Rawlings ND, Morton FR, Kok CY, Kong J, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2008;36:D320–D325. doi: 10.1093/nar/gkm954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Timmer JC, et al. Structural and kinetic determinants of protease substrates. Nature Struct. Mol. Biol. 2009;16:1101–1108. doi: 10.1038/nsmb.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davie EW, Fujikawa K. Basic mechanisms in blood coagulation. Annu. Rev. Biochem. 1975;44:799–829. doi: 10.1146/annurev.bi.44.070175.004055. [DOI] [PubMed] [Google Scholar]
- 8.Davie EW. A brief historical review of the waterfall/cascade of blood coagulation. J. Biol. Chem. 2003;278:50819–50832. doi: 10.1074/jbc.X300009200. [DOI] [PubMed] [Google Scholar]
- 9.Peden JC, Jr, Mc Farland JA. Use of the plasma thrombin time to assess the adequacy of in vivo neutralization of heparin: comparative studies following operations employing extracorporeal circulation. Blood. 1959;14:1230–1236. [PubMed] [Google Scholar]
- 10.Bennett B, Ratnoff OD. The normal coagulation mechanism. Med. Clin. North Am. 1972;56:95–104. doi: 10.1016/s0025-7125(16)32425-7. [DOI] [PubMed] [Google Scholar]
- 11.Turk B, Turk D, Salvesen GS. Regulating cysteine protease activity: essential role of protease inhibitors as guardians and regulators. Curr. Pharm. Des. 2002;8:1623–1637. doi: 10.2174/1381612023394124. [DOI] [PubMed] [Google Scholar]
- 12.Van den Steen PE, et al. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9) Crit. Rev. Biochem. Mol. Biol. 2002;37:375–536. doi: 10.1080/10409230290771546. [DOI] [PubMed] [Google Scholar]
- 13.McQuibban GA, et al. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289:1202–1206. doi: 10.1126/science.289.5482.1202. [DOI] [PubMed] [Google Scholar]
- 14.Overall CM, Dean RA. Degradomics: systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer Metastasis Rev. 2006;25:69–75. doi: 10.1007/s10555-006-7890-0. [DOI] [PubMed] [Google Scholar]
- 15.Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus-independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J. Cell Biol. 2009;185:11–19. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cauwe B, Van den Steen PE, Opdenakker G. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit. Rev. Biochem. Mol. Biol. 2007;42:113–185. doi: 10.1080/10409230701340019. [DOI] [PubMed] [Google Scholar]
- 17.Wolf K, Muller R, Borgmann S, Brocker EB, Friedl P. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood. 2003;102:3262–3269. doi: 10.1182/blood-2002-12-3791. [DOI] [PubMed] [Google Scholar]
- 18.Uhlmann F, Wernic D, Poupart MA, Koonin EV, Nasmyth M. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell. 2000;103:375–386. doi: 10.1016/s0092-8674(00)00130-6. [DOI] [PubMed] [Google Scholar]
- 19.Kraft C, Gmachl M, Peters JM. Methods to measure ubiquitin-dependent proteolysis mediated by the anaphase-promoting complex. Methods. 2006;38:39–51. doi: 10.1016/j.ymeth.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 20.Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishnaswamy S. Exosite-driven substrate specificity and function in coagulation. J. Thromb. Haemost. 2005;3:54–67. doi: 10.1111/j.1538-7836.2004.01021.x. [DOI] [PubMed] [Google Scholar]
- 22.Rijken DC, Lijnen HR. New insights into the molecular mechanisms of the fibrinolytic system. J. Thromb. Haemost. 2009;7:4–13. doi: 10.1111/j.1538-7836.2008.03220.x. [DOI] [PubMed] [Google Scholar]
- 23.Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nature Rev. Drug Discov. 2007;6:480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- 24.Dusing R, Sellers F. ACE inhibitors, angiotensin receptor blockers and direct renin inhibitors in combination: a review of their role after the ONTARGET trial. Curr. Med. Res. Opin. 2009;25:2287–2301. doi: 10.1185/03007990903152045. [DOI] [PubMed] [Google Scholar]
- 25.Maisey HC, Doran KS, Nizet V. Recent advances in understanding the molecular basis of group B Streptococcus virulence. Expert Rev. Mol. Med. 2008;10:e27. doi: 10.1017/S1462399408000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imamura T, Potempa J, Travis J. Activation of the kallikrein–kinin system and release of new kinins through alternative cleavage of kininogens by microbial and human cell proteinases. Biol. Chem. 2004;385:989–996. doi: 10.1515/BC.2004.129. [DOI] [PubMed] [Google Scholar]
- 27.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Invest. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 28.Green DR, Evan GI. A matter of life and death. Cancer Cell. 2002;1:19–30. doi: 10.1016/s1535-6108(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 29.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nature Rev. Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 30.Schimmer AD, et al. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell. 2004;5:25–35. doi: 10.1016/s1535-6108(03)00332-5. [DOI] [PubMed] [Google Scholar]
- 31.Troy CM, Salvesen GS. Caspases on the brain. J. Neurosci. Res. 2002;69:145–150. doi: 10.1002/jnr.10294. [DOI] [PubMed] [Google Scholar]
- 32.Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nature Rev. Cancer. 2007;7:800–808. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- 33.Lu X, Lu D, Scully M, Kakkar V. ADAM proteins — therapeutic potential in cancer. Curr. Cancer Drug Targets. 2008;8:720–732. doi: 10.2174/156800908786733478. [DOI] [PubMed] [Google Scholar]
- 34.Palermo C, Joyce JA. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol. Sci. 2008;29:22–28. doi: 10.1016/j.tips.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 35.Mattos C, et al. Multiple solvent crystal structures: probing binding sites, plasticity and hydration. J. Mol. Biol. 2006;357:1471–1482. doi: 10.1016/j.jmb.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 36.Kim D, et al. (2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-α]pyrazin -7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2005;48:141–151. doi: 10.1021/jm0493156. [DOI] [PubMed] [Google Scholar]
- 37.Kim D, et al. Discovery of potent and selective dipeptidyl peptidase IV inhibitors derived from β-aminoamides bearing subsituted triazolopiperazines. J. Med. Chem. 2008;51:589–602. doi: 10.1021/jm070330v. [DOI] [PubMed] [Google Scholar]
- 38.Leiting B, et al. Catalytic properties and inhibition of proline-specific dipeptidyl peptidases, II, IV and VII. Biochem. J. 2003;371:525–532. doi: 10.1042/BJ20021643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rano TA, et al. A combinatorial approach for determining protease specificities: application to interleukin-1β converting enzyme (ICE) Chem. Biol. 1997;4:149–155. doi: 10.1016/s1074-5521(97)90258-1. [DOI] [PubMed] [Google Scholar]
- 40.Wood WJ, Patterson AW, Tsuruoka H, Jain RK, Ellman JA. Substrate activity screening: a fragment-based method for the rapid identification of nonpeptidic protease inhibitors. J. Am. Chem. Soc. 2005;127:15521–15527. doi: 10.1021/ja0547230. [DOI] [PubMed] [Google Scholar]
- 41.Thompson LA, Ellman JA. Synthesis and applications of small molecule libraries. Chem. Rev. 1996;96:555–600. doi: 10.1021/cr9402081. [DOI] [PubMed] [Google Scholar]
- 42.Schneider EL, Craik CS. Positional scanning synthetic combinatorial libraries for substrate profiling. Methods Mol. Biol. 2009;539:59–78. doi: 10.1007/978-1-60327-003-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, et al. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J. Biol. Chem. 2005;280:28766–28774. doi: 10.1074/jbc.M500588200. [DOI] [PubMed] [Google Scholar]
- 44.Backes BJ, Harris JL, Leonetti F, Craik CS, Ellman JA. Synthesis of positional-scanning libraries of fluorogenic peptide substrates to define the extended substrate specificity of plasmin and thrombin. Nature Biotechnol. 2000;18:187–193. doi: 10.1038/72642. [DOI] [PubMed] [Google Scholar]
- 45.Harris JL, et al. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc. Natl Acad. Sci. USA. 2000;97:7754–7759. doi: 10.1073/pnas.140132697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drag M, et al. Positional-scanning fluorigenic substrate libraries reveal unexpected specificity determinants of DUBs (deubiquitinating enzymes) Biochem. J. 2008;415:367–375. doi: 10.1042/BJ20080779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snipas SJ, Drag M, Stennicke HR, Salvesen GS. Activation mechanism and substrate specificity of the Drosophila initiator caspase DRONC. Cell Death Differ. 2008;15:938–945. doi: 10.1038/cdd.2008.23. [DOI] [PubMed] [Google Scholar]
- 48.Choe Y, et al. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J. Biol. Chem. 2006;281:12824–12832. doi: 10.1074/jbc.M513331200. [DOI] [PubMed] [Google Scholar]
- 49.Brak K, Doyle PS, McKerrow JH, Ellman JA. Identification of a new class of nonpeptidic inhibitors of cruzain. J. Am. Chem. Soc. 2008;130:6404–6410. doi: 10.1021/ja710254m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inagaki H, et al. Characterization and optimization of selective, nonpeptidic inhibitors of cathepsin S with an unprecedented binding mode. J. Med. Chem. 2007;50:2693–2699. doi: 10.1021/jm070111+. [DOI] [PubMed] [Google Scholar]
- 51.Melnikova I. Hepatitis C therapies. Nature Rev. Drug Discov. 2008;7:799–800. [Google Scholar]
- 52.Hu J, Van den Steen PE, Dillen C, Opdenakker G. Targeting neutrophil collagenase/matrix metalloproteinase-8 and gelatinase B/matrix metalloproteinase-9 with a peptidomimetic inhibitor protects against endotoxin shock. Biochem. Pharmacol. 2005;70:535–544. doi: 10.1016/j.bcp.2005.04.047. [DOI] [PubMed] [Google Scholar]
- 53.Hu J, Fiten P, Van den Steen PE, Chaltin P, Opdenakker G. Simulation of evolution-selected propeptide by high-throughput selection of a peptidomimetic inhibitor on a capillary DNA sequencer platform. Anal. Chem. 2005;77:2116–2124. doi: 10.1021/ac048631p. [DOI] [PubMed] [Google Scholar]
- 54.Piccard H, et al. “Reverse degradomics”, monitoring of proteolytic trimming by multi-CE and confocal detection of fluorescent substrates and reaction products. Electrophoresis. 2009;30:2366–2377. doi: 10.1002/elps.200800698. [DOI] [PubMed] [Google Scholar]
- 55.Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nature Biotechnol. 2001;19:661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 56.Turk BE, et al. MMP-20 is predominately a tooth-specific enzyme with a deep catalytic pocket that hydrolyzes type V collagen. Biochemistry. 2006;45:3863–3874. doi: 10.1021/bi052252o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turk BE, et al. The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nature Struct. Mol. Biol. 2004;11:60–66. doi: 10.1038/nsmb708. [DOI] [PubMed] [Google Scholar]
- 58.Watzke A, et al. Selective activity-based probes for cysteine cathepsins. Angew. Chem. Int. Edn Engl. 2008;47:406–409. doi: 10.1002/anie.200702811. [DOI] [PubMed] [Google Scholar]
- 59.Matthews DJ, Wells JA. Substrate phage: selection of protease substrates by monovalent phage display. Science. 1993;260:1113–1117. doi: 10.1126/science.8493554. [DOI] [PubMed] [Google Scholar]
- 60.Hansen M, et al. A urokinase-type plasminogen activator-inhibiting cyclic peptide with an unusual P2 residue and an extended protease binding surface demonstrates new modalities for enzyme inhibition. J. Biol. Chem. 2005;280:38424–38437. doi: 10.1074/jbc.M505933200. [DOI] [PubMed] [Google Scholar]
- 61.Scholle MD, et al. Mapping protease substrates by using a biotinylated phage substrate library. Chembiochem. 2006;7:834–838. doi: 10.1002/cbic.200500427. [DOI] [PubMed] [Google Scholar]
- 62.Timmer JC, Salvesen GS. Caspase substrates. Cell Death Differ. 2007;14:66–72. doi: 10.1038/sj.cdd.4402059. [DOI] [PubMed] [Google Scholar]
- 63.Impens F, et al. MS-driven protease substrate degradomics. Proteomics. 2010;10:1284–1296. doi: 10.1002/pmic.200900418. [DOI] [PubMed] [Google Scholar]
- 64.Chen X, et al. Inhibitors of Plasmodium falciparum methionine aminopeptidase 1b possess antimalarial activity. Proc. Natl Acad. Sci. USA. 2006;103:14548–14553. doi: 10.1073/pnas.0604101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pellecchia M, et al. Perspectives on NMR in drug discovery: a technique comes of age. Nature Rev. Drug Discov. 2008;7:738–745. doi: 10.1038/nrd2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Erlanson DA, et al. In situ assembly of enzyme inhibitors using extended tethering. Nature Biotechnol. 2003;21:308–314. doi: 10.1038/nbt786. [DOI] [PubMed] [Google Scholar]
- 67.Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rydel TJ, et al. The structure of a complex of recombinant hirudin and human α-thrombin. Science. 1990;249:277–280. doi: 10.1126/science.2374926. [DOI] [PubMed] [Google Scholar]
- 69.Wada CK. The evolution of the matrix metalloproteinase inhibitor drug discovery program at Abbott laboratories. Curr. Top. Med. Chem. 2004;4:1255–1267. doi: 10.2174/1568026043388015. [DOI] [PubMed] [Google Scholar]
- 70.Wang J, et al. Tumor suppression by a rationally designed reversible inhibitor of methionine amino-peptidase-2. Cancer Res. 2003;63:7861–7869. [PubMed] [Google Scholar]
- 71.Forino M, et al. Efficient synthetic inhibitors of anthrax lethal factor. Proc. Natl Acad. Sci. USA. 2005;102:9499–9504. doi: 10.1073/pnas.0502733102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hardy JA, Lam J, Nguyen JT, O’Brien T, Wells JA. Discovery of an allosteric site in the caspases. Proc. Natl Acad. Sci. USA. 2004;101:12461–12466. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Herman GA, Stein PP, Thornberry NA, Wagner JA. Dipeptidyl peptidase-4 inhibitors for the treatment of type 2 diabetes: focus on sitagliptin. Clin. Pharmacol. Ther. 2007;81:761–767. doi: 10.1038/sj.clpt.6100167. [DOI] [PubMed] [Google Scholar]
- 74.McKerrow JH, Engel JC, Caffrey CR. Cysteine protease inhibitors as chemotherapy for parasitic infections. Bioorg Med. Chem. 1999;7:639–644. doi: 10.1016/s0968-0896(99)00008-5. [DOI] [PubMed] [Google Scholar]
- 75.Renatus M, et al. Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure. 2006;14:1293–1302. doi: 10.1016/j.str.2006.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mikolajczyk J, et al. Small ubiquitin-related modifier (SUMO)-specific proteases: profiling the specificities and activities of human SENPs. J. Biol. Chem. 2007;282:26217–26224. doi: 10.1074/jbc.M702444200. [DOI] [PubMed] [Google Scholar]
- 77.Drag M, Mikolajczyk J, Krishnakumar IM, Huang Z, Salvesen GS. Activity profiling of human deSUMOylating enzymes (SENPs) with synthetic substrates suggests an unexpected specificity of two newly characterized members of the family. Biochem. J. 2008;409:461–469. doi: 10.1042/BJ20070940. [DOI] [PubMed] [Google Scholar]
- 78.Reyes-Turcu FE, et al. The ubiquitin binding domain ZnF UBP recognizes the C-terminal diglycine motif of unanchored ubiquitin. Cell. 2006;124:1197–1208. doi: 10.1016/j.cell.2006.02.038. [DOI] [PubMed] [Google Scholar]
- 79.Kornacker MG, et al. An inhibitor binding pocket distinct from the catalytic active site on human β-APP cleaving enzyme. Biochemistry. 2005;44:11567–11573. doi: 10.1021/bi050932l. [DOI] [PubMed] [Google Scholar]
- 80.Shahian T, et al. Inhibition of a viral enzyme by a small-molecule dimer disruptor. Nat. Chem. Biol. 2009;5:640–646. doi: 10.1038/nchembio.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schweizer A, et al. Inhibition of caspase-2 by a designed ankyrin repeat protein: specificity, structure, and inhibition mechanism. Structure. 2007;15:625–636. doi: 10.1016/j.str.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 82.Thornberry NA, et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 83.Silverman GA, et al. The serpins are an expanding superfamily of structurally similar but funtionally diverse proteins: evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem. 2001;276:33293–33296. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- 84.Brennan S. Revisiting α1-antitrypsin therapy in cystic fibrosis: can it still offer promise? Eur. Respir. J. 2007;29:229–230. doi: 10.1183/09031936.00159606. [DOI] [PubMed] [Google Scholar]
- 85.Stoop AA, Craik CS. Engineering of a macromolecular scaffold to develop specific protease inhibitors. Nature Biotechnol. 2003;21:1063–1068. doi: 10.1038/nbt860. [DOI] [PubMed] [Google Scholar]
- 86.Dennis MS, Herzka A, Lazarus RA. Potent and selective Kunitz domain inhibitors of plasma kallikrein designed by phage display. J. Biol. Chem. 1995;270:25411–25417. doi: 10.1074/jbc.270.43.25411. [DOI] [PubMed] [Google Scholar]
- 87.Levy JH, O’Donnell PS. The therapeutic potential of a kallikrein inhibitor for treating hereditary angioedema. Expert Opin. Investig. Drugs. 2006;15:1077–1090. doi: 10.1517/13543784.15.9.1077. [DOI] [PubMed] [Google Scholar]
- 88.Xuan JA, et al. Antibodies neutralizing hepsin protease activity do not impact cell growth but inhibit invasion of prostate and ovarian tumor cells in culture. Cancer Res. 2006;66:3611–3619. doi: 10.1158/0008-5472.CAN-05-2983. [DOI] [PubMed] [Google Scholar]
- 89.Sun J, Pons J, Craik CS. Potent and selective inhibition of membrane-type serine protease 1 by human single-chain antibodies. Biochemistry. 2003;42:892–900. doi: 10.1021/bi026878f. [DOI] [PubMed] [Google Scholar]
- 90.Devy L, et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009;69:1517–1526. doi: 10.1158/0008-5472.CAN-08-3255. [DOI] [PubMed] [Google Scholar]
- 91.Lazarus RA, Olivero AG, Eigenbrot C, Kirchhofer D. Inhibitors of tissue factor. Factor VIIa for anticoagulant therapy. Curr. Med. Chem. 2004;11:2275–2290. doi: 10.2174/0929867043364568. [DOI] [PubMed] [Google Scholar]
- 92.Chang MW, et al. Identification of broad-based HIV-1 protease inhibitors from combinatorial libraries. Biochem J. 2010;429:527–532. doi: 10.1042/BJ20091645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brenke R, et al. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics. 2009;25:621–627. doi: 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bogoyevitch MA, Fairlie DP. A new paradigm for protein kinase inhibition: blocking phosphorylation without directly targeting ATP binding. Drug Discov. Today. 2007;12:622–633. doi: 10.1016/j.drudis.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 95.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature Rev. Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 97.Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nature Med. 2002;8:1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 98.Girolami A, Randi ML, Gavasso S, Lombardi AM, Spiezia F. The occasional venous thromboses seen in patients with severe (homozygous) FXII deficiency are probably due to associated risk factors: a study of prevalence in 21 patients and review of the literature. J. Thromb. Thrombolysis. 2004;17:139–143. doi: 10.1023/B:THRO.0000037670.42776.cd. [DOI] [PubMed] [Google Scholar]
- 99.Baglia FA, Walsh PN. Thrombin-mediated feedback activation of factor XI on the activated platelet surface is preferred over contact activation by factor XIIa or factor XIa. J. Biol. Chem. 2000;275:20514–20519. doi: 10.1074/jbc.M000464200. [DOI] [PubMed] [Google Scholar]
- 100.Renné T, Gailani D. Role of factor XII in hemostasis and thrombosis: clinical implications. Expert Rev. Cardiovasc. Ther. 2007;5:733–741. doi: 10.1586/14779072.5.4.733. [DOI] [PubMed] [Google Scholar]
- 101.Duncan RC, Wijeyewickrema LC, Pike RN. The initiating proteases of the complement system: controlling the cleavage. Biochimie. 2008;90:387–395. doi: 10.1016/j.biochi.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 102.Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 2004;384:201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nature Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 104.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nature Rev. Mol. Cell Biol. 2007;5:405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 105.LeMosy EK, Tan YQ, Hashimoto C. Activation of a protease cascade involved in patterning the Drosophila embryo. Proc. Natl Acad. Sci. USA. 2001;98:5055–5060. doi: 10.1073/pnas.081026598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 107.Overall CM, Kleifeld O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br. J. Cancer. 2006;94:941–946. doi: 10.1038/sj.bjc.6603043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jensen C, Herold P, Brunner HR. Aliskiren: the first renin inhibitor for clinical treatment. Nature Rev. Drug Discov. 2008;7:399–410. doi: 10.1038/nrd2550. [DOI] [PubMed] [Google Scholar]
- 109.Szelke M, et al. Potent new inhibitors of human renin. Nature. 1982;299:555–557. doi: 10.1038/299555a0. [DOI] [PubMed] [Google Scholar]
- 110.Clozel JP, Fischli W. Discovery of remikiren as the first orally active renin inhibitor. Arzneimittelforschung. 1993;43:260–262. [PubMed] [Google Scholar]
- 111.Sureshkumar KK. Renin inhibition with aliskiren in hypertension: focus on aliskiren/hydrochlorothiazide combination therapy. Vasc. Health Risk Manag. 2008;4:1205–1220. doi: 10.2147/vhrm.s3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Thornberry NA, Weber AE. Discovery of Januvia (sitagliptin), a selective dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Curr. Top. Med. Chem. 2007;7:557–568. doi: 10.2174/156802607780091028. [DOI] [PubMed] [Google Scholar]
- 113.Weber AE. Dipeptidyl peptidase IV inhibitors for the treatment of diabetes. J. Med. Chem. 2004;47:4135–4141. doi: 10.1021/jm030628v. [DOI] [PubMed] [Google Scholar]
- 114.Liu Y, et al. Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem. Biol. 2003;10:837–846. doi: 10.1016/j.chembiol.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 115.Stack CM, et al. Characterization of the Plasmodium falciparum M17 leucyl aminopeptidase. A protease involved in amino acid regulation with potential for antimalarial drug development. J. Biol. Chem. 2007;282:2069–2080. doi: 10.1074/jbc.M609251200. [DOI] [PubMed] [Google Scholar]
- 116.Cunningham E, Drag M, Kafarski P, Bell A. Chemical target validation studies of aminopeptidase in malaria parasites using α-aminoalkylphosphonate and phosphonopeptide inhibitors. Antimicrob. Agents Chemother. 2008;52:3221–3228. doi: 10.1128/AAC.01327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Maric S, et al. The M17 leucine aminopeptidase of the malaria parasite Plasmodium falciparum: the importance of active site metal ions in the binding of substrates and inhibitors. Biochemistry. 2009;48:5435–5439. doi: 10.1021/bi9003638. [DOI] [PubMed] [Google Scholar]
- 118.Skinner-Adams TS, et al. Identification of phosphinate dipeptide analog inhibitors directed against the Plasmodium falciparum M17 leucine aminopeptidase as lead antimalarial compounds. J. Med. Chem. 2007;50:6024–6031. doi: 10.1021/jm070733v. [DOI] [PubMed] [Google Scholar]
- 119.Kuebler PS. Method of treating stroke with thrombolytic agent. 20080107641 US patent. 2007
- 120.Rojkjaer RG. Pharmaceutical composition comprising factor VII polypeptides and TAFI polypeptides. 20030118574 US patent. 2002
- 121.Varadarajan N, Georgiou G, Iverson BL. An engineered protease that cleaves specifically after sulfated tyrosine. Angew. Chem. Int. Ed Engl. 2008;47:7861–7863. doi: 10.1002/anie.200800736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Varadarajan N, Rodriguez S, Hwang BY, Georgiou G, Iverson BL. Highly active and selective endopeptidases with programmed substrate specificities. Nature Chem. Biol. 2008;4:290–294. doi: 10.1038/nchembio.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Madison EL, Thanos C, Ruggles SW, Coughlin S. Modified factor VII polypeptides and uses thereof. 20090098103 US patent. 2008
- 124.Tanaka F. Catalytic antibodies as designer proteases and esterases. Chem. Rev. 2002;102:4885–4906. doi: 10.1021/cr010180a. [DOI] [PubMed] [Google Scholar]
- 125.Reverter D, Lima CD. A basis for SUMO protease specificity provided by analysis of human Senp2 and a Senp2–SUMO complex. Structure. 2004;12:1519–1531. doi: 10.1016/j.str.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 126.Friedrich R, Steinmetzer T, Huber R, Sturzebecher J, Bode W. The methyl group of Nα(Me)Arg-containing peptides disturbs the active-site geometry of thrombin, impairing efficient cleavage. J. Mol. Biol. 2002;316:869–874. doi: 10.1006/jmbi.2001.5394. [DOI] [PubMed] [Google Scholar]
- 127.Lupardus PJ, Shen A, Bogyo M, Garcia KC. Small molecule-induced allosteric activation of the Vibrio cholerae RTX cysteine protease domain. Science. 2008;322:265–268. doi: 10.1126/science.1162403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schecter I, Berger M. On the size of the active site in proteases. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 129.Matheson AJ, Goa KL. Desirudin: a review of its use in the management of thrombotic disorders. Drugs. 2000;60:679–700. doi: 10.2165/00003495-200060030-00012. [DOI] [PubMed] [Google Scholar]
- 130.Kakar P, Watson T, Lip GY. Drug evaluation: rivaroxaban, an oral, direct inhibitor of activated factor X. Curr. Opin. Investig. Drugs. 2007;8:256–265. [PubMed] [Google Scholar]
- 131.Eriksson BI, Smith H, Yasothan U, Kirkpatrick P. Dabigatran etexilate. Nature Rev. Drug Discov. 2008;7:557–558. doi: 10.1038/nrd2622. [DOI] [PubMed] [Google Scholar]
- 132.Matsuo T, Koide M, Kario K. Development of argatroban, a direct thrombin inhibitor, and its clinical application. Semin. Thromb. Hemost. 1997;23:517–522. doi: 10.1055/s-2007-996129. [DOI] [PubMed] [Google Scholar]