

Oxygen activated Cu-ZSM-5 has been recently shown to selectively oxidize methane to methanol at low temperatures1 by means of a mono(μ-oxo)dicopper(II) species, [Cu2O]2+.2 The geometric and electronic structure of this reactive core was unambiguously assigned using resonance Raman (rR) spectroscopy and density functional theory (DFT) and represents a new species in inorganic chemistry. DFT calculations reproduced the low reaction barrier and kinetic isotope effect (KIE) measured experimentally and showed that the low barrier for H-atom abstraction from CH4 reflects the strong [Cu2O-H]2+ bond in the initial product and a frontier molecular orbital (FMO) that polarizes to an oxyl (O-·) along the reaction coordinate. Interestingly, a binuclear Cu site has recently been demonstrated to be the reactive site in particulate methane monooxygenase (pMMO), an enzyme that also oxidizes methane to methanol.3 In this study we observe an oxygen precursor to the formation of the [Cu2O]2+ species in Cu-ZSM-5 and, using rR spectroscopy, define its structure as a side-on bridged μ-(η2-η2) peroxo dicopper(II) core, [Cu2(O2)]2+. Absorption and rR data show the conversion of [Cu2(O2)]2+ into [Cu2O]2+, while O2 temperature programmed desorption (O2-TPD) experiments provide insight into how this conversion occurs upon heating.
Na-ZSM-5 (VAW, Si/Al=12) samples were ion-exchanged with aqueous solutions of varied Cu(II)-acetate concentrations.4 The samples were initially calcined under O2 at 450°C for 2h (5°C/min, 50ml/min), followed by He flow overnight (50ml/min). This treatment results in the auto-reduction of the Cu sites in Cu-ZSM-5.5-7 Fiber optic UV-vis spectroscopy was used to monitor spectral changes of Cu-ZSM-5 at ambient and elevated temperatures, and rR measurements were performed to obtain the electronic and geometric structure information regarding the Cu/O2 species in Cu-ZSM-5. MS was used to monitor the O-isotope distribution in O2-TPD experiments.
When pre-reduced Cu-ZSM-5 (He at 450°C; Cu/Al=0.5) was exposed to O2 at room temperature (RT) an absorption band at ∼29,000 cm-1 is rapidly formed (Figure 1A). After ∼2 min in O2 flow, the intensity increase of this absorption band levels off. This band is also observed in a Cu-ZSM-5 sample with Cu/Al=0.3 and is essentially absent in samples with Cu/Al<0.2 (see Figures S1A and B). After full formation of the 29,000 cm-1 band, the sample was flushed in He to remove excess O2 at RT. Subsequent heating of Cu-ZSM-5 (Cu/Al=0.3) in He atmosphere resulted in the UV-vis spectral changes shown in Figure 1B. Starting at ∼175°C and higher temperatures, the formation of the 22,700 cm-1 band, associated with the reactive [Cu2O]2+ core, is observed along with the parallel disappearance of the 29,000 cm-1 band. This occurs with heating in either He or O2 atmosphere. The [Cu2O]2+ species has also been shown to form in the presence of N2O at 100°C, 1,2 and, in fact, the [Cu2O]2+ core still forms even at RT with N2O. However, unlike with O2, when pre-reduced Cu-ZMS-5 is exposed to N2O at RT, no 29,000 cm-1 band is formed, and thus there is no formation of the precursor.
Figure 1.

UV-vis absorption spectra of a pre-reduced Cu-ZSM-5 (in He at 450°C) during A) O2 treatment at RT. Time interval between spectra 10 sec in the first 2 min, then every 50 sec for 10 min B) subsequent heating from 25°C to 375°C in He atmosphere (temperature interval between spectra is 25°C).
The rR spectum of the oxygen precursor species formed at RT obtained with laser excitation at 363.8 nm (27,473 cm-1) is shown in Figure 2A. Vibrational features are observed at 269 and 736 cm-1 that are not present using laser excitation outside of the 29,000 cm-1 band, proving that they are resonance enhanced by the species responsible for this absorption feature. When the RT treatment of the auto-reduced Cu-ZSM-5 sample is performed with isotope labeled 18O2, the 736 cm-1 feature shifts to 695 cm-1 (Δ18O2=41 cm-1) while the 269 cm-1 feature is isotope insensitive. These vibrational frequencies and isotope perturbation pattern are characteristic of those of μ-(η2-η2) peroxo dicopper(II) species.8 Thus, we assign the 736 and 269 cm-1 features to the O-O stretch (νO-O) and the Cu-Cu stretch (νCu-Cu) of the μ-(η2-η2) peroxo dicopper(II) moiety, respectively. The 29,000 cm-1 absorption band is thus assigned as a peroxo π*σ to Cu(II) charge transfer (CT) transition. Upon heating the rR sample, the 363.8 nm rR μ-(η2:η2) peroxo dicopper(II) precursor features go away (Figure S3). In parallel, excitation at 457.9 nm (21,834 cm-1) leads to the enhancement of the vibrational features in Figure 2B (see Figure S4 for comparison of 457.9 nm rR spectra of the RT precursor and the [Cu2O]2+ species). These have been assigned in reference 2 as the isotope sensitive, intense symmetric (456 cm-1) and weak antisymmetric (870 cm-1) stretching vibrations characteristic of the μ-oxo-bridged [Cu2O]2+ species. These results parallel the absorption changes and show that the side-on bridged peroxo dicopper(II) species converts to the [Cu2O]2+ species reactive in the selective oxidation of methane to methanol.
Figure 2.

A) rR spectra (363.8 nm) of 16O2 (black) and 18O2 (blue) precursor formed at RT and B) rR spectra (457.9 nm) of reactive site formed by heating the O2 precursor rR samples.
An important issue in this conversion is the fate of the second O atom as only one O atom remains in the reactive [Cu2O]2+ intermediate. He treatment of the reactive intermediate at temperatures above 350°C results in the disappearance of its characteristic 22,700 cm-1 absorption feature with release of O2. Thus, a second O atom recombines with the bridging O atom from the reactive intermediate, and through microscopic reversibility, the desorbing O2 can contain information on the conversion of [Cu2(O2)]2+ into [Cu2O]2+.
An O2-TPD study in He flow was performed after treatment of Cu-ZSM-5 with 18O2 at 240°C. From our previous study, reaction of Cu-ZSM-5 and 18O2 at 240°C results in formation of pure 18O labeled reactive sites (i.e. [CuII-18O-CuII]2+).2 Figure 3 follows the desorbing O2 isotopes, 16O2, 16,18O2 and 18O2, upon subsequent heating in He flow. Although the site was formed with pure 18O2, little 18O2 desorbs. In the temperature range where the 22,700 cm-1 band disappears (between 350°C and 420°C), corresponding to the loss of the [CuII-18O-CuII]2+ species, the ratio of 16,18O2/18O2 released is greater than 10. This shows that the second 18O atom initially present in the peroxo precursor does not recombine with the bridging 18O atom of [Cu2O]2+ upon O2 desorption. As shown in Figure 3, the majority of bridging 18O recombines with 16O, which originates from the zeolite lattice, resulting in dominantly 16,18O2 desorption. This also indicates that the second 18O atom does not form a second [CuII-18O-CuII]2+ species as this would result in desorption of 18O2. A reference TPD experiment without the initial O2 treatment at 240°C showed no O2 desorption in this temperature region. The large fraction of 16O2 observed in Figure 3 is thus not the result of destruction of the zeolite lattice. At higher temperature the fraction of desorbing 16,18O2 atoms decreases and mainly 16O2 desorbs. This represents migration-recombination through the zeolite lattice of abundantly present O atoms deposited on other remote Cu sites in Cu-ZSM-5, 4,9 resulting in isotope scrambling of the 16Olattice and 18O.10-12
Figure 3.

MS signal of 16O2, 16,18O2 and 18O2 as a function of temperature during O2-TPD (2°C/min in He) of activated Cu-ZSM-5 (Cu/Al=0.5, Si/Al=12). Note that the peak in the 16,18O2 desorption profile corresponds to loss of the 22,700 cm-1 absorption feature.
The high incorporation of lattice 16O into O2 desorbed from [Cu218O]2+ (T < 420°C) indicates that the reverse occurs upon formation of the [Cu2O]2+ reactive species from the [Cu2(O2)]2+ precursor. Thus, the high 16,18O2/18O2 desorption ratio reflects the competition between newly formed 18O lattice sites and equivalent and more prevalent 16O lattice sites.
Two additional electrons are required to cleave the O-O bond of [Cu2(O2)]2+. Experimental and computational data showed that the Cu's of the reactive intermediate are Cu2+ and not Cu3+, and DFT calculations of an initial [Cu2O]4+ resulted in delocalization of the additional holes into the lattice, creating [Cu2O]2+ and an electron deficient lattice.2 Also, the electron donor and acceptor capabilities of zeolite lattices have been demonstrated experimentally,13-15 indicating that spectator Cu+ ions in ion-exchange sites can donate the electrons required to reduce the precursor and form the [Cu2O]2+ reactive species.
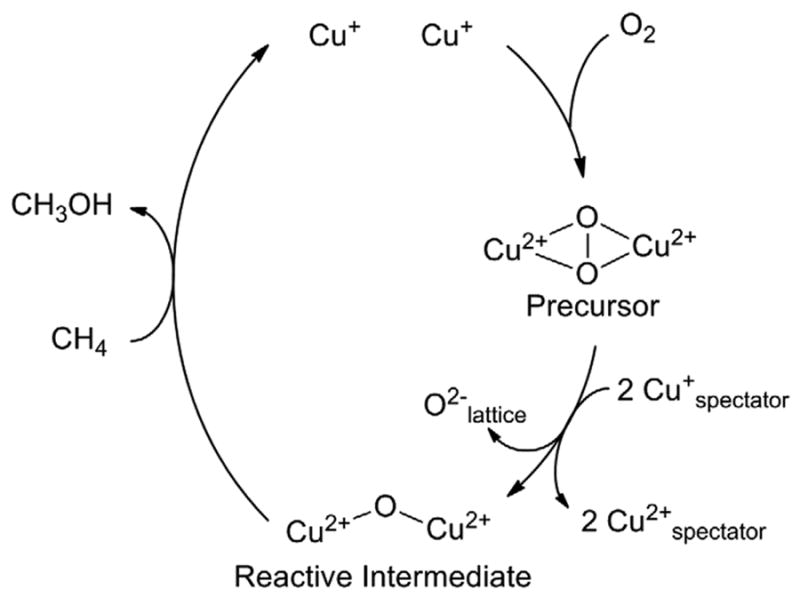
In summary, we have characterized a RT O2 precursor in the formation of [Cu2O]2+, a reactive site capable of the low temperature, selective oxidation of methane to methanol in oxygen activated Cu-ZSM-5. In combination, UV-vis absorption and rR data show the precursor is a μ-(η2:η2) peroxo dicopper(II) core (with an absorption band at 29,000 cm-1) and that this species converts directly into the [Cu2O]2+ reactive intermediate (with an absorption band at 22,700 cm-1). Scheme 1 presents a summary of this process. We propose that the spectator Cu+ ions in the ion-exchange sites provide the required electrons to cleave the bridging peroxo O-O bond. 18O2-TPD experiments showed incorporation of the second 18O atom into the zeolite lattice upon formation of the [Cu2O]2+ reactive intermediate. This study defines the mechanism of oxo-reactive site formation in Cu-ZSM-5. While the [Cu2O]2+ core has been shown to be highly reactive in methane oxidation, we are actively pursuing other reactive Cu/O2 species in oxygen activated Cu-ZSM-5, and are currently investigating the relative reactivity of the precursor and the [Cu2O]2+ intermediate.
Scheme 1.
Supplementary Material
Acknowledgments
P.J.S. acknowledges the I.W.T., F.W.O. and K.U.Leuven for graduate and post-doctoral fellowships and J.S.W acknowledges the NIH for a traineeship. R.G.H. is a Gerhard Casper Stanford Graduate Fellow. This research was supported by the G.O.A. and the Long Term Structural Funding-Methusalem Funding by the Flemish Government (R.A.S., B.F.S.) and by the NIH Grant DK-31450 (E.I.S.).
Footnotes
Supporting Information Available: Experimental procedures, UV-vis and resonance Raman spectra of formation of precursor and conversion into reactive intermediate. This material is available free of charge via the Internet at http://pubs.acs.org
Contributor Information
Robert A. Schoonheydt, Email: robert.schoonheydt@biw.kuleuven.be.
Bert F. Sels, Email: bert.sels@biw.kuleuven.be.
Edward I. Solomon, Email: edward.solomon@stanford.edu.
References
- 1.Groothaert MH, Smeets PJ, Sels BF, Jacobs PA, Schoonheydt RA. J Am Chem Soc. 2005;127:1394–1395. doi: 10.1021/ja047158u. [DOI] [PubMed] [Google Scholar]
- 2.Woertink JS, Smeets PJ, Groothaert MH, Vance MA, Sels BF, Schoonheydt RA, Solomon EI. Proc Natl Acad Sci U S A. 2009;106:18908–18913. doi: 10.1073/pnas.0910461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balasubramanian R, Smith SM, Rawat S, Yatsunyk LA, Stemmler TL, Rosenzweig AC. Nature. 465:115–119. doi: 10.1038/nature08992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smeets PJ, Groothaert MH, van Teeffelen RM, Leeman H, Hensen EJM, Schoonheydt RA. J Catal. 2007;245:358–368. [Google Scholar]
- 5.Iwamoto M, Furukawa H, Mine Y, Uemura F, Mikuriya SI, Kagawa S. J Am Chem Soc. 1986:1272–1273. [Google Scholar]
- 6.Da Costa P, Moden B, Meitzner GD, Lee DK, Iglesia E. Phys Chem Chem Phys. 2002;4:4590–4601. [Google Scholar]
- 7.Smeets PJ, Woertink JS, Sels BF, Solomon EI, Schoonheydt RA. Inorg Chem. 2010;49:3573–3583. doi: 10.1021/ic901814f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baldwin MJ, Root DE, Pate JE, Fujisawa K, Kitajima N, Solomon EI. J Am Chem Soc. 1992;114:10421–10431. [Google Scholar]
- 9.Pirngruber GD, Pieterse JAZ. J Catal. 2006;237:237–247. [Google Scholar]
- 10.Fu CM, Korchak VN, Hall WK. J Catal. 1981;68:166–171. [Google Scholar]
- 11.Pirngruber GD, Roy PK, Prins R. J Catal. 2007;246:147–157. [Google Scholar]
- 12.Novakova J, Schwarze M, Sobalik Z. Catalysis Letters. 2005;104:157–162. [Google Scholar]
- 13.Corma A, Garcia H. Chem Rev. 2002;102:3837–3892. doi: 10.1021/cr010333u. [DOI] [PubMed] [Google Scholar]
- 14.Garcia H, Roth HD. Chem Rev. 2002;102:3947–4007. doi: 10.1021/cr980026x. [DOI] [PubMed] [Google Scholar]
- 15.Yoon KB. Chem Rev. 1993;93:321–339. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.