

Abstract
After CNS injuries, axon growth inhibitors from the myelin and the scar tissue at the injury site are considered major impediments to axon regeneration. The presence of several classes of inhibitors with multiple members in each class suggests functional redundancy in growth inhibition. To test redundancy within the myelin inhibitory pathway, we analyzed raphe spinal serotonergic (5-HT) axon regeneration in mice deficient in two major myelin inhibitors, Nogo and MAG, and their common receptor NgR1 (or NgR). After a complete transection spinal cord injury, there was no significant enhancement of 5-HT axon regeneration beyond the injury site in either Nogo/MAG/NgR1 triple mutants or NgR1 single mutants. Occasional, genotype-independent traversal of 5-HT axons through GFAP-positive tissue bridges at the injury site implicates GFAP-negative lesion areas as especially inhibitory to 5-HT axons. To assess the contribution of class 3 Semaphorins that are expressed by GFAP-negative meningeal fibroblasts at the injury site, we analyzed mice deficient in PlexinA3 and PlexinA4, two key receptors for class 3 Semaphorins, with or without additional NgR1 deletion. No enhanced regeneration of 5-HT or corticospinal axons was detected in PlexinA3/PlexinA4 double mutants or PlexinA3/PlexinA4/NgR1 triple mutants through a complete transection injury. In contrast with previous reports, these data demonstrate that attenuating myelin or Semaphorin-mediated inhibition of axon growth is insufficient to promote 5-HT axon regeneration and further indicate that even attenuating both classes of inhibitory influences is insufficient to promote regeneration of injured axons through a complete transection spinal cord injury.
Introduction
A major hypothesis for the failure of CNS axon regeneration is the presence of axon growth inhibitors in the CNS environment that actively block regeneration (Yiu and He, 2006). Prototypical myelin-derived axon growth inhibitors, including Nogo (or Rtn4), myelin-associated glycoprotein (MAG), and oligodendrocyte myelin glycoprotein (OMgp), have been vigorously pursued as therapeutic targets. Although sharing no sequence homology, these three molecules can bind to common receptors NgR1 (or NgR) and PirB (paired Ig-like receptor B) (Fournier et al., 2001; Atwal et al., 2008). Signal-transducing coreceptors include p75NTR, TROY (tumor necrosis factor receptor superfamily member 19), and LINGO-1 (leucine-rich repeat and Ig domain containing, Nogo Receptor-interacting protein), with downstream effectors such as Rho and ROCK (Rho-associated kinase) (Yiu and He, 2006). Importantly, Nogo and MAG may also signal through other receptors: both may signal through an integrin-dependent mechanism (Goh et al., 2008; Hu and Strittmatter, 2008), whereas MAG can signal through gangliosides and NgR2, an NgR1 homolog (Venkatesh et al., 2005; Williams et al., 2008; Wörter et al., 2009) (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Despite rapid progress in elucidating the molecular pathway associated with the prototypical myelin inhibitors, their contribution to regeneration in vivo remains poorly understood.
Another class of proposed inhibitors is the chemorepulsive axon guidance molecules. Although the role of these molecules during development is well documented, their role as inhibitors of axon regeneration in the adult CNS remains to be investigated (Yaron and Zheng, 2007). Among these, class 3 Semaphorins are distinctly expressed by meningeal fibroblasts (De Winter et al., 2002) that reside in glial fibrillary acidic protein (GFAP)-negative (GFAP−) regions and form distinct boundaries with the GFAP-positive (or GFAP+) astrocytic scar at the injury site (Herrmann et al., 2010). Class 3 Semaphorins signal through Neuropilin (ligand binding)–Plexin (signal transducing) receptor complexes (Yaron and Zheng, 2007). Disrupting this signaling pathway may enable injured axons to overcome class 3 Semaphorin-mediated inhibition and to extend through the inhibitory meningeal fibroblasts and the astrocyte–meningeal fibroblast border.
Here we test the hypothesis that axon growth inhibitors are functionally redundant in blocking axon regeneration by genetic analyses. To test redundancy within the myelin inhibitory pathway, we analyzed mice deficient in Nogo/MAG/NgR1. To test redundancy between myelin-mediated and class 3 Semaphorin-mediated inhibition, we analyzed mice deficient in PlexinA3/PlexinA4 (two key signal-transducing receptors for class 3 Semaphorins) (Yaron et al., 2005) as well as NgR1. To our knowledge, this represents the first functional test on targeting multiple classes of inhibitory influences in the CNS to promote spinal axon regeneration. Our data indicate that even targeting multiple components of the myelin inhibitory pathway or multiple classes of inhibitory influences is insufficient to enhance regeneration of injured axons, including serotonergic axons through a complete transection spinal cord injury.
Materials and Methods
All single mutants have been described previously: Nogo-A,B (Zheng et al., 2003), MAG (Li et al., 1994), NgR1 (Zheng et al., 2005), PlexinA3 (Cheng et al., 2001), and PlexinA4 (Yaron et al., 2005). NgR1 mutants were assessed in C57BL/6 background; Nogo/MAG/NgR1 triple mutants and mice carrying Plexin mutations were assessed in mixed genetic background, including 129S7 and C57BL/6. For Plexin/NgR1 mutants, single-mutant mice were bred to each other to obtain triple heterozygous mice (males carried PlexinA3 −/Y), which were intercrossed to obtain homozygous mutants and wild-type (WT) siblings. The respective genotypes were then intercrossed among themselves to give more homozygous mutant and WT cousins for experimentation.
Spinal cord surgeries.
Six- to eight-week-old female mice were subjected to a T8 complete transection injury. After laminectomy to expose the spinal cord, the dura was punctured bilaterally with a 30 gauge needle, and the cord was transected throughout the entire width and depth with iridectomy microscissors. A microknife was then used to trace the cord, pressing against the lateral and ventral sides of the vertebral cavity to ensure completeness of the lesion. Two weeks before they were killed, mice in the Plexin triple-mutant groups received stereotaxic injection of biotinylated dextran amine (BDA) (10%; Invitrogen) to trace the corticospinal tract (CST) (0.7 mm deep into the right sensorimotor cortex at the following six sites: 0.1, 0.6, and 1.1 mm posterior; 1.0 and 1.4 mm lateral, all in reference to bregma, 0.2 μl/site). Six weeks after injury, mice were perfused with 4% paraformaldehyde transcardially, and spinal cord tissues were dissected, postfixed, cryoprotected, and embedded. Sections, 20 μm, were cut serially from an 8 mm block surrounding the injury site in the sagittal plane and immunostained in a high-salt buffer (Lee et al., 2007) for GFAP (Invitrogen) and 5-hydroxytryptamine (5-HT) (Immunostar). Streptavidin-546 was used to detect BDA-traced CST axons. All experimental procedures were approved by the University of California, San Diego Institutional Animal Care and Use Committee.
Quantification.
NIH ImageJ software was used. In the myelin triple-mutant study, the region caudal to injury (500 μm from the GFAP+ lesion border) was quantified for total thresholded 5-HT immunoreactivity. In the Plexin triple-mutant study, the regions rostral, at, and caudal to the injury site were quantified for percentage area positive for 5-HT and neurofilament immunoreactivity. CST defect in PlexinA3/PlexinA4 mutants was quantified by crossing the mutants to the Thy1–YFP-H transgenic line (Feng et al., 2000) in which CST axons (and other neuronal/axonal populations) are labeled with yellow fluorescent protein (YFP). The number of ectopic CST axons that fail to decussate was determined from transverse brainstem sections and expressed as a percentage of the total YFP-labeled CST axons before midline crossing.
Results
The presence of multiple myelin-derived axon growth inhibitors and receptors indicates a high level of redundancy in the prototypical myelin inhibitory pathway, which may explain the lack of a robust and consistent regeneration phenotype in mice lacking any one inhibitor or receptor (Zheng et al., 2006; Lee et al., 2009). We hypothesized that disrupting multiple key components of the myelin inhibitory pathway would remove much of the redundancy in the system (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) and consequently would be sufficient to elicit robust axon regeneration in the CNS. We tested this hypothesis by generating Nogo-A,B/MAG/NgR1 triple-mutant mice (referred to as Nogo/MAG/NgR1 mutants below for simplicity) by breeding single-mutant lines described previously (see Materials and Methods). We chose to combine these three mutations for several reasons: (1) Nogo-A and NgR1 are the founding members of the inhibitory ligands and receptors, respectively, of the prototypical myelin inhibitory pathway, and there is most evidence for a role of these two molecules in blocking axon regeneration based on studies with neutralizing antibodies, peptides, or receptor bodies (Schnell and Schwab, 1990; GrandPré et al., 2002; Li et al., 2004; Liebscher et al., 2005); (2) among the three Nogo isoforms (Nogo-A,B,C), Nogo-A is the primary isoform expressed by oligodendrocytes and therefore the most relevant to myelin inhibition (Chen et al., 2000); (3) deleting Nogo-A would eliminate the inhibitory signal from its N-terminal region (or amino-Nogo) in addition to that from its C-terminal Nogo-66 region that signals through NgR1 (Fournier et al., 2001); (4) deleting MAG would eliminate its inhibitory signal through its NgR2 and ganglioside receptors; and (5) deleting Nogo-A and MAG would eliminate their inhibitory signals through integrins or PirB. Thus, the only known inhibitory signal presumably left intact in the prototypical myelin inhibitory pathway would be from OMgp to PirB in these triple mutants.
We chose the raphe spinal serotonergic (5-HT) tract as the primary axonal tract to assess regeneration because of its known ability for injury-induced axonal growth (Saruhashi et al., 1996). We reasoned that focusing on an axonal population prone to regenerative growth would make it more likely to detect any regeneration-promoting effect of attenuating the inhibitory influences. Indeed, a previous study reported enhanced 5-HT axon regeneration in an NgR1 mutant line (Kim et al., 2004). We thus included a (different) NgR1 null mutant (Zheng et al., 2005) in our study as a reference point to examine the degree of any 5-HT axon regeneration.
Because 5-HT axons descend throughout the dorsal and ventral spinal cord (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), only a complete transection can ensure the elimination of all 5-HT axons at the injury site en route to caudal spinal cord. NgR1 mutants, Nogo/MAG/NgR1 triple-mutants, along with their respective WT controls were subjected to a complete transection spinal cord injury at thoracic level 8 (T8) and allowed to survive for 6 weeks (see Materials and Methods). Sagittal sections around the injury site were immunostained for GFAP to verify lesion effectiveness and 5-HT to detect serotonergic axons (Fig. 1). In all mice, the injury sites encompassed a GFAP− lesion epicenter that was surrounded by strongly GFAP-immunoreactive areas indicative of reactive astrogliosis, typical for penetrating spinal injuries in mice (Herrmann et al., 2010).
Figure 1.
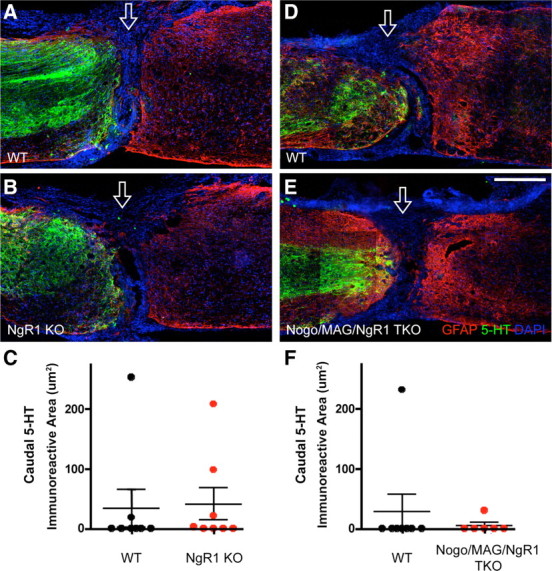
Lack of enhanced 5-HT axon regeneration after a complete transection spinal cord injury in NgR1 single-mutant and Nogo/MAG/NgR1 triple-mutant mice. A, B, D, E, Representative sagittal sections immunostained for 5-HT (green) and GFAP (red) along with 4′,6-diamidino-2-phenylindole (DAPI) nuclear staining (blue) in completely transected spinal cord of an NgR1 mutant (B), a Nogo/MAG/NgR1 triple-mutant (E), and respective WT controls (A, D). Open arrow indicates injury site. KO, Knock-out (mutant). Scale bar, 500 μm. C, F, Quantification of 5-HT immunoreactivity caudal to the injury. n = 8 for WT and 8 for NgR1 knock-outs (C); n = 8 for WT and 6 for Nogo/MAG/NgR1 triple knock-outs (TKO). No statistically significant differences, two-way ANOVA.
In both WT and NgR1 mutant mice, rostral to the injury, 5-HT axons were present primarily in the gray matter until they approached the injury site, where they often spread into the white matter, likely reflecting axon sprouting (Fig. 1A,B). Regardless of the genotype, 5-HT axons could be detected in areas of strong GFAP immunoreactivity, often stopping just before the GFAP− lesion epicenter. However, no axons penetrated the injury site except in two WT mice and three NgR1 mutants. Those that did appeared to have traveled through the GFAP+ tissue bridges at the injury epicenter (Fig. 2). In contrast to published data also with a complete transection injury but in a different NgR1 mutant line (Kim et al., 2004), the caudal 5-HT axons seen here were few in number, did not extend far past the injury site, and were present in a subset of the mice regardless of the genotype. Quantification of 5-HT axons caudal to the injury site did not reveal a statistically significant difference between WT controls and NgR1 mutants (Fig. 1C).
Figure 2.
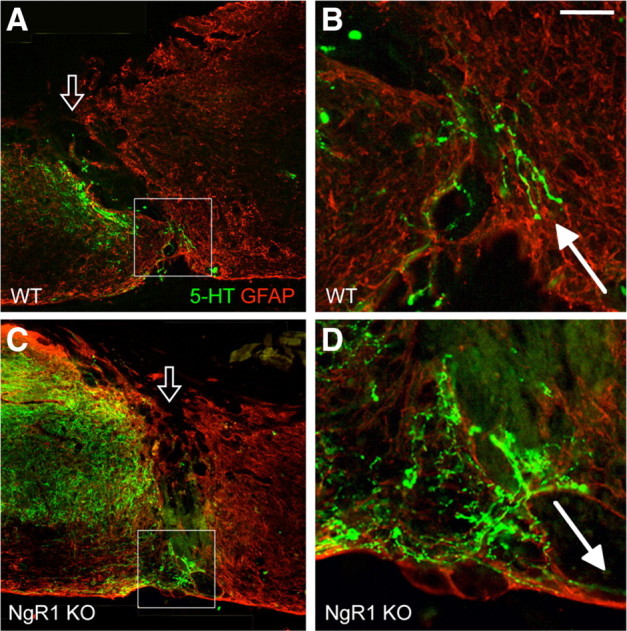
5-HT axons occasionally traversed the injury site via GFAP+ bridges regardless of the genotype. A, C, Representative images of 5-HT axons traversing the GFAP+ bridges at the injury site (open arrow) on spinal cord sagittal sections in one WT mouse (A) and one NgR1 mutant (B). B, D, High-magnification images of the boxed area shown in A and C, respectively. Filled arrows indicate 5-HT axons that had just passed beyond the lesion site, apparently through GFAP+ bridges. Scale bar: A, C, 250 μm; B, D, 60 μm.
Likewise, Nogo/MAG/NgR1 triple mutants, along with their WT controls, exhibited no or few 5-HT axons caudal to the injury site (Fig. 1D,E). One mouse from each genotype exhibited few short 5-HT-positive axons caudal to the injury site, and again, these axons appeared to have traveled through the injury site by way of GFAP+ tissue bridges (data not shown). Quantification of 5-HT axons caudal to the injury site did not reveal a significant difference between WT controls and Nogo/MAG/NgR1 triple mutants (Fig. 1F). Thus, deleting NgR1, or Nogo-A,B, MAG, and NgR1 altogether, did not lead to enhanced 5-HT axon regeneration beyond a complete transection spinal cord injury in mice.
The observation that a small number of 5-HT axons traversed the GFAP+ tissue bridges regardless of the genotype in these experiments indicates that the GFAP− injury area is particularly inhibitory to 5-HT axon growth. Among the proposed axon growth inhibitors, class 3 Semaphorins are distinctly expressed by GFAP− meningeal fibroblasts at the injury site after complete spinal transection (De Winter et al., 2002). Class 3 Semaphorins signal through receptor complexes containing Neuropilins as the ligand-binding moieties and Plexins as the signal-transducing moieties (Yaron and Zheng, 2007). A previous attempt to decipher the role of class 3 Semaphorins in spinal axon regeneration using virally delivered short hairpin RNA (shRNA) knockdown of Neuropilins was unsuccessful as a result of apparent shRNA-mediated cytotoxicity (Ehlert et al., 2010). To determine whether attenuating class 3 Semaphorin-mediated growth inhibition promotes axon regeneration, we characterized mice deficient in PlexinA3 and PlexinA4, two key signal-transducing receptors for class 3 Semaphorins, and Sema3F and Sema3A in particular (Yaron et al., 2005; Tran et al., 2009). To test for any synergistic effect of attenuating myelin-mediated inhibition and Semaphorin-mediated inhibition, we also generated and characterized triple-mutant mice that lack PlexinA3, PlexinA4, and NgR1 altogether (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material).
Six weeks after a complete transection injury, 5-HT axons in all mice regardless of genotype approached the injury site, which was outlined using GFAP immunoreactivity (Fig. 3A–C). Again, 5-HT axons often stopped just before—but did not penetrate—the GFAP− lesion epicenter. Occasionally, 5-HT axons were found within the injury site associated with GFAP+ bridges. In rare instances, 5-HT axons protruded into the caudal spinal cord through GFAP+ bridges. However, this occurred in a subset of the mice regardless of the genotype: 4 of 17 WT mice, 3 of 9 PlexinA3/PlexinA4 double mutants, and 2 of 9 PlexinA3/PlexinA4/NgR1 triple mutants (an example from a WT mouse shown in supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Quantification of 5-HT immunoreactivity rostral, at, and caudal to the injury site did not reveal any significant differences among the three genotypes (Fig. 3D).
Figure 3.
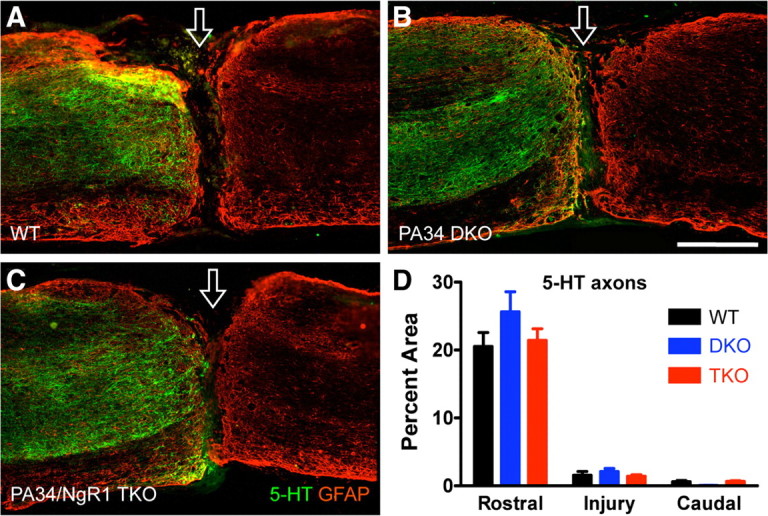
Lack of enhanced 5-HT axon regeneration after a complete transection injury in PlexinA3/PlexinA4 double and PlexinA3/PlexinA4/NgR1 triple mutants. A–C, Representative sagittal sections immunostained for 5-HT (green) and GFAP (red) in completely transected spinal cord. Open arrow indicates injury site. PA34 DKO, PlexinA3/PlexinA4 double knock-out; PA34/NgR1 TKO, PlexinA3/PlexinA4/NgR1 triple knock-out. Scale bar, 500 μm. D, Quantification of 5-HT immunoreactivity rostral, at, and caudal to injury. n = 17 for WT, 9 for PA34 double knock-outs, and 9 for PA34/NgR1 triple knock-outs. No significant differences, two-way repeated-measures ANOVA, Bonferroni's post hoc test.
To determine whether other axonal populations might behave differently, we next examined the regeneration of the CST, which is often assessed using a dorsal hemisection injury model (Zheng et al., 2006). However, PlexinA4 mutants exhibit an altered trajectory in the CST because of a developmental defect. In WT mice, the main CST bundle crosses the midline at medullary pyramids and then descends in the ventral part of the dorsal column in the spinal cord (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). In all PlexinA3/PlexinA4 double mutants (n = 16) as well as PlexinA4 single mutants (n = 5), a significant portion of CST axons fail to cross the midline at medullary pyramids and consequently remain ipsilateral in the spinal cord (supplemental Fig. 3C–F, available at www.jneurosci.org as supplemental material) (and data not shown), as described previously (Faulkner et al., 2008; Rünker et al., 2008). These ectopic CST axons were readily detected by either surgical tracing with BDA (supplemental Fig. 3C,D, available at www.jneurosci.org as supplemental material) or genetic tracing using the Thy1–YFP-H transgenic line (Feng et al., 2000) (supplemental Fig. 3E,F, available at www.jneurosci.org as supplemental material). Using the genetic tracer, we determined the percentage of ectopic CST axons in the PlexinA3/PlexinA4 mutants to be ∼33% (21, 33, and 44% in three animals analyzed).
Because these aberrant CST axons reside in the ventrolateral white matter (arrow in supplemental Fig. 3D, available at www.jneurosci.org as supplemental material), only a complete transection injury can eliminate these axons en route to the caudal spinal cord. After complete transection, CST axons in both WT and PlexinA3/PlexinA4/NgR1 mutants retracted from the injury site, typical for this axonal population (Fig. 4A,B). Not a single CST axon traversed the injury site, let alone reaching the caudal segment in nine PlexinA3/PlexinA4/NgR1 triple mutants along with 17 WT controls.
Figure 4.
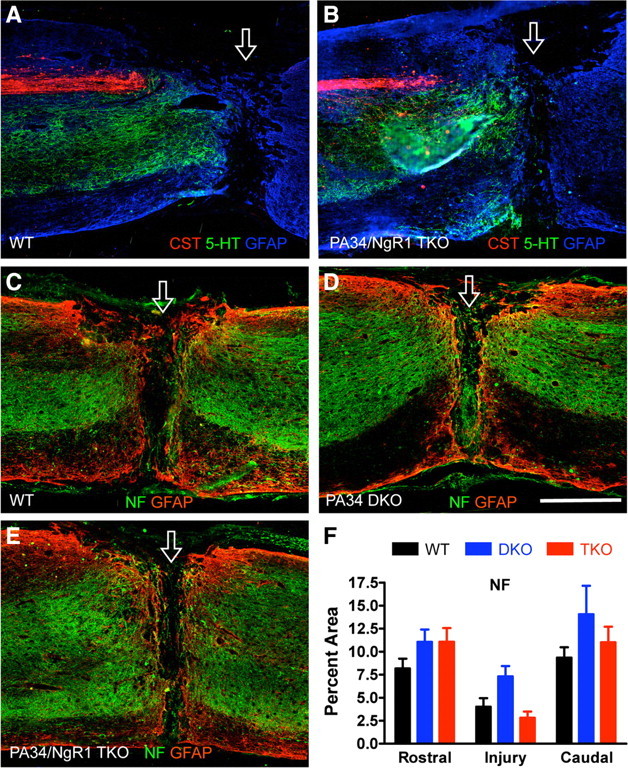
Lack of enhanced CST and neurofilament-positive axon regeneration after a complete transection injury in PlexinA3/PlexinA4/NgR1 triple-mutant mice. A, B, Representative spinal cord sagittal sections around a complete transection injury site stained for traced CST axons and 5-HT axons. C–E, Representative spinal cord sagittal sections stained for neurofilament (NF) around a complete transection injury in different genotypes. Open arrow indicates injury site. Scale bar, 500 μm. F, Quantification of neurofilament immunoreactivity rostral, at, and caudal to the injury site. n = 17 for WT (6 traced for CST), 9 for PA34 double knock-outs (DKO), and 9 for PA34/NgR1 triple knock-outs (TKO). No significant differences, two-way repeated-measures ANOVA, Bonferroni's post hoc test.
To assess the regenerative growth of a more general axon population, we analyzed neurofilament immunoreactivity that is widely used as a general axonal marker to detect possible changes in the amount of axons present at or around the injury site (White et al., 2008). Neurofilament-positive axons were present in similar densities among the different genotypes rostral, at, and caudal to the injury site (Fig. 4C–F). Thus, deleting PlexinA3/PlexinA4 or PlexinA3/PlexinA4/NgR1 altogether did not lead to enhanced 5-HT or CST regeneration across a complete transection injury or detectable alteration of general axonal growth into and around the lesion as assessed by neurofilament immunoreactivity.
Discussion
Our data indicate that genetically deleting three key components of the myelin inhibitory pathway, Nogo, MAG, and NgR1, is insufficient to promote 5-HT axon regeneration through a complete transection spinal cord injury. Furthermore, genetically attenuating two classes of inhibitory influences, by deleting NgR1, PlexinA3, and PlexinA4, is insufficient to enhance regeneration of 5-HT or CST axons, or growth of a more general, neurofilament-positive axonal population at and around the injury site. Together, these data illustrate, for the first time to our knowledge, that even targeting more than one class of inhibitory influences in the injured adult CNS is insufficient to promote significant axon regeneration after a complete spinal transection.
In the absence of Nogo/MAG/NgR1, other myelin inhibitory components, such as OMgp and PirB, might be sufficient to inhibit regeneration. In addition, other classes of inhibitory influences remain in Nogo/MAG/NgR1 mutants, including chondroitin sulfate proteoglycans derived from the GFAP+ astroglial scar (Bradbury et al., 2002; Silver and Miller, 2004). In this regard, 5-HT axons appear to be capable of traversing the GFAP+ bridges of the injury site, indicating that, compared with GFAP− cells, GFAP+ astroglia present a less inhibitory—perhaps even growth-permissive—substrate for 5-HT axons. This is consistent with previous observations that GFAP+ astroglia may express growth-promoting molecules, although they are known to produce growth inhibitors when activated (Shearer et al., 2003). This observation prompted us to examine the role of class 3 Semaphorins known to be expressed by GFAP− meningeal fibroblasts (De Winter et al., 2002) by genetically deleting two key receptors for class 3 Semaphorins, PlexinA3, and PlexinA4 (Suto et al., 2005; Yaron et al., 2005; Tran et al., 2009). Because PlexinA4 has also been implicated in Sema6A- and Sema6B-mediated axon guidance (Suto et al., 2005; Faulkner et al., 2008; Rünker et al., 2008), our result has implications beyond the class 3 Semaphorins. The lack of enhanced 5-HT and CST axon regeneration in PlexinA3/PlexinA4/NgR1 triple mutants indicates that attenuating both Semaphorin and myelin-mediated inhibition is insufficient to promote axon regeneration through a complete transection.
The role of Sema3A in CNS repair has been interrogated with a small molecular inhibitor, SM-216289 (or xanthofulvin) (Kaneko et al., 2006). When administered in rats, this inhibitor has been shown to elicit a plethora of beneficial effects, including axon preservation and/or regeneration, remyelination, reduced apoptosis and lesion cavity, enhanced angiogenesis, and improved functional recovery after a complete transection spinal cord injury. Although CST axons remained refractory to regeneration, SM-216289-treated rats showed enhanced 5-HT axon growth into the lesion site but not substantially beyond. It will be interesting to determine whether the multiple beneficial effects of SM-216289 observed are mediated exclusively through Sema3A because SM-216289 also binds to other molecular targets, including matrix metalloprotease and epidermal growth factor receptor.
The results of our NgR1 single mutants directly contrasts with published data reporting enhanced 5-HT axon regeneration of another NgR1 mutant line also using a complete transection model (Kim et al., 2004). Although different mutants were analyzed, both are NgR1 null (Kim et al., 2004; Zheng et al., 2005). Genetic background may be a factor. However, both mutations were made in the 129S7 background and backcrossed to C57BL/6 for three or more times. Different surgical techniques may also contribute to variability. Future studies are required to clarify this discrepancy.
In summary, our data illustrate the limitations of targeting the myelin-derived and Semaphorin-mediated inhibitory influences to promote spinal axon regeneration after a complete transection injury. Whether class 3 Semaphorins modulate injury-induced axonal growth in a less severe injury model and whether receptors other than PlexinA3 and PlexinA4 may mediate their inhibitory action in the adult CNS remain unknown. Regardless, the current study questions the potential of therapeutic strategies that focus on neutralizing these inhibitory molecules as the primary measure to treat spinal cord injury when axon regeneration is the goal.
Footnotes
This work was supported by grants from the International Spinal Research Trust (London, UK), the Roman Reed Spinal Cord Injury Research Fund of California, and National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant NS054734. J.K.L. was supported by National Research Service Award Postdoctoral Fellowship F32NS056697. F.X. was supported by a Christopher and Dana Reeve Foundation Postdoctoral Fellowship. We thank Marc Tessier-Lavigne and Avraham Yaron for the Plexin mutants and Andrea Chan and Yuhong Zhu for technical assistance.
References
- Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y, Shatz C, Tessier-Lavigne M. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science. 2008;322:967–970. doi: 10.1126/science.1161151. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, Christ F, Schwab ME. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403:434–439. doi: 10.1038/35000219. [DOI] [PubMed] [Google Scholar]
- Cheng HJ, Bagri A, Yaron A, Stein E, Pleasure SJ, Tessier-Lavigne M. Plexin-A3 mediates semaphorin signaling and regulates the development of hippocampal axonal projections. Neuron. 2001;32:249–263. doi: 10.1016/s0896-6273(01)00478-0. [DOI] [PubMed] [Google Scholar]
- De Winter F, Oudega M, Lankhorst AJ, Hamers FP, Blits B, Ruitenberg MJ, Pasterkamp RJ, Gispen WH, Verhaagen J. Injury-induced class 3 semaphorin expression in the rat spinal cord. Exp Neurol. 2002;175:61–75. doi: 10.1006/exnr.2002.7884. [DOI] [PubMed] [Google Scholar]
- Ehlert EM, Eggers R, Niclou SP, Verhaagen J. Cellular toxicity following application of adeno-associated viral vector-mediated RNA interference in the nervous system. BMC Neurosci. 2010;11:20. doi: 10.1186/1471-2202-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner RL, Low LK, Liu XB, Coble J, Jones EG, Cheng HJ. Dorsal turning of motor corticospinal axons at the pyramidal decussation requires plexin signaling. Neural Dev. 2008;3:21. doi: 10.1186/1749-8104-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fournier AE, GrandPre T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- Goh EL, Young JK, Kuwako K, Tessier-Lavigne M, He Z, Griffin JW, Ming GL. β1-integrin mediates myelin-associated glycoprotein signaling in neuronal growth cones. Mol Brain. 2008;1:10. doi: 10.1186/1756-6606-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GrandPré T, Li S, Strittmatter SM. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Shah RR, Chan AF, Zheng B. EphA4 deficient mice maintain astroglial-fibrotic scar formation after spinal cord injury. Exp Neurol. 2010;223:582–598. doi: 10.1016/j.expneurol.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Strittmatter SM. The N-terminal domain of Nogo-A inhibits cell adhesion and axonal outgrowth by an integrin-specific mechanism. J Neurosci. 2008;28:1262–1269. doi: 10.1523/JNEUROSCI.1068-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Iwanami A, Nakamura M, Kishino A, Kikuchi K, Shibata S, Okano HJ, Ikegami T, Moriya A, Konishi O, Nakayama C, Kumagai K, Kimura T, Sato Y, Goshima Y, Taniguchi M, Ito M, He Z, Toyama Y, Okano H. A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat Med. 2006;12:1380–1389. doi: 10.1038/nm1505. [DOI] [PubMed] [Google Scholar]
- Kim JE, Liu BP, Park JH, Strittmatter SM. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron. 2004;44:439–451. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Lee JK, Johnson CS, Wrathall JR. Up-regulation of 5-HT2 receptors is involved in the increased H-reflex amplitude after contusive spinal cord injury. Exp Neurol. 2007;203:502–511. doi: 10.1016/j.expneurol.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Chan AF, Luu SM, Zhu Y, Ho C, Tessier-Lavigne M, Zheng B. Reassessment of corticospinal tract regeneration in Nogo-deficient mice. J Neurosci. 2009;29:8649–8654. doi: 10.1523/JNEUROSCI.1864-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Tropak MB, Gerlai R, Clapoff S, Abramow-Newerly W, Trapp B, Peterson A, Roder J. Myelination in the absence of myelin-associated glycoprotein. Nature. 1994;369:747–750. doi: 10.1038/369747a0. [DOI] [PubMed] [Google Scholar]
- Li S, Liu BP, Budel S, Li M, Ji B, Walus L, Li W, Jirik A, Rabacchi S, Choi E, Worley D, Sah DW, Pepinsky B, Lee D, Relton J, Strittmatter SM. Blockade of Nogo-66, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein by soluble Nogo-66 receptor promotes axonal sprouting and recovery after spinal injury. J Neurosci. 2004;24:10511–10520. doi: 10.1523/JNEUROSCI.2828-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebscher T, Schnell L, Schnell D, Scholl J, Schneider R, Gullo M, Fouad K, Mir A, Rausch M, Kindler D, Hamers FP, Schwab ME. Nogo-A antibody improves regeneration and locomotion of spinal cord-injured rats. Ann Neurol. 2005;58:706–719. doi: 10.1002/ana.20627. [DOI] [PubMed] [Google Scholar]
- Rünker AE, Little GE, Suto F, Fujisawa H, Mitchell KJ. Semaphorin-6A controls guidance of corticospinal tract axons at multiple choice points. Neural Dev. 2008;3:34. doi: 10.1186/1749-8104-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saruhashi Y, Young W, Perkins R. The recovery of 5-HT immunoreactivity in lumbosacral spinal cord and locomotor function after thoracic hemisection. Exp Neurol. 1996;139:203–213. doi: 10.1006/exnr.1996.0094. [DOI] [PubMed] [Google Scholar]
- Schnell L, Schwab ME. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269–272. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- Shearer MC, Niclou SP, Brown D, Asher RA, Holtmaat AJ, Levine JM, Verhaagen J, Fawcett JW. The astrocyte/meningeal cell interface is a barrier to neurite outgrowth which can be overcome by manipulation of inhibitory molecules or axonal signalling pathways. Mol Cell Neurosci. 2003;24:913–925. doi: 10.1016/j.mcn.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Suto F, Ito K, Uemura M, Shimizu M, Shinkawa Y, Sanbo M, Shinoda T, Tsuboi M, Takashima S, Yagi T, Fujisawa H. Plexin-a4 mediates axon-repulsive activities of both secreted and transmembrane semaphorins and plays roles in nerve fiber guidance. J Neurosci. 2005;25:3628–3637. doi: 10.1523/JNEUROSCI.4480-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TS, Rubio ME, Clem RL, Johnson D, Case L, Tessier-Lavigne M, Huganir RL, Ginty DD, Kolodkin AL. Secreted semaphorins control spine distribution and morphogenesis in the postnatal CNS. Nature. 2009;462:1065–1069. doi: 10.1038/nature08628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh K, Chivatakarn O, Lee H, Joshi PS, Kantor DB, Newman BA, Mage R, Rader C, Giger RJ. The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J Neurosci. 2005;25:808–822. doi: 10.1523/JNEUROSCI.4464-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RE, Yin FQ, Jakeman LB. TGF-alpha increases astrocyte invasion and promotes axonal growth into the lesion following spinal cord injury in mice. Exp Neurol. 2008;214:10–24. doi: 10.1016/j.expneurol.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G, Wood A, Williams EJ, Gao Y, Mercado ML, Katz A, Joseph-McCarthy D, Bates B, Ling HP, Aulabaugh A, Zaccardi J, Xie Y, Pangalos MN, Walsh FS, Doherty P. Ganglioside inhibition of neurite outgrowth requires Nogo receptor function: identification of interaction sites and development of novel antagonists. J Biol Chem. 2008;283:16641–16652. doi: 10.1074/jbc.M802067200. [DOI] [PubMed] [Google Scholar]
- Wörter V, Schweigreiter R, Kinzel B, Mueller M, Barske C, Böck G, Frentzel S, Bandtlow CE. Inhibitory activity of myelin-associated glycoprotein on sensory neurons is largely independent of NgR1 and NgR2 and resides within Ig-Like domains 4 and 5. PLoS One. 2009;4:e5218. doi: 10.1371/journal.pone.0005218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaron A, Zheng B. Navigating their way to the clinic: emerging roles for axon guidance molecules in neurological disorders and injury. Dev Neurobiol. 2007;67:1216–1231. doi: 10.1002/dneu.20512. [DOI] [PubMed] [Google Scholar]
- Yaron A, Huang PH, Cheng HJ, Tessier-Lavigne M. Differential requirement for Plexin-A3 and -A4 in mediating responses of sensory and sympathetic neurons to distinct class 3 Semaphorins. Neuron. 2005;45:513–523. doi: 10.1016/j.neuron.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Ho C, Li S, Keirstead H, Steward O, Tessier-Lavigne M. Lack of enhanced spinal regeneration in Nogo-deficient mice. Neuron. 2003;38:213–224. doi: 10.1016/s0896-6273(03)00225-3. [DOI] [PubMed] [Google Scholar]
- Zheng B, Atwal J, Ho C, Case L, He XL, Garcia KC, Steward O, Tessier-Lavigne M. Genetic deletion of the Nogo receptor does not reduce neurite inhibition in vitro or promote corticospinal tract regeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:1205–1210. doi: 10.1073/pnas.0409026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Lee JK, Xie F. Genetic mouse models for studying inhibitors of spinal axon regeneration. Trends Neurosci. 2006;29:640–646. doi: 10.1016/j.tins.2006.09.005. [DOI] [PubMed] [Google Scholar]