

Abstract
Doxorubicin remains an important part of chemotherapy regimens in the clinic and is considered as an effective agent in the treatment of acute lymphoblastic leukemia (ALL). Although the cellular responses induced by doxorubicin treatment have been investigated for years, the precise mechanisms underlying its cytotoxicity and therapeutic activity remain unclear. Here we utilized mass spectrometry, together with stable isotope labeling by amino acids in cell culture (SILAC), to analyze comparatively the protein expression in Jurkat-T cells before and after treatment with a clinically relevant concentration of doxorubicin. We were able to quantify 1066 proteins in Jurkat-T cells with both forward and reverse SILAC measurements, among which 62 were with significantly altered levels of expression induced by doxorubicin treatment. These included the up-regulation of core histones, heterogeneous nuclear ribonucleoproteins and superoxide dismutase 2 as well as the down-regulation of hydroxymethylglutaryl-CoA synthase and farnesyl diphosphate synthase. The latter two are essential enzymes for cholesterol biosynthesis. We further demonstrated that the doxorubicin-induced growth inhibition of Jurkat-T cells could be rescued by treatment with cholesterol, supporting that doxorubicin exerts its cytotoxic effect, in part, via suppressing the expression of hydroxymethylglutaryl-CoA synthase and farnesyl diphosphate synthase, thereby inhibiting the endogenous production of cholesterol. The results from the present study provided important new knowledge for gaining insights into the molecular mechanisms of action of doxorubicin.
Introduction
The antitumor drug doxorubicin (Dox) has been widely used in the clinic for the treatment of a broad spectrum of solid tumors and hematological malignancies since the early 1960s1. However, the exact mechanism of action of doxorubicin is still somewhat unclear. Doxorubicin is known to intercalate into DNA2, 3, which prohibits the progression of topoisomerase II, an enzyme unwinding DNA for replication and transcription4. Doxorubicin stabilizes the topoisomerase II complex after it has broken the DNA chain for replication, preventing the DNA double helix from being resealed and thereby terminating DNA replication and transcription. Doxorubicin can also induce the generation of free oxygen radicals and lipid peroxidation. A major adverse side effect associated with the use of Dox in the clinic is the onset of cardiomyopathy and heart failure5. Several reports suggest that Dox-induced apoptosis plays an important role in its cardiotoxicity that is linked with the formation of reactive oxygen species (ROS) derived from the redox activation of Dox6–8. Recent studies have focused on Dox-induced apoptotic signaling mechanisms9.
Mass spectrometry (MS)-based proteomics allows for the identification and quantification of a large number of proteins in complex samples. Two-dimensional gel electrophoresis (2-DE) is a traditional technique for studying the effects of drug treatments on protein expression. In 2-DE, quantification is achieved by recording differences in the stained spot intensities of proteins derived from two states of cell populations or tissues10. A number of proteomic studies underlying the effect of treatments with different anti-cancer drugs, such as cisplatin11, 12, etoposide13 and all-trans retinoic acid14, have been performed by using mass spectrometry for protein identification and 2-DE for protein quantification. The combination of 2-DE with mass spectrometry has also been used previously for assessing the doxorubicin-induced alterations in protein expression in human hepatoma cells15, MCF-7 breast tumor cells16, and mouse heart.17
Other than 2-DE, several stable isotope labeling-based quantification strategies, such as isotope-coded affinity tag (ICAT)18, isobaric tags for relative and absolute quantitation (iTRAQ)19 and stable isotope labeling by amino acids in cell culture (SILAC)20, have been developed for MS-based analysis of differential protein expression. Among these isotope-labeling strategies, SILAC is a metabolic labeling method, which is simple, efficient, and can facilitate almost complete heavy isotope incorporation. It is very suitable for the comparative study of protein expression in cells with and without drug treatment15, 21. With the use of SILAC, accurate results could be obtained with minimal bias, facilitating relative quantification of subtle changes in protein abundance.20
In this study, we employed LC-MS/MS, in conjunction with SILAC, to examine quantitatively the perturbation of protein expression in cultured Jurkat-T human acute lymphoblastic leukemia (ALL) cells upon doxorubicin treatment. A total of 1066 proteins were quantified in both forward and reverse SILAC measurements, among which 62 were significantly altered after doxorubicin treatment. Importantly, we observed, for the first time, the perturbation of cholesterol biosynthesis induced by doxorubicin treatment.
Materials and Methods
Materials
Heavy lysine and arginine ([13C6,15N2]-L-lysine and [13C6,15N4]-L-arginine) were purchased from Cambridge Isotope Laboratories (Andover, MA). All reagents unless otherwise stated were from Sigma (St. Louis, MO).
Cell Culture
Jurkat-T cells, obtained from ATCC (Manassas, VA), were cultured in Iscove’s modified minimal essential medium (IMEM) supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA) and penicillin (100 IU/mL). Cells were maintained in a humidified atmosphere with 5% CO2 at 37ºC, with medium renewal at every 2 or 3 days depending on cell density. For SILAC experiments, the IMEM medium without L-lysine or L-arginine was custom-prepared according to the ATCC formulation. The complete light and heavy IMEM media were prepared by the addition of light or heavy lysine and arginine, along with dialyzed FBS, to the above lysine, arginine-depleted medium. The Jurkat-T cells were cultured in heavy IMEM medium for at least 5 cell doublings to achieve complete isotope incorporation as described by Mann and coworkers22, 23. They demonstrated that culturing cells in heavy medium for five population doublings could result in full heavy isotope incorporation.
Doxorubicin Treatment and Cell Lysate Preparation
In forward SILAC experiment, Jurkat-T cells, at a density of approximately 7.5×105 cells/mL, cultured in light medium were treated with 1 μM doxorubicin (Sigma) for 24 hrs, whereas the cells cultured in heavy medium were untreated. Reverse SILAC experiments were also performed in which the cells cultured in the heavy and light medium were treated with doxorubicin and mock-treated, respectively (Figure 1). After 24 hrs, the light and heavy isotope-labeled cells were collected by centrifugation at 300g, and washed three times with ice-cold PBS.
Figure 1.

Flowcharts of forward and reverse SILAC combined with LC-MS/MS for the comparative analysis of protein expression in Jurkat-T cells upon doxorubicin treatment (A). Shown in (B) is the distribution of expression ratios (treated/untreated) for the quantified proteins.
The cell pellets were then resuspended in the CelLyticTM M cell lysis reagent for 30 min with occasional vortexing. Cell lysates were centrifuged at 12,000 g at 4°C for 30 min, and the resulting supernatants were collected. To the supernatant was subsequently added a protease inhibitor cocktail, and the protein concentrations of the cell lysates were determined by using Quick Start Bradford Protein Assay kit (Bio-Rad, Hercules, CA).
SDS-PAGE Separation and In-gel Digestion
The light and heavy cell lysates were combined at 1:1 ratio (w/w), denatured by boiling in Laemmli loading buffer for 5 min and separated by 12% SDS-PAGE with a 4% stacking gel. The gel was stained with Coomassie blue; after destaining, the gel was cut into 20 bands, in-gel reduced with dithiothreitol and alkylated with iodoacetamide. The proteins were digested in-gel with trypsin (Promega, Madison, WI) for overnight, after which peptides were extracted from gels with 5% acetic acid in H2O and then with 5% acetic acid in CH3CN/H2O (1:1, v/v). The resulting peptide mixtures were dried and stored at −20°C for further analysis.
Exogenous Cholesterol Addition and Cell Viability Assay
The cholesterol-BSA complex was prepared following a previously published method24. A 10-ml aliquot of 1% cholesterol in ethanol was added to the same volume of doubly-distilled water under continuous magnetic stirring at room temperature. The milk-like solution was then centrifuged at 2,000 g for 10 min. The supernatant was discarded, and the pellet was resuspended in a 10-ml solution containing 0.25 M sucrose and 1 mM EDTA (pH 7.3). The resulting white emulsion was stirred gently, to which was slowly added 4 g of BSA at room temperature. Once the BSA was completely dissolved, the pH of the solution was adjusted to 7.3 with Tris, and the solution was then centrifuged at 4°C at 12,000 g for 10 min. The supernatant was collected and used for cholesterol incorporation.
Jurkat-T cells were resuspended in IMEM medium and seeded in 6-well plates at a density of ~ 4×105 cells/mL. To the cells was added doxorubicin solution until its concentration reached 1 μM. The cholesterol-BSA complex solution was added subsequently to the wells containing the control, or doxorubicin-treated cells until cholesterol concentrations reached 30 mg/L or 60 mg/L. After 12 or 24 hrs of treatment, cells were stained with trypan blue, and counted on a hemocytometer to measure cell viability.
Extraction and Determination of the Cellular Level of Cholesterol
Cells that had been washed 3 times with 0.85% NaCl solution in 10 mM sodium acetate were extracted with chloroform:methanol:water (2:1.1:0.9, v/v/v) as specified by Madden25. The chloroform (bottom) layer was washed 3 times with methanol and water (5:4, v/v). The bottom layer was then collected, placed in a test tube, and evaporated to dryness with dry N2 gas. The cholesterol level was determined by HPLC using methanol as mobile phase, and a UV detector, with the wavelength of detection being set at 205 nm, was employed for monitoring the effluent26.
LC-MS/MS for Protein Identification and Quantification
On-line LC-MS/MS analysis was performed on an Agilent 6510 Q-TOF system coupled with an Agilent HPLC-Chip Cube MS interface (Agilent Technologies, Santa Clara, CA). The sample injection, enrichment, desalting, and HPLC separation were carried out automatically on the Agilent HPLC Chip with an integrated trapping column (160 nL) and a separation column (Zorbax 300SB-C18, 75 μm×150 mm, 5 μm in particle size). The peptide mixture was first loaded onto the trapping column with a solvent mixture of 0.1% formic acid in CH3CN/H2O (2:98, v/v) at a flow rate of 4 μL/min, which was delivered by an Agilent 1200 capillary pump. The peptides were then separated with a 90-min linear gradient of 2–60% acetonitrile in 0.1% formic acid and at a flow rate of 300 nL/min, which was delivered by an Agilent 1200 Nano pump.
The Chip spray voltage (VCap) was set as 1950 V and varied depending on the chip conditions. The temperature and flow rate of the drying gas were 325ºC and 4 L/min, respectively. Nitrogen was used as the collision gas; the collision energy followed an equation with a slope of 3 V/100 Da and an offset of 2.5 V. MS/MS experiments were carried out in the data-dependent scan mode with a maximum of five MS/MS scans following each MS scan. The m/z ranges for MS and MS/MS were 300–2000 and 60–2000, and the acquisition rates were 6 and 3 spectra/s, respectively.
Data Processing
Agilent MassHunter workstation software (Version B.01.03) was used to extract the MS and MS/MS data. The data were converted to m/z Data files with MassHunter Qualitative Analysis. Bioworks 3.2 was used for protein identification by searching the m/z Data files against the human IPI protein database (version 3.21) and its reversed complement. The maximum number of miss-cleavages for trypsin was set as two per peptide. Cysteine carbamidomethylation was set as a fixed modification. Methionine oxidation as well as lysine (+8 Da) and arginine (+10 Da) mass shifts introduced by heavy isotope labeling were considered as variable modifications. The mass tolerances for MS and MS/MS were 100 ppm and 0.6 Da, respectively. The searching results were then filtered with DTASelect software, developed by Yates and coworkers27, to achieve a protein false discovery rate <1%.
Protein quantification was carried out using Census28. The ratio obtained for each individual protein was then normalized against the average ratio for all quantified proteins. This “multi-point” normalization strategy assumes that the ratios for the majority of proteins are not affected by the drug treatment, facilitating the use of the average ratio of all quantified proteins to re-scale the data. This has been widely employed to eliminate the inaccuracy during sample mixing introduced by protein quantification with the Bradford assay29, 30. The quantification was based on three independent SILAC and LC-MS/MS experiments, which included two forward and one reverse SILAC labeling experiments, and the proteins reported here could be quantified in both forward and reverse SILAC experiments. Some peptides identified in only 1 or 2 trials of QTOF analysis could be quantified in all three trials, where the accurate mass of peptide ions, retention time, and the numbers of isotope-labeled lysine and/or arginine in the peptide were employed as criteria to locate the light/heavy peptide pairs for the quantification.
Results and Discussion
Doxorubicin Treatment, Protein Identification and Quantification
To gain insights into the molecular pathways perturbed by doxorubicin treatment, we employed SILAC together with LC-MS/MS to assess the doxorubicin-induced differential expression of the whole proteome of Jurkat-T cells. It has been shown that the pharmacokinetics of doxorubicin are highly variable, but the peak plasma concentration of doxorubicin is in the low-to-high31, 32 micromolar range for ALL patients treated with doxorubicin. To perform the proteomic experiments with the optimal dose of doxorubicin, we initially determined the dose-dependent survival rate of Jurkat-T cells upon doxorubicin treatment. We observed, based on trypan blue exclusion assay, a less than 5% cell death after a 24-hr treatment with 1 μM doxorubicin, whereas a significant reduction in cell viability (by ~25%) was induced by a 24-hr treatment with 3 μM doxorubicin. Thus, 1 μM doxorubicin was selected for the subsequent experiments to minimize the apoptosis-induced alteration in protein expression.
Jurkat-T cells were cultured in both light and heavy media. After treating with doxorubicin, the cells were lysed, and the lysates were combined and subsequently fractionated with SDS-PAGE. After in-gel digestion, the proteins were identified and quantified by LC-MS/MS. To obtain reliable quantification results, we conducted triplicate SILAC experiments, with two sets of forward labeling and one set of reverse labeling (Figure 1A and the Materials and Methods section). In total 1306 proteins were identified from either treated or untreated sample, among which 1066 proteins were quantified. Details of all the quantified proteins can be found in supplemental Table S1.
For screening the significantly changed proteins, we considered only the quantification results for those proteins that could be quantified in all three experiments or in two experiments, which included both the forward and reverse SILAC. Figure 2 depicts the representative quantification result of peptide IGVFSYGSGLAATLYSLK from hydroxymethylglutaryl-CoA synthase (HMG-CoA synthase). As can be seen, in both forward and reverse SILAC experiments, this peptide showed significant down-regulation upon treatment with doxorubicin, supporting the down-regulation of the protein from which the peptide is derived (Figure 2A, 2B). In addition, the MS/MS results revealed the unambiguous identification of this peptide (Figure 2C, 2D).
Figure 2.

Representative ESI-MS and MS/MS data revealing the doxorubicin-induced down-regulation of HMG-CoA synthase. Shown are the MS for the [M+2H]2+ ions of HMG-CoA synthase peptide IGVFSYGSGLAATLYSLK and IGVFSYGSGLAATLYSLK* (‘K*’ designates the heavy lysine) from the forward (A) and reverse (B) SILAC experiments. Depicted in (C) and (D) are the MS/MS for the [M+2H]2+ ions of IGVFSYGSGLAATLYSLK and IGVFSYGSGLAATLYSLK*, respectively.
The distribution of the changes in protein expression levels induced by doxorubicin treatment is shown in Figure 1B. Among the 1066 quantified proteins, most did not exhibit significant changes. The average ratio of all proteins was around 1 and the average relative standard deviation of ratios for all quantified proteins was 18%. Thus, we chose a ratio of 1.5 as threshold for screening the significantly changed proteins21, 33. It turned out that a total of 62 proteins displayed significant changes upon doxorubicin treatment (the ratio of treated/untreated was greater than 1.5 or less than 0.67), with 35 and 27 being up- and down-regulated, respectively. The quantification results for the proteins with significant changes are summarized in Table 1, and the detailed information about the quantified peptides are listed in Table S2.
Table 1.
Proteins quantified with more than 1.5 fold changes, with IPI numbers, protein names, average ratios and S.D. listed (Peptides used for the quantification of individual proteins are listed in Table S2).
| IPI Number | Protein Name | Ratio (treated/untreated) |
|---|---|---|
| A. Histones and HMG | ||
| IPI00026272.1 | Histone H2A type 1-B | 2.52 ± 0.28 |
| IPI00171611.5 | H3 histone family, member M | 2.67 ± 0.93 |
| IPI00217465.4 | Histone H1.2 | 2.75 ± 0.56 |
| IPI00018534.3 | Histone H2B | 3.39 ± 0.39 |
| IPI00453473.5 | Histone H4 | 3.48 ± 1.09 |
| IPI00419258.3 | High mobility group protein B1 | 0.52 ± 0.04 |
| B. hnRNPs | ||
| IPI00216049.1 | hnRNP K, splice isoform 1 | 1.82 ± 0.28 |
| IPI00028888.1 | hnRNP D0, splice isoform 1 | 1.85 ± 0.33 |
| IPI00025054.1 | hnRNPU, splice isoform long | 1.97 ± 0.02 |
| IPI00176692.7 | PREDICTED: similar to hnRNP A1 | 2.19 ± 0.72 |
| IPI00334587.1 | hnRNP A/B, splice isoform 4 | 2.51 ± 0.34 |
| IPI00027834.3 | hnRNP L, isoform a | 2.59 ± 0.42 |
| IPI00171903.1 | hnRNP M, isoform a | 3.01 ± 0.81 |
| IPI00334587.1 | hnRNP A/B, splice isoform 2 | 3.18 ± 0.59 |
| IPI00216592.1 | hnRNP C1/C2, splice isoform C1 | 3.38 ± 0.47 |
| IPI00027569.1 | hnRNP C-like 1 | 3.97 ± 0.39 |
| IPI00215965.1 | hnRNP A1, isoform b | 4.15 ± 0.30 |
| IPI00386854.5 | hnRNP A2B1 protein | 4.38 ± 0.87 |
| IPI00396378.3 | hnRNP A2/B1, splice isoform B1 | 4.47 ± 0.23 |
| C. translation related factors | ||
| IPI00023048.3 | Elongation factor 1δ | 0.54 ± 0.13 |
| IPI00005154.1 | Structure-specific recognition protein 1 | 0.56 ± 0.04 |
| IPI00216587.8 | 40S ribosomal protein S8 | 0.64 ± 0.05 |
| D. Enzymes | ||
| IPI00008475.1 | Hydroxymethylglutaryl-CoA synthase, cytoplasmic | 0.49 ± 0.15 |
| IPI00103732.1 | Thymidylate synthase | 0.52 ± 0.05 |
| IPI00218371.3 | Ribose-phosphate pyrophosphokinase III | 0.54 ± 0.14 |
| IPI00101405.1 | Farnesyl diphosphate synthase | 0.60 ± 0.02 |
| IPI00025874.2 | Dolichyl-diphosphooligosaccharide--protein glycosyltransferase 67 kDa subunit precursor | 0.63 ± 0.17 |
| IPI00155168.2 | Protein tyrosine phosphatase, receptor type, C | 0.64 ± 0.09 |
| IPI00020454.1 | Deoxycytidine kinase | 0.66 ± 0.06 |
| IPI00027382.1 | Serine/threonine-protein kinase PAK 3, splice isoform 2 | 0.67 ± 0.02 |
| IPI00014808.1 | Platelet-activating factor acetylhydrolase IB γ subunit | 0.67 ± 0.02 |
| IPI00305978.4 | Aflatoxin B1 aldehyde reductase member 2 | 0.67 ± 0.09 |
| IPI00029997.1 | 6-phosphogluconolactonase | 0.67 ± 0.14 |
| IPI00011118.2 | Ribonucleoside-diphosphate reductase M2 subunit | 1.62 ± 0.24 |
| IPI00022314.1 | Superoxide dismutase [Mn], mitochondrial precursor | 2.17 ± 0.69 |
| E. Others | ||
| IPI00007423.1 | Acidic leucine-rich nuclear phosphoprotein 32 family member B | 0.51 ± 0.04 |
| IPI00003527.4 | Ezrin-radixin-moesin-binding phosphoprotein 50 | 0.55 ± 0.06 |
| IPI00027430.1 | Leukosialin precursor | 0.57 ± 0.14 |
| IPI00411356.5 | Vacuolar sorting protein 4a | 0.60 ± 0.02 |
| IPI00002214.1 | Importin α2 subunit | 0.60 ± 0.14 |
| IPI00004656.1 | β2-microglobulin precursor | 0.62 ± 0.02 |
| IPI00033494.3 | Myosin regulatory light chain | 0.64 ± 0.05 |
| IPI00027493.1 | 4F2 cell-surface antigen heavy chain | 0.65 ± 0.01 |
| IPI00010133.1 | Coronin-1A | 0.66 ± 0.04 |
| IPI00006932.1 | LUC7-like 2 | 0.66 ± 0.12 |
| IPI00301263.2 | CAD protein | 0.67 ± 0.05 |
| IPI00027341.1 | Macrophage capping protein | 0.67 ± 0.10 |
| IPI00298363.2 | Far upstream element-binding protein 2 | 1.62 ± 0.10 |
| IPI00215638.5 | ATP-dependent RNA helicase A | 1.58 ± 0.14 |
| IPI00017617.1 | Probable ATP-dependent RNA helicase DDX5 | 1.60 ± 0.37 |
| IPI00024913.1 | Splice isoform long of ES1 protein homolog, mitochondrial precursor | 1.63 ± 0.26 |
| IPI00017297.1 | Matrin-3 | 1.64 ± 0.31 |
| IPI00479786.2 | KH-type splicing regulatory protein | 1.78 ± 0.20 |
| IPI00220740.1 | Nucleophosmin, splice isoform 2 | 1.89 ± 0.48 |
| IPI00418471.5 | Vimentin | 1.97 ± 0.83 |
| IPI00005198.2 | Interleukin enhancer-binding factor 2 | 2.23 ± 0.15 |
| IPI00221354.1 | RNA-binding protein FUS, splice isoform short | 2.34 ± 0.56 |
| IPI00304596.3 | Non-POU domain-containing octamer-binding protein | 2.35 ± 0.52 |
| IPI00179964.5 | Polypyrimidine tract-binding protein 1, splice isoform 1 | 2.40 ± 0.63 |
| IPI00430812.4 | Zinc finger protein 9 | 2.70 ± 1.42 |
| IPI00301936.3 | ELAV-like protein 1 | 2.97 ± 0.69 |
| IPI00010740.1 | Splice isoform long of splicing factor, proline- and glutamine-rich | 3.75 ± 0.88 |
Histone proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs) are up-regulated upon doxorubicin treatment
Among the differentially expressed proteins, all four core histones were substantially up-regulated in doxorubicin-treated Jurkat-T cells (Table 1). In this respect, histone proteins adopt many sites of post-translational modifications (PTMs), especially on their N-terminal tails34. Doxorubicin treatment may perturb the PTMs of histone proteins, which might result in inaccurate quantifications of their expression levels. Indeed, the exposure of SKN-SH human neuroblastoma cells to doxorubicin could promote acetylation on histone H335. Thus, to avoid the inaccurate quantification of histones introduced by doxorubicin-induced changes in PTMs, we only chose those peptides that do not contain any known PTMs for the quantification34.
Although the mechanisms through which the histone proteins and hnRNPs are up-regulated upon doxorubicin treatment remain unknown, many studies showed that doxorubicin could induce single- and double-strand breaks in DNA mediated by mammalian DNA topoisomerase II and modulate DNA methylation in mammalian cells36, 37. The substantially increased expression of histones might reflect the considerable change in chromatin structure induced by doxorubicin treatment35.
Aside from the substantial up-regulation of histone proteins, doxorubicin treatment led to a systematic down-regulation of several important groups of proteins involved in translation (Table 1). In this context, ribosomal proteins and translation elongation factors were all modestly down-regulated upon doxorubicin treatment (Table S3); the ratios (treated/untreated) for these proteins were 0.85 and 0.79, respectively. These results are in accordance with the previous findings made by Cui et al.17 and with the doxorubicin-induced growth inhibition of Jurkat-T cells (vide infra).
Doxorubicin induced the down-regulation of HMG-CoA synthase and farnesyl diphosphate synthase (FDP synthase)
Our LC-MS/MS quantification results showed that HMG-CoA synthase and FDP synthase were down-regulated by approximately 50% upon doxorubicin treatment. Both enzymes are required for the biosynthesis of cholesterol from acetoacetyl-CoA in human cells25. Therefore, we reason that the intracellular cholesterol concentration should be decreased upon doxorubicin treatment. To test this, we extracted cholesterol from Jurkat-T cells and measured its concentration by HPLC analysis. As displayed in Table 2, treatment of cells with doxorubicin resulted in a statistically significant reduction of the cellular cholesterol content from 53 μg to 35 μg of cholesterol per 107 culture cells. The content of cholesterol returned to a similar level to that of untreated cells at 24 hrs after the addition of exogenous cholesterol. Therefore, this result is in keeping with the doxorubicin-induced suppression of HMG-CoA synthase and FDP synthase, as revealed by quantitative proteomic experiment.
Table 2.
The cellular concentrations of cholesterol in Jurkat-T cells 24 hrs after treatment with none (control), 1 μM doxorubicin (Dox) or both Dox and cholesterol (Cho).
| Cholesterol content (μg/107 cells) | |
|---|---|
| Control cells | 53 ± 2 |
| Cells treated with Dox | 35 ± 1 |
| Cells treated with Dox and Cho | 49 ± 2 |
Cholesterol has been proposed to play an etiological role in various cancers including leukemia38–40. Leukemia cells were reported to have enhanced rates of cholesterol synthesis41, 42, and these cells lack feedback inhibition of cholesterogenesis42. Lymphocytes and leukemia cells also appear to have a special requirement for endogenously synthesized cholesterol for proliferation; specific inhibition of endogenous cholesterol biosynthesis, despite the presence of exogenous cholesterol in the serum-containing growth medium, results in growth inhibition43, 44. Thus, the inhibition of HMG-CoA synthase and FDP synthase may afford an effective route for cancer treatment and prevention.
Based on these previous findings, we reason that the decreased expression of the two important cholesterol biosynthesis enzymes and the resulting diminished cellular cholesterol content may account partly for the cytotoxic effect of doxorubicin. To test this hypothesis, we assessed whether the proliferation of Jurkat-T cells can be perturbed by doxorubicin treatment and how this perturbation is affected by externally added cholesterol. It turned out that exposure to doxorubicin inhibited the proliferation of Jurkat-T cells, and addition of cholesterol to the culture medium indeed rescued Jurkat-T cells from the doxorubicin-induced growth inhibition (Figure 3A&B). This result, corroborated with the doxorubicin-induced down-regulation of HMG-CoA synthase and FDP synthase and its resultant decrease in intracellular cholesterol concentration, supports that doxorubicin may induce the cytotoxic effect through inhibiting endogenous cholesterol biosynthesis via suppressing the expression of these two enzymes.
Figure 3.
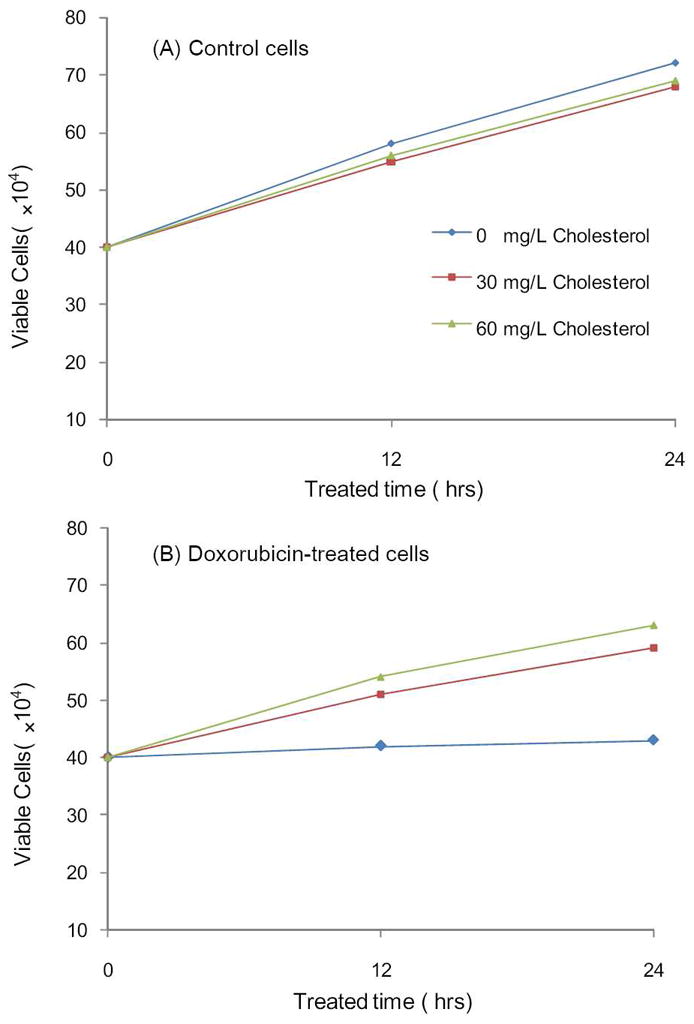
The viabilities of Jurkat-T cells after 12 and 24 hrs of treatment with 0, 30 mg/L or 60 mg/L cholesterol alone (A), or together with 1 μM doxorubicin (B).
Doxorubicin induced the alteration in expression of other important enzymes
Doxorubicin treatment also gave rise to considerable changes in the expression levels of some other important enzymes, including thymidylate synthase, splice isoform 2 of serine/threonine-protein kinase PAK 3 (PAK 3), protein tyrosine phosphatase-receptor type C (PTPRC), Mn-superoxide dismutase (Mn-SOD, or SOD2), and aflatoxin B1 aldehyde reductase member 2 (Table 1). These proteins play pivotal roles in different cellular pathways, and the functions for some of them are discussed below.
We next conducted protein interaction network and pathway analysis using the Ingenuity Pathway Analysis (IPA) software45. Proteins with more than a 1.5-fold change in expression upon the drug treatment were considered for the analysis. Networks represent a highly interconnected set of proteins derived from the input data set. Biological functions and processes were attributed to networks by mapping the proteins in the network to functions in the Ingenuity ontology. We found that several pathways were altered upon doxorubicin treatment, including cholesterol biosynthesis, regulation of actin-based motility by Rho, pyrimidine metabolism, etc. (see Table S4)
PAK 3 and myosin light chain, which participate in the regulation of actin-based motility by Rho, were down-regulated upon doxorubicin treatment. Signaling to the cytoskeleton can lead to diverse effects on cellular activity, including changes in cell shape, migration, proliferation and survival. PAK3 is the critical effector that regulates directly the behavior of the actin cytoskeleton by linking to Rho GTPases46, 47. Signaling through different pathways can lead to the formation of distinct actin-dependent structures whose coordinated assembly/disassembly is important for directed cell migration and other cellular behaviors. Migration is also regulated by signaling to myosin, which participates in leading edge actin dynamics and enables retraction of the rear of the cell. In this work, we observed a ~40% reduction in the expression of PAK 3 and myosin light chain upon doxorubicin treatment, suggesting that doxorubicin treatment may influence the actin polymerization and cell motility.
Three enzymes involved in pyrimidine metabolism, including thymidylate synthase (TYMS), deoxycytidine kinase (DCK) and carbamoyl phosphate synthetase II (CAD), exhibited diminished expression upon doxorubicin treatment. TYMS induces the generation of thymidine monophosphate (dTMP), which is subsequently phosphorylated to thymidine triphosphate and participates in DNA synthesis and repair48. DCK is required for the phosphorylation of several deoxyribonucleosides and their analogs, and this enzyme is clinically important because of its association with drug resistance and sensitivity49. CAD catalyzes the cytosolic production of carbamoyl phosphate which is important in pyrimidine biosynthesis. The decreased expression of these three enzymes, as underscored by the SILAC-based quantification experiment, suggests that doxorubicin treatment might also inhibit cell growth through decreased DNA synthesis.
PTPRC (CD45) and leukosialin precursor (CD43) are proteins involved in the stage of pro-B cell and have been shown to be an essential regulator of T- and B-cell antigen receptor signaling. CD45 is important in promoting cell survival by modulating integrin-mediated FAK/ERK signaling in Jurkat-T cells and is involved in a distinct signal transduction pathway, which is separated from T cell receptor signaling and influences T cell immune responses50, 51. In this study, we observed that CD45 was down-regulated by approximately 35% upon doxorubicin treatment, indicating that exposure to doxorubicin may compromise cell survival through decreasing the expression of CD45 and CD43.
We also found that the level of SOD2 was substantially elevated upon doxorubicin treatment. SOD2 is known to be an important antioxidant defense protein in nearly all cells exposed to oxygen and it converts superoxide to hydrogen peroxide and O2, thereby protecting cells from superoxide toxicity. It is known that doxorubicin can induce ROS formation52; thus, the up-regulation of SOD2 likely reflects the cells’ adaptive response to doxorubicin-induced ROS formation.
It is worth noting that doxorubicin perturbed the expression of some proteins in Jurkat-T cells in the same trend as what we observed recently for HL-60 cells treated with arsenite21. For instance, histones were up-regulated and translation factors were down-regulated under both circumstances. These results suggest that these two anti-leukemic drugs may both perturb chromatin structure and inhibit protein translation. In addition, arsenite treatment could induce the down-regulation of fatty acid synthase in HL-60 cells, whereas exposure to doxorubicin could lead to decrease in cholesterol biosynthesis in Jurkat-T cells. Furthermore, the respective growth inhibition of HL-60 and Jurkat-T cells induced by arsenite and doxorubicin could be rescued by the addition of the final products of these two pathways (i.e., palmitate and cholesterol), which suggested the importance of lipids in leukemia cell survival and may provide a useful rationale for developing novel anti-leukemic therapy.
Conclusions
Doxorubicin is a well-established and clinically successful anticancer drug for ALL treatment53. In this study, we employed SILAC, together with LC-MS/MS, and assessed quantitatively the doxorubicin-induced perturbation of protein expression in a widely studied ALL cell line, namely, Jurkat-T cells. Our results revealed that the drug treatment led to the up- or down-regulation of many important proteins, including HMG-CoA synthase, FDP synthase, SOD2, thymidylate synthase, etc. In addition, most ribosomal proteins and translation elongation factors were modestly down-regulated upon the treatment.
Among the proteins whose expressions were perturbed by doxorubicin, the down-regulation of HMG-CoA synthase and FDP synthase, which are essential enzymes for cholesterol biosynthesis, were of particular importance. Cholesterol is required to establish and maintain cell membranes; it regulates membrane fluidity over the range of physiological temperatures. Cholesterol also functions in intracellular transport and cell signaling. More importantly, cholesterol has been proposed to play an etiological role in various cancers including leukemia38. Endogenous cholesterol biosynthesis is required for lymphocyte proliferation and differentiation43. As a result, cholesterol biosynthesis is markedly enhanced in leukemia cells and inhibition of this pathway has been suggested for cancer treatment41, 42. Our cell survival data revealed that the doxorubicin-induced growth inhibition of Jurkat-T cells could be rescued by cholesterol, the final product of HMG-CoA synthase and FDP synthase. This finding is reminiscent of ALL cell growth inhibition induced by dexamethasone, a specific inhibitor for cholesterol synthesis25. Thus, our result underscored that the inhibition of HMG-CoA synthase and FDP synthase may constitute a novel mechanism for doxorubicin-induced cytotoxic effect.
The current study improves our understanding of mechanisms of doxorubicin-induced anticancer effect, and confirms that the SILAC-based quantitative proteomic analysis is a powerful tool for revealing protein expression related to the effect of an antitumor drug (i.e., doxorubicin) on tumor growth. This approach opens the door for discovering novel molecular pathways that are altered by doxorubicin and defines potential therapeutic targets for the treatment of ALL and other human cancers.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01 CA 116522).
Abbreviations
- ALL
acute lymphoblastic leukemia
- Dox
doxorubicin
- FDP synthase
farnesyl diphosphate synthase
- hnRNP
heterogeneous nuclear ribonucleoprotein
- HMG-CoA synthase
hydroxymethylglutaryl-CoA synthase
- MS
mass spectrometry
- PTM
post-translational modification
- ROS
reactive oxygen species
- SILAC
stable isotope labeling by amino acids in cell culture
- 2-DE
two-dimensional gel electrophoresis
Footnotes
Supporting Information Available: Tables for quantification results. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dimarco A, Gaetani M, Scarpina B. A new antibiotic with antitumor activity. Cancer Chemother Rep. 1969;53:33–37. [PubMed] [Google Scholar]
- 2.Fornari FA, Randolph JK, Yalowich JC, Ritke MK, Gewirtz DA. Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells. Mol Pharmacol. 1994;45:649–656. [PubMed] [Google Scholar]
- 3.Momparler RL, Karon M, Siegel SE, Avila F. Effect of adriamycin on DNA, RNA and protein synthesis in cell-free systems and intact cells. Cancer Res. 1976;36:2891–2895. [PubMed] [Google Scholar]
- 4.Fornari FA, Jarvis WD, Grant S, Orr MS, Randolph JK, White FKH, Mumaw VR, Lovings ET, Freeman RH, Gewirtz DA. Induction of differentiation and growth arrest associated with nascent (non-oligosomal) DNA fragmentation and reduced c-myc expression in MCF-7 human breast tumor cells after continuous exposure to a sublethal concentration of doxorubicin. Cell Growth Differ. 1994;5:723–733. [PubMed] [Google Scholar]
- 5.Buzdar AU, Marcus C, Smith TL, Blumenschein GR. Early and delayed clinical cardiotoxicity of doxorubicin. Cancer. 1985;55:2761–2765. doi: 10.1002/1097-0142(19850615)55:12<2761::aid-cncr2820551206>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 6.Sawyer DB, Fukazawa R, Arstall MA, Kelly RA. Daunorubicin-induced apoptosis in rat cardiac myocytes is inhibited by dexrazoxane. Cir Res. 1999;84:257–265. doi: 10.1161/01.res.84.3.257. [DOI] [PubMed] [Google Scholar]
- 7.Kalyanaraman B, Perezreyes E, Mason RP. Spin-trapping and direct electron spin resonance investigations of the redox metabolism of quinone anticancer drugs. Biochim Biophys Acta. 1980;630:119–130. doi: 10.1016/0304-4165(80)90142-7. [DOI] [PubMed] [Google Scholar]
- 8.Kotamraju S, Konorev EA, Joseph J, Kalyanaraman B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen - Role of reactive oxygen and nitrogen species. J Biol Chem. 2000;275:33585–33592. doi: 10.1074/jbc.M003890200. [DOI] [PubMed] [Google Scholar]
- 9.Wang L, Ma WQ, Markovich R, Chen JW, Wang PH. Regulation of cardiomyocyte apoptotic signaling by insulin-like growth factor I. Cir Res. 1998;83:516–522. doi: 10.1161/01.res.83.5.516. [DOI] [PubMed] [Google Scholar]
- 10.Klose J, Kobalz U. Two-dimensional electrophoresis of proteins: an updated protocol and implications for a functional analysis of the genome. Electrophoresis. 1995;16:1034–1059. doi: 10.1002/elps.11501601175. [DOI] [PubMed] [Google Scholar]
- 11.Castagna A, Antonioli P, Astner H, Hamdan M, Righetti SC, Perego P, Zunino F, Righetti PG. A proteomic approach to cisplatin resistance in the cervix squamous cell carcinoma cell line A431. Proteomics. 2004;4:3246–3267. doi: 10.1002/pmic.200400835. [DOI] [PubMed] [Google Scholar]
- 12.Sinha P, Poland J, Kohl S, Schnolzer M, Helmbach H, Hutter G, Lage H, Schadendorf D. Study of the development of chemoresistance in melanoma cell lines using proteome analysis. Electrophoresis. 2003;24:2386–2404. doi: 10.1002/elps.200305456. [DOI] [PubMed] [Google Scholar]
- 13.Urbani A, Poland J, Bernardini S, Bellincampi L, Biroccio A, Schnolzer M, Sinha P, Federici G. A proteomic investigation into etoposide chemo-resistance of neuroblastoma cell lines. Proteomics. 2005;5:796–804. doi: 10.1002/pmic.200401147. [DOI] [PubMed] [Google Scholar]
- 14.Bertagnolo V, Grassilli S, Bavelloni A, Brugnoli F, Piazzi M, Candiano G, Petretto A, Benedusi M, Capitani S. Vav1 modulates protein expression during ATRA-induced maturation of APL-derived promyelocytes: A proteomic-based analysis. J Proteome Res. 2008;7:3729–3736. doi: 10.1021/pr7008719. [DOI] [PubMed] [Google Scholar]
- 15.Hammer E, Bien S, Salazar MG, Steil L, Scharf C, Hildebrandt P, Schroeder HWS, Kroemer HK, Volker U, Ritter CA. Proteomic analysis of doxorubicin-induced changes in the proteome of HepG2 cells combining 2-D DIGE and LC-MS/MS approaches. Proteomics. 2010;10:99–114. doi: 10.1002/pmic.200800626. [DOI] [PubMed] [Google Scholar]
- 16.Chen ST, Pan LT, Tsai YC, Huang CM. Proteomics reveals protein profile changes in doxorubicin - treated MCF-7 human breast cancer cells. Cancer Lett. 2002;181:95–107. doi: 10.1016/s0304-3835(02)00025-3. [DOI] [PubMed] [Google Scholar]
- 17.Cui Y, Piao CS, Ha KC, Kim DS, Lee GH, Kim HK, Chae SW, Lee YC, Park SJ, Yoo WH, Kim HR, Chae HJ. Measuring adriamycin-induced cardiac hemodynamic dysfunction with a proteomics approach. Immunopharmacol Immunotoxicol. 2010;32:376–386. doi: 10.3109/08923970903440168. [DOI] [PubMed] [Google Scholar]
- 18.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotech. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 19.Ross PL, Huang YLN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 21.Xiong L, Wang YS. Quantitative proteomic analysis reveals the perturbation of multiple cellular pathways in HL-60 cells induced by arsenite treatment. J Proteome Res. 2010;9:1129–1137. doi: 10.1021/pr9011359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ong SE, Kratchmarova I, Mann M. Properties of 13C-substituted arginine in stable isotope labeling by amino acids in cell culture (SILAC) J Proteome Res. 2002;2:173–181. doi: 10.1021/pr0255708. [DOI] [PubMed] [Google Scholar]
- 23.Ong SE, Foster LJ, Mann M. Mass spectrometric-based approaches in quantitative proteomics. Nat Methods. 2003;29:124–130. doi: 10.1016/s1046-2023(02)00303-1. [DOI] [PubMed] [Google Scholar]
- 24.Martinez F, Eschegoyen S, Briones R, Cuellar A. Cholesterol increase in mitochondria: a new method of cholesterol incorporation. J Lipid Res. 1988;29:1005–1011. [PubMed] [Google Scholar]
- 25.Madden EA, Bishop EJ, Fiskin AM, Melnykovych G. Possible role of cholesterol in the susceptibility of human acute lymphoblastic leukemia cell line to dexamethasone. Cancer Res. 1986;46:617–622. [PubMed] [Google Scholar]
- 26.Osada K, Ravandi A, Kuksis A. Rapid analysis of oxidized cholesterol derivatives by high-performance liquid chromatography combined with diode-array ultraviolet and evaporative laser light-scattering detection. J Am Oil Chem Soc. 1999;76:863–71. [Google Scholar]
- 27.Tabb DL, McDonald WH, Yates JR. DTASelect and contrast: Tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park SK, Venable JD, Xu T, Yates JR. A quantitative analysis software tool for mass spectrometry–based proteomics. Nat Methods. 2008;5:319–322. doi: 10.1038/nmeth.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romijn EP, Christis C, Wieffer M, Gouw JW, Fullaondo A, van der Sluijs P, Braakman I, Heck AJR. Expression clustering reveals detailed coexpression patterns of functionally related proteins during B cell differentiation - A proteomic study using a combination of one-dimensional gel electrophoresis, LC-MS/MS, and stable isotope labeling by amino acids in cell culture (SILAC) Mol Cell Proteomics. 2005;4:1297–1310. doi: 10.1074/mcp.M500123-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Uitto PM, Lance BK, Wood GR, Sherman J, Baker MS, Molloy MP. Comparing SILAC and two-dimensional gel electrophoresis image analysis for profiling urokinase plasminogen activator signaling in ovarian cancer cells. J Proteome Res. 2007;6:2105–2112. doi: 10.1021/pr060638v. [DOI] [PubMed] [Google Scholar]
- 31.Eksborg S, Strandler HS, Edsmyr F, Naslund I, Tahvanainen P. Pharmacokinetic study of IV infusions of adriamycin. Eur J Clin Pharmacol. 1985;28:205–212. doi: 10.1007/BF00609693. [DOI] [PubMed] [Google Scholar]
- 32.Frost BM, Eksborg S, Bjork O, Abrahamsson J, Behrendtz M, Castor A, Forestier E, Lonnerholm G. Pharmacokinetics of doxorubicin in children with acute lymphoblastic leukemia: Multi-institutional collaborative study. Med Ped Oncol. 2002;38:329–337. doi: 10.1002/mpo.10052. [DOI] [PubMed] [Google Scholar]
- 33.Yu KH, Barry CG, Austin D, Busch CM, Sangar V, Rustgi AK, Blair IA. Stable isotope dilution multidimensional liquid chromatography-tandem mass spectrometry for pancreatic cancer serum biomarker discovery. J Proteome Res. 2009;8:1565–1576. doi: 10.1021/pr800904z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lennartsson A, Ekwall K. Histone modification patterns and epigenetic codes. Biochim Biophys Acta. 2009;1790:863–868. doi: 10.1016/j.bbagen.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 35.Rebbaa A, Zheng X, Chu F, Mirkin BL. The role of histone acetylation versus DNA damage in drug-induced senescence and apoptosis. Cell Death Diff. 2006;13:1960–1967. doi: 10.1038/sj.cdd.4401895. [DOI] [PubMed] [Google Scholar]
- 36.Dejeux E, Ronneberg JA, Solvang H, Bukholm I, Geisler S, Aas T, Gut IG, Borresen-Dale AL, Lonning PE, Kristensen VN, Tost J. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol Cancer. 2010;9:68. doi: 10.1186/1476-4598-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226:466–468. doi: 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- 38.Chen HW, Kandutsch AA, Heiniger HJ. The role of cholesterol in malignancy. 1978 doi: 10.1159/000401203. [DOI] [PubMed] [Google Scholar]
- 39.Siperstein MD. Feedback control of cholesterol synthesis in normal, malignant, and premalignant tissues. Proc Can Canc Conf. 1967;7:152–162. [PubMed] [Google Scholar]
- 40.Sabine JR. Progressive loss of cellular metabolic controls during hepatic carcinogenesis 1976 [Google Scholar]
- 41.Philippot JR, Cooper AG, Wallach DFH. Regulation of cholesterol biosynthesis by normal and leukemic (L2C) guinea pig lymphocytes. Proc Natl Acad Sci USA. 1977;74:956–960. doi: 10.1073/pnas.74.3.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heiniger HJ, Chen HW, Applegate OL, Schacter LP, Schacter BZ, Anderson PN. Elevated synthesis of cholesterol in human leukemic cells. J Mol Med. 1976;1:109–116. [Google Scholar]
- 43.Heiniger HJ, Marshall JD. Cholesterol synthesis in polyclonally activated cytotoxic lymphocytes and its requirement for differentiation and proliferation. Proc Natl Acad Sci USA. 1982;79:3823–3827. doi: 10.1073/pnas.79.12.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madden EA, Melnykovych G, Fiskin AM. Compactin (ML-236B) reduces the content of filipin-cholesterol complexes in the plasma membrane of chronic lymphocytic leukemia cells. Exp Cell Res. 1984;153:91–98. doi: 10.1016/0014-4827(84)90451-8. [DOI] [PubMed] [Google Scholar]
- 45.Nilsson CL, Dillon R, Devakumar A, Shi SDH, Greig M, Rogers JC, Krastins B, Rosenblatt M, Kilmer G, Major M, Kaboord BJ, Sarracino D, Rezai T, Prakash A, Lopez M, Ji YJ, Priebe W, Lang FF, Colman H, Conrad CA. Quantitative phosphoproteomic analysis of the STAT3/IL-6/HIF1 alpha signaling network: An initial study in GSC11 glioblastoma stem cells. J Proteome Res. 2010;9:430–443. doi: 10.1021/pr9007927. [DOI] [PubMed] [Google Scholar]
- 46.Ahmadian MR, Wittinghofer A, Schmidt G. The actin filament architecture: Tightly regulated by the cells, manipulated by pathogens. EMBO Rep. 2002;3:214–218. doi: 10.1093/embo-reports/kvf057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bokoch GM. Biology of the p21-activated kinases. Ann Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- 48.Kaneda S, Nalbantoglu J, Takeishi K, Shimizu K, Gotoh O, Seno T, Ayusawa D. Structural and functional analysis of the human thymidylate synthase gene. J Biol Chem. 1990;265:20277–20284. [PubMed] [Google Scholar]
- 49.Hazra S, Sabini E, Ort S, Konrad M, Lavie A. Extending thymidine kinase activity to the catalytic repertoire of human deoxycytidine kinase. Biochemistry. 2009;48:1256–1263. doi: 10.1021/bi802062w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuchs A, Atkinson JP, Fremeaux-Bacchi V, Kemper C. CD46-induced human Treg enhance B-cell responses. Eur J Immunol. 2009;39:3097–3109. doi: 10.1002/eji.200939392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bijian K, Zhang L, HSS Collagen-mediated survival signaling is modulated by CD45 in Jurkat T cells. Mol Immunol. 2007;44:3682–3690. doi: 10.1016/j.molimm.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 52.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 53.Muggia FM, Green MD. New anthracycline antitumor antibiotics. Crit Rev Oncol/Hematol. 1991;11:43–64. doi: 10.1016/1040-8428(91)90017-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.