

Abstract
Protein hydroxylation at proline and lysine residues is known to have important effects on cellular functions, such as the response to hypoxia. However, for protein hydroxylation at tyrosine residues (called protein-bound 3,4-dihydroxy-phenylalanine (PB-DOPA) has not been carefully examined. Here we report the first proteomics screening of the PB-DOPA protein substrates and their sites in E. coli and human mitochondria by nano-LC/MS/MS and protein sequence alignment using the PTMap algorithm. Our study identified 67 novel PB-DOPA sites in 43 E. coli proteins, and 9 novel PB-DOPA sites in 7 proteins from HeLa mitochondria. Bioinformatics analysis indicates that the structured region is more favored than the unstructured regions of proteins for the PB-DOPA modification. The PB-DOPA substrates in E. coli were dominantly enriched in proteins associated with carbohydrate metabolism. Our study showed that PB-DOPA may be involved in regulation of the specific activity of certain evolutionarily conserved proteins such as superoxide dismutase and glyceraldehyde 3-phosphate dehydrogenase, suggesting the conserved nature of the modification among distant biological species. The substrate proteins identified in this study offer a rich source for hunting their regulatory enzymes, and for further characterization of the possible contributions of this modification to cellular physiology and human diseases.
Keywords: Protein hydroxylation; protein-bound 3,4-dihydroxy-phenylalanine; PB-DOPA; E. coli; human mitochondria; tyrosine oxidation
INTRODUCTION
Protein hydroxylation has been found in multiple amino acid residues, including proline, lysine, aspartate, asparagine, tryptophan, and tyrosine 1. Members of the Fe(II)- and 2-oxoglutarate-dependent family of dioxygenases can catalyze these hydroxylation reactions 2. Emerging evidence suggest that this family of post-translational modifications (PTMs) play structural roles, and have important cellular functions. For example, prolyl and asparaginyl hydroxylation are central to the regulation of the transcription factor HIFα, the master regulator of the cellular hypoxic response pathway 3, 4. This protein is associated with diverse physiological and pathophysiological conditions, such as angiogenesis, myocardial ischemia, and neuronal ischemia 5. U2AF65 (U2 small nuclear ribonucleoprotein auxiliary factor 65-kilodalton subunit) can be lysine-5-hydroxylated by the Fe(II) and Jmid6 (2-oxoglutarate-dependent dioxygenase Jumonji domain-6 protein); and this modification has a selective effect on the RNA splicing 6. Hydroxylation of proline in the newly synthesized collagen polypeptide chains is essential for the formation of the triple-helical molecules 7. Given the critical roles of the hydroxylation reactions at proline, lysine, and asparagine residues in regulating proteins’ and cellular functions, it is anticipated that hydroxylation at the tyrosine residue will also play a role in cellular physiology.
The tyrosine residue can be oxidized to the protein-bound 3,4-dihydroxyphenylalanine residue (PB-DOPA, or 3-hydroxytyrosine) by enzyme-catalyzed reaction. PB-DOPA can be generated by oxidation of tyrosine residue by tyrosinase or tyrosine oxidase, which are present in neuronal cells and melanocytes of skin, hair follicles, and pigmented epithelial cells of the retina 8. PB-DOPA can be also produced by the enzymatic reactions of bacterial tyrosinase as cresolase activity, and by autooxidation in Streptomyces species 9, 10. Moreover, PB-DOPA could be generated from L-DOPA 11, 12 which is a precursor to the biological pigment melanin and is also the precursor to the neurotransmitters dopamine, norepinephrine, and epinephrine 8, 13, 14. In addition, the L-DOPA has been used in clinics for treating Parkinson’s diseases and dopamine-responsive dystonia, in order to increase dopamine concentration in the brains of patients 15. Limited studies suggest that PB-DOPA is associated with cellular functions.
Determining the substrates of PTMs, and their modification sites, are typically the first steps in study of PTM biology. The history of investigations into protein lysine acetylation provides a good example of substrate identification and functional characterization of a PTM, which the study of protein tyrosine hydroxylation is likely to follow. Lysine acetylation (KAc) was initially identified in histones in 1964 16. The first non-histone substrate protein, p53, was identified in 1997 17. Initially, KAc was thought to be restricted to nuclei because of its initial identification and characterization within histones and transcription factors. However, this paradigm was challenged after the discovery of the first KAc cytosolic proteins 18, 19. Identification of diverse substrates in both cytosolic and mitochondrial fractions 20, and demonstration of the class III histone deacetylases in mitochondria 19, conclusively established the substrate and functional diversities of this PTM. A proteomics screen in mitochondria gave the surprising result that more than 20% of mitochondrial proteins are lysine acetylated in mammalian cells 20, indicating the role of this modification in mitochondria and metabolism 20.
Here, we presented the first systematic screening of PB-DOPA substrates and sites in E. coli and HeLa mitochondria by protein separation, nano-LC/MS/MS, and the PTMap for sequence alignment. Our studies identified 67 novel sites of PB-DOPA in 43 E. coli proteins and 9 novel sites of PB-DOPA in 7 HeLa mitochondrial proteins, present in 2.5% and 0.5% of proteins in E. coli and HeLa mitochondria, respectively. We verified the identification of PB-DOPA by the purified recombinant proteins and MS/MS of the corresponding synthetic peptides. Spectral counting comparison between the modified peptides and their corresponding unmodified counterparts showed that the PTM stoichiometry can be as high as 32.7%. In addition, the E. coli proteins bearing PB-DOPA are highly enriched in cellular metabolism, especially carbohydrate metabolism. This study therefore revealed a previously unexpected role of PB-DOPA in prokaryotic biochemical pathways, and provided evidence that PB-DOPA is an abundant and evolutionarily conserved modification.
EXPERIMENTAL PROCEDURE
Materials
Modified porcine trypsin was purchased from Promega Inc. (Madison, WI). LB-medium was purchased from MP Biomedicals LLC (Solon, Ohio) and Ni-NTA Agarose was a product of QIAGEN GmBH (Valencia, CA). Trichloroacetic acid (TCA) was purchased from Sigma-Aldrich, Inc. (St. Louis, MO). LC/MS grade water and acetonitrile (ACN) were purchased from Honeywell Burdick & Jackson (Muekegon, MI). All other chemicals were of the highest purity available or analytical grade. All the peptides used in this study were synthesized by Genemed Synthesis Inc. (San Antonio, TX).
Preparation of E. coli lysate and mitochondrial fractions from HeLa S3 cell
E. coli K-12 DH10B (Invitrogen, Carlsbad, CA) was cultured in LB media to OD600 = 0.7. The cells were harvested and washed twice with cold PBS. The cell pellet was lysed in NETN buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 0.5% NP40, 20 mM beta-mercaptoethanol, pH 8.0, with the protease cocktail inhibitors) by sonication. The lysate was dialysed against 10 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer, pH 6.0 for 1.5 hrs at 4 °C and then centrifuged at 20,000 x g for 20 minutes. Supernatant was collected for HPLC separation.
Mitochondria were isolated from HeLa S3 cells (Biovest International Inc. Minneapolis, MN) using a discontinuous Percoll gradient as previously reported 21. The mitochondria were lysed by HIM buffer (200 mM mannitol, 70 mM sucrose, 1 mM EGTA, 10 mM HEPES, pH 7.5, with the protease cocktail inhibitors) containing 1% n-dodecyl-D-maltoside on ice with frequent vortexing for 30 mins. Samples were centrifuged at 50,000 x g for 30 mins. The supernatant was collected for HPLC fractionation.
Fractionation of protein lysates
A preparative HPLC system (Shimadzu Scientific Instruments, Kyoto, Japan) was used for protein fractionation. 10-20 mg of a protein lysate of interest was loaded onto a PolyCATWAX column (200 × 9.4mm, 5 m, 1000Å, PolyLC Inc., Columbia, MD). The proteins were eluted using a HPLC gradient from 0% buffer B to 100% buffer B in buffer A for 56 min (mobile buffer A: 10 mM MES, pH 6.0; and mobile buffer B: 10 mM MES, 800 mM NaCl, pH 6.0) at a flow rate of 4 ml/min at 4 °C. HPLC elution was collected every 2 min. The proteins in each fraction were precipitated by conventional trichloroacetic acid (TCA)/acetone method and air dried.
Nano-HPLC/mass spectrometric analysis
The tryptic digest from each fraction was analyzed in an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). The peptides were separated in a home-made capillary HPLC column (100 mm length × 75 μm internal diameter, 5 μm particle size, 100 Å pore diameter) with Jupiter C12 resin (Phenomenex, St. Torrance, CA) using a gradient from 2% to 30% solvent B in solvent A (mobile phase A: 0% ACN in 0.1% formic acid; and mobile phase B: 100% ACN in 0.1% formic acid) for 100 min. The eluted peptides were directly electrosprayed into the mass spectrometer using a nanospray source. The MS was operated in the data-dependent mode to automatically switch between Orbitrap-MS and LTQ-MS/MS (MS2) acquisition. Survey full scan MS spectra (from m/z 350-1800) were acquired in the Orbitrap with resolution R=30,000 at m/z=400. The 10 most intense ions were sequentially isolated for fragmentation in the linear ion trap using collsionally induced dissociation. The following parameters were specified: dynamic exclusion: 36 seconds; the repeat count: 2, and the exclusion window: +2 and −1 Da.
Protein sequencing alignment
All MS/MS spectra were searched against the Refseq protein sequence database for E. coli sub str. K12DH10B (4127 sequences) using Mascot and PTMap software 22. The specific parameters for protein sequence database searching included tyrosine hydroxylation, methionine oxidation, and cysteine alkylation as a variable modification, trypsin as the digesting enzyme, three allowed missing cleavages, and mass error of 15 ppm for precursor ions and 0.6 Da for fragment ions. Charge states of +2 and +3 were considered for parent ions. The ions of the single charge state was not considered, because single charged ions represent lower quality data than ions with +2 and +3 charges. If more than one spectrum was assigned to a peptide, only the spectrum with the highest PTMap score was selected for manual analysis. False discovery rate was estimated using reverse-decoy database strategy by appending a reversed database to the forward database 23. All peptides bearing PB-DOPA identified with peptide score of PTMap > 1.0 (FDR < 5%) were manually examined with the rules described previously 24.
PTM stoichiometry (%) used in this study is the ratio of the number of MS/MS spectra for the peptide bearing the PB-DOPA residue divided by the number of MS/MS spectra for the both peptides bearing the corresponding Tyr site, namely the unmodified peptides with or without missing cleavages (cutoff PTMap score > 0.5, FDR < 1%) and modified peptides (cutoff PTMap score > 1.0, FDR < 5%).
Bioinformatic analysis
Secondary structures of E.coli proteins were predicted using GOR algorithm 25. Biological processes enrichment analysis was performed using Gostats package in the statistical environment R. Probability values (p) of all enriched processes (p<0.01) were first converted into L values: L = -Log10(p) and then Z scores were calculated using the following formula: Z = [L-Ave(L)] / Std(L), where Ave(L) and Std(L) refer to the average and the standard deviation of all L values. Clustering of the biological processes was performed by one-way hierarchical clustering function in Genesis software 26. Evolution conservation analysis was performed using blast search of each PB-DOPA protein against the RefSeq protein database of the six model species – yeast (taxonomy ID: 4932), C. elegans (taxonomy ID: 6239), Drosophila (taxonomy ID: 7227), zebra fish (taxonomy ID: 7955), mouse (taxonomy ID: 10090), human (taxonomy ID: 9606).
MS/MS of synthetic peptides bearing a PB-DOPA residue
The peptides bearing a PB-DOPA residue were synthesized by Genemed Synthesis Inc (San Antonio, TX). All synthetic peptides were verified by MS and MS/MS using the LTQ-XL (Thermo Fisher Scientific, Waltham, MA) coupled with a nano-HPLC system (Agilent 1200 series Nanoflow, Agilent Technologies, Santa Clara, CA). During HPLC/MS/MS analysis, loading of a synthetic peptide was adjusted close to that of its corresponding in vivo peptides. Extensive washing was carried out to avoid peptide carry-over. To verify coelution of the in vivo peptide and its synthetic counterpart, the in vivo peptide bearing a PB-DOPA residue and its corresponding synthetic peptide were monitored by extracted ion chromatogram of specific product ion. The nano-HPLC/mass spectrometric analysis was carried out using the procedure described in Materials and Methods.
Expression and purification of the recombinant proteins
The bacteria strain of interest containing a plasmid expressing an His6-tagged protein of interest, originally developed by National BioResource Project in Japan 27, was grown in LB media with chloramphenicol (30 μg/ml). The cells were treated with isopropyl β-D-1-thiogalactopyranoside (IPTG) (0.04 mM) to stimulate the protein expression for 3 hr, and harvested by centrifugation at 5,000 x g at 4 °C for 20 min. The pellet was washed with 5 ml of cold PBS, resuspended in a lysis buffer (10 mM imidazole with proteinase inhibitor cocktail) and sonicated four times at 4 °C for 10 s each time. Unbroken cells and cellular debris were removed by centrifugation at 20,000 x g at 4 °C for 20 min. Finally, the His6-tagged protein was purified under non-denaturing conditions by a standard procedure using Ni-NTA agarose (Qiagen, Valencia, CA). The purified proteins were resolved in SDS PAGE. The protein band of interest was excised, and in-gel digested for nano-HPLC/mass spectrometric analysis.
In-gel digestion of purified proteins with trypsin
The purified His6-tagged proteins were separated by SDS-PAGE and visualized by staining with colloidal Coomassie blue. The protein bands of interest were excised, and in-gel digested using the clean protocol previously described 28.
RESULTS
Strategy
To identify protein-bearing PB-DOPA residues, the E. coli and HeLa mitochondrial protein lysate were separated into 55 fractions in a mixed-bed PolyCATWAX column (Figure 1A). The proteins in each fraction were precipitated using the TCA/acetone method, and tryptically digested 29. The resulting proteolytic peptides were analyzed by nano-HPLC/LTQ Orbitrap mass spectrometer. The MS/MS data were first analyzed by Mascot algorithm to identify proteins. The MS/MS data and the identified proteins were then used as inputs, and analyzed by PTMap algorithm to identify PB-DOPA peptides and sites (Figures 1B and 1C). The candidate peptides bearing the PB-DOPA residue were further examined manually as previously described, to ensure the exclusive localization of oxidation sites in tyrosine instead of in other adjacent amino acid residues 24, 30.
Figure 1.

Strategy. Procedure for global screening of PB-DOPA in E. coli and HeLa mitochondria (A), MS (B) and MS/MS spectra of an PB-DOPA-containing peptide (LEQYOHFDR) from ribosomal hibernation promoting factor in E. coli (C).
The analysis identified 1,733 and 1,355 proteins from E. coli and HeLa mitochondrial protein lysates, respectively. Among these proteins, we identified 67 PB-DOPA sites in 43 E. coli proteins, and 9 sites in 7 HeLa mitochondrial proteins (Tables 1 and 2, Supplementary_table 1 and Supplementary_figures 1 and 2). These results suggest that about 2.5% and 0.5% proteins were tyrosine hydroxylated in E. coli and HeLa mitochondria, respectively. Given the fact that a peptide bearing a PB-DOPA typically has lower abundance, and will be more difficult to detect in HPLC/MS/MS analysis than its corresponding unmodified counterpart, the real abundance of PB-DOPA should be significantly higher than the percentage calculated above.
Table 1.
List of peptides bearing PB-DOPA in E. coli lysate
| No | Sequencesa | Stochiometryb |
GI number | Protein Information | |
|---|---|---|---|---|---|
| Scan # | % | ||||
| 1 | KLSYTGEVK* | 2/8 | 20.0% | 170079671 | transaldolase_B |
| 2 | LSYDTEASIAK* | 1/7 | 12.5% | 170079671 | transaldolase_B |
| 3 | YLEHR | 1/31 | 3.1% | 170079751 | pyruvate_dehydrogenase,_decarboxylase_component_E1 |
| 4 | EQVAYYKEDEK* | 6/37 | 13.9% | 170079751 | pyruvate_dehydrogenase,_decarboxylase_component_E1 |
| 5 | YAMIGDPTGALTR | 1/39 | 2.5% | 170080288 | alkyl_hydroperoxide_reductase,_C22_subunit |
|
| |||||
| 6 | HYGALQGLNK* | 5/29 | 14.7% | 170080415 | phosphoglyceromutase_1 |
| 7 | AIDFSDGYYK* | 4/17 | 19.0% | 170080470 | glutamine_ABC_transporter_periplasmic_binding_protein |
| 8 | SGTGSVDYAK* | 2/7 | 22.2% | 170080470 | glutamine_ABC_transporter_periplasmic_binding_protein |
| 9 | ADAVLHDTPNILYFIK* | 3/29 | 9.4% | 170080470 | glutamine_ABC_transporter_periplasmic_binding_protein |
| 10 | QFPNIDNAYMELGTNR* | 3/23 | 11.5% | 170080470 | glutamine_ABC_transporter_periplasmic_binding_protein |
|
| |||||
| 11 | ATNLLYTR* | 14/100 | 12.3% | 170080471 | Fe-binding_and_storage_protein |
| 12 | YAIVANDVR | 6/87 | 6.5% | 170080471 | Fe-binding_and_storage_protein |
| 13 | YTSVDQLK* | 2/9 | 18.2% | 170080522 | arginine_ABC_transporter_periplasmic-binding_protein |
| 14 | IEYVYQSAEQLR | 3/40 | 7.0% | 170080661 | glucose-1-phosphatase/inositol_phosphatase |
| 15 | YQQEPGVSGPLK | 1/44 | 2.2% | 170080661 | glucose-1-phosphatase/inositol_phosphatase |
|
| |||||
| 16 | NVAKPLVSYIDK* | 1/10 | 9.0% | 170080661 | glucose-1-phosphatase/inositol_phosphatase |
| 17 | TVTYDFER | 1/21 | 4.5% | 170080787 | isocitrate_dehydrogenase,_specific_for_NADP+ |
| 18 | YYQGTPSPVK | 1/46 | 2.1% | 170080787 | isocitrate_dehydrogenase,_specific_for_NADP+ |
| 19 | LKDGEDPGYTLYDLSER | 1/87 | 1.1% | 170081164 | glutamate_decarboxylase_B,_PLP-dependent |
| 20 | YLSDHPK | 1/63 | 1.6% | 170081164 | glutamate_decarboxylase_B,_PLP-dependent |
|
| |||||
| 21 | TPEGYASGSLGPTTAGR | 1/22 | 4.3% | 170081276 | fumarate_hydratase,_aerobic_class_I |
| 22 | DALAPHISAETIEYHYGK | 1/33 | 2.9% | 170081319 | superoxide_dismutase,_Fe |
| 23 | HHQTYVTNLNNLIK* | 6/21 | 22.2% | 170081319 | superoxide_dismutase,_Fe |
| 24 | EITSTDDFYR* | 2/2 | 100% | 170081336 | pyruvate_kinase_I |
| 25 | LNFSHGDYAEHGQR | 1/7 | 12.5% | 170081336 | pyruvate_kinase_I |
|
| |||||
| 26 | AATYEQIK* | 2/5 | 28.6% | 170081435 | glyceraldehyde-3-phosphate_dehydrogenase_A |
| 27 | LVSWYDNETGYSNK | 1/35 | 2.8% | 170081435 | glyceraldehyde-3-phosphate_dehydrogenase_A |
| 28 | VSQASDSYYYR | 2/55 | 3.5% | 170081885 | hypothetical_protein_ECDH10B_2427 |
| 29 | HLVDLYQQQGVEK* | 4/22 | 15.4% | 170082073 | transaldolase_A |
| 30 | TYQQQVAK | 2/38 | 5.0% | 170082161 | serine_hydroxymethyltransferase |
|
| |||||
| 31 | AGYAEDEVVAVSK | 1/41 | 2.4% | 170082188 | pyruvate_formate_lyase_subunit |
| 32 | LGDIEYR | 2/34 | 5.6% | 170082188 | pyruvate_formate_lyase_subunit |
| 33 | YPQLTIR | 1/116 | 0.9% | 170082188 | pyruvate_formate_lyase_subunit |
| 34 | DAGYTAVISHR | 2/124 | 1.6% | 170082351 | enolase |
| 35 | QLGVSYFLER | 2/51 | 3.8% | 170082411 | 5-keto_4-deoxyuronate_isomerase |
|
| |||||
| 36 | HYHESR* | 2/16 | 11.1% | 170082464 | glycine_decarboxylase,_PLP-dependent,_subunit |
| 37 | ANEAYLQGQLGNPK | 1/35 | 2.8% | 170082482 | fructose-bisphosphate_aldolase,_class_II |
| 38 | DSVSYGVVK | 1/19 | 5.0% | 170082482 | fructose-bisphosphate_aldolase,_class_II |
| 39 | YYDPR* | 3/15 | 16.7% | 170082482 | fructose-bisphosphate_aldolase,_class_II |
| 40 | DSQEYVSK | 2/28 | 6.7% | 170082482 | fructose-bisphosphate_aldolase,_class_II |
|
| |||||
| 41 | SLYEADLVDEAKR* | 4/35 | 10.3% | 170082483 | phosphoglycerate_kinase |
| 42 | EVHIEGYTPEDKK* | 1/10 | 9.1% | 170082514 | YggX |
| 43 | IPLLIHQPSYNLLNR* | 3/24 | 8.1% | 170082547 | aldo-keto_reductase |
| 44 | IYEAAR* | 1/9 | 10.0% | 170082555 | alcohol_dehydrogenase,_NAD(P)-dependent_ |
| 45 | DLADKYGK* | 1/5 | 20.0% | 170082556 | 2,5-diketo-D-gluconate_reductase_A |
|
| |||||
| 46 | AYSEAVK | 1/15 | 6.3% | 170082572 | quinol_monooxygenase |
| 47 | YTQLIER | 2/22 | 8.3% | 170082702 | 30S_ribosomal_subunit_protein_S15 |
| 48 | LEQYFDR* | 2/7 | 22.2% | 170082737 | ribosomal_hibernation_promoting_factor |
| 49 | YADEVVR | 2/25 | 7.4% | 170082825 | 50S_ribosomal_subunit_protein_L6_ |
| 50 | KNPQKNLYTFK | 3/96 | 3.0% | 170083020 | acid-resistance_protein |
|
| |||||
| 51 | LVSSAGTGHFYTTTK | 1/32 | 3.0% | 170083144 | 50S_ribosomal_subunit_protein_L33 |
| 52 | NIFGYQYTIPTHQGR | 8/153 | 5.0% | 170083210 | tryptophanase/L-cysteine_desulfhydrase |
| 53 | YADMLAMSAK | 2/100 | 2.0% | 170083210 | tryptophanase/L-cysteine_desulfhydrase |
| 54 | AYREEAIIK | 1/124 | 0.8% | 170083210 | tryptophanase/L-cysteine_desulfhydrase |
| 55 | FAENAYFIK | 6/135 | 4.3% | 170083210 | tryptophanase/L-cysteine_desulfhydrase |
|
| |||||
| 56 | GDEAYSGSR | 1/67 | 1.5% | 170083210 | tryptophanase/L-cysteine_desulfhydrase |
| 57 | SYYALAESVK* | 9/75 | 10.7% | 170083210 | tryptophanase/L-cysteine_desulfhydrase |
| 58 | VLYGALPR* | 2/11 | 15.4% | 170083249 | sugar_ABC_transporter_ATP-binding_protein |
| 59 | IAGDYIAK* | 16/91 | 15.0% | 170083251 | sugar_ABC_transporter_periplasmic-binding_protein |
| 60 | YPVDLK* | 17/35 | 32.7% | 170083251 | sugar_ABC_transporter_periplasmic-binding_protein |
|
| |||||
| 61 | RPDYIK | 1/26 | 3.7% | 170083379 | superoxide_dismutase,_Mn |
| 62 | EAGVYMR | 1/42 | 2.3% | 170083445 | 50S_ribosomal_subunit_protein_L10 |
| 63 | EVVEQEYR* | 4/21 | 16.0% | 170083482 | glucosephosphate_isomerase |
| 64 | VNYGVTVLPTFK* | 2/5 | 28.6% | 170083491 | maltose_ABC_transporter_periplasmic_substrate- binding_protein |
| 65 | YGYQKDQAEK* | 9/75 | 10.7% | 170083502 | stress_response_protein |
|
| |||||
| 66 | QQIEEATSDYDREK | 17/251 | 6.3% | 170083592 | Cpn60_chaperonin_GroEL,_large_subunit_of_GroESL |
| 67 | AQIAHFFEHYK | 1/30 | 3.2% | 170083670 | inorganic_pyrophosphatase |
Hydroxylation sites are denoted in bold, and underline.
PTM stoichiometry (%) is the ratio of the number of MS/MS spectra for the peptide bearing the PB-DOPA residue divided by the number of MS/MS spectra for the peptides bearing the corresponding Tyr site, namely the unmodified peptides with or without missing cleavages (cutoff PTMap score > 0.5, FDR < 1%) and modified peptides (cutoff PTMap score > 1.0, FDR < 5%).
denotes PB-DOPA peptides with high stoichiometry (≥10%).
Table 2.
List of proteins and peptides bearing PB-DOPA identified in HeLa cell mitochondrial fraction
| No | Proteins | IPI# | Sequences a | Stochiometry b |
|
|---|---|---|---|---|---|
| Spectra # (Modified/Unmodified |
% | ||||
| 1 | Annexin A2 isoform 1 | IPI00418169 | TPAQYDASELK | 1/42 | 2.4 |
|
| |||||
| 2 | Electron transfer flavoprotein subunit alpha | IPI00010810 | VLVAQHDVYK* | 1/9 | 10.0 |
|
| |||||
| 3 | Inosine-5′-monophosphate dehydrogenase 2 | IPI00291510 | VSEYAR* | 1/7 | 12.5 |
|
| |||||
| 4 | Glyceraldehyde-3-phosphate dehydrogenase | IPI00219018 | YDDIKK | 5/71 | 6.6 |
| YDNSLK* | 2/12 | 14.3 | |||
|
| |||||
| 5 | Protein disulfide-isomerase A3 | IPI00025252 | YKELGEK | 3/44 | 6.4 |
| LAPEYEAAATR | 1/55 | 1.8 | |||
|
| |||||
| 6 | Catalase | IPI00465436 | LFAYPDTHR | 5/62 | 7.5 |
|
| |||||
| 7 | Superoxide dismutase 2, mitochondrial | IPI00022314 | HHAAYVNNLNVTEEK* | 2/19 | 9.5 |
Hydroxylation sites are denoted in bold and underline.
PTM stoichiometry (%) is the ratio of the number of MS/MS spectra for the peptide bearing the PB-DOPA residue divided by the number of MS/MS spectra for the both peptides bearing the corresponding Tyr site, namely the unmodified peptides with or without missing cleavages (cutoff PTMap score > 0.5, FDR < 1%) and modified peptides (cutoff PTMap score > 1.0, FDR < 5%).
denote PB-DOPA peptides with high stoichiometry (≥10%).
Verification of the PB-DOPA sites by HPLC/MS/MS analysis of the recombinant proteins and synthetic peptides
To confirm the in vivo PB-DOPA sites, we expressed and purified two His6-tagged proteins bearing the PB-DOPA residues in E. coli (Figure 2 and Table 3), superoxide dismutase Fe (GI 170081319) and pyruvate dehydrogenase, decarboxylase component E1 (GI 170079751). The isolated proteins were resolved in SDS-PAGE, followed by in-gel digestion and nano-HPLC/MS/MS analysis. All four peptides bearing PB-DOPA that had been identified from the two proteins in global screening, were confirmed in the corresponding recombinant proteins (Table 3 and Supplementary_figure 3).
Figure 2.
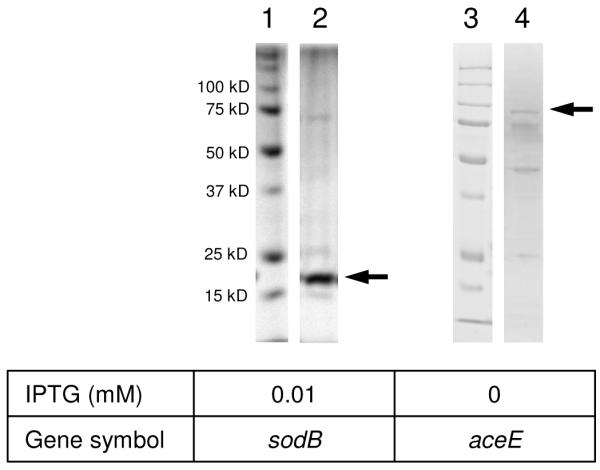
The SDS-PAGE (10-12%) profiles of His6-tagged protein purification in E. coli. Lanes 1 and 3: molecular weight marker (15-100 kDa), respectively; lane 2, sodB; lane 4, aceE.
Table 3.
Peptides bearing PB-DOPA in the His6-tagged purified proteins
| Gene symbol |
Protein name | GI number | Coveragea | Verified peptides bearing PB-DOPAb | Unmodified peptide |
|---|---|---|---|---|---|
| sodB | Superoxide dismutase Fe | 170081319 | 52.6% | HHQTYVTNLNNLIK | + |
| DALAPHISAETIEYHYGK | + | ||||
|
| |||||
| aceE | Pyruvate dehydrogenase, | 170079751 | 52.6% | EQVAYYKEDEK | + |
| Decarboxylase component E1 | YLEHR | + | |||
The coverage was calculated by PTMap. PTMap cutoff score is 0.5 for unmodified peptides.
Hydroxylation sites are denoted in bold and underline.
To further confirm the tyrosine hydroxylation sites in E. coli, we also performed synthetic peptide verification (Figures 3 and 4, and Supplementary_figures 4-6). Two PB-DOPA peptides identified in E. coli lysate were synthesized, namely IAGDYOHIAK in sugar ABC transporter periplasmic-binding protein, and DALAPHISAETIEYOHHYGK in superoxide dismutase Fe. The result showed that the MS/MS spectra of the PB-DOPA peptides from E. coli lysate were exactly matched to the MS/MS spectra of their synthetic counterparts (Figure 3A-C and Supplementary_figure 5A-C), and the in vivo PB-DOPA peptides co-eluted with the synthetic counterparts in HPLC (Figure 3D-F and Supplementary_figure 5D-F). We further verified 8 PB-DOPA peptides identified in HeLa mitochondria by MS/MS spectra matching using synthetic peptides (Supplementary_figure 2). Taken together, our results conclusively confirmed the identification of peptides bearing the PB-DOPA residue.
Figure 3.

Identification and verification of the peptide bearing PB-DOPA (IAGDYOHIAK). Tandem mass spectra (A-C). Tryptic peptide ion from fractionated E. coli lysate (A), synthetic peptide corresponding to the sequences identified in A (B) and mixture of A and B (C). Extracted ion chromatograms of focus ion scans (D-F). Tryptic peptide ion from fractionated E. coli lysate (D), synthetic peptide corresponding to the sequences identified in D (E) and mixture of D and E (F).
Figure 4.

Identification and verification of the peptide bearing PB-DOPA (HHQTYOHVTNLNNLIK). Tandem mass spectra (A-C). Tryptic peptide ion from His6-tagged purified protein (A), synthetic peptide corresponding to the sequences identified in A (B) and mixture of A and B (C). Extracted ion chromatograms of focus ion scans (D-F). Tryptic peptide ion from His6-tagged purified protein (D), synthetic peptide corresponding to the sequences identified in D (E) and mixture of D and E (F).
Evolutionarily conserved PB-DOPA sites in E. coli and human mitochondria
Superoxide dismutase (SOD) is a metal-binding enzyme that catalyzes critical reactions that destroy radicals in the cell. The enzyme is highly redox-sensitive, and involved in catalyzing the dismutation of the superoxide radical anion to prevent the formation of highly aggressive ROS 31-34. In the present study, we identified Y34 (HHQTYOHVTNLNNLIK) as one of the two PB-DOPA sites on the E. coli SOD. The modification site was confirmed by MS/MS analysis of the His6-tagged recombinant protein, and by MS/MS and co-elution experiments of its corresponding synthetic peptide (Figure 4 and Supplementary_figures 7B-C). Previous studies have shown that the Y34 in E. coli SOD is an active site of the enzyme (Supplementary_figure 7A) acting as a proton donor for catalysis 35, promoting heat stability and facilitating substrate binding in both Fe-SODs and Mn-SODs 36, 37. Consequently, it is highly likely that the hydroxylation at Y34 plays a role in regulating its enzymatic activity.
Interestingly, we also identified the PB-DOPA modification at the same residue in human SOD2 from HeLa mitochondria, which is the human ortholog of bacterial SOD. (Supplementary_figure 7A). To verify the identification, we confirmed the MS/MS fragmentation pattern of the PB-DOPA peptide in SOD2 from mitochondria with the corresponding synthetic peptide (Supplementary_figures 7D-E). Previous studies have shown that mutagenesis on Y34, the conserved site in human SOD2, has resulted in decreased enzyme activity 32, 33. Our data therefore revealed an important PB-DOPA site that is evolutionarily conserved, and critical for cellular stress response.
In addition to Y34 in SOD, we also identified an evolutionarily conserved PB-DOPA site on glyceraldehyde 3-phosphate dehydrogenase (GAPDH) – Y256 of GAPDH in E. coli (AATYOHEQIK) (GI 170081435) (Supplementary_figure 8A) and Y253 of GAPDH in HeLa mitochondrial (YOHDDIK) (IPI00219018). GAPDH is an essential enzyme in glycolysis that can catalyze the reaction to convert glyceraldehyde 3-phosphate to D-1,3-bisphospho-glycerate. Accordingly, our data revealed an evolutionarily conserved PB-DOPA site in a protein that is associated with carbohydrate metabolism processes. Identification of the evolutionarily conserved PB-DOPA sites suggests the possibility of a common mechanism that regulates the PTM pathway among a variety of biological species.
We further performed evolution-conservation analysis for all of PB-DOPA sites identified in E. coli lysate among six model species – S. crevisiae, C. elegans, Drosophila, zebra fish, mouse and human. The analysis revealed that among 67 PB-DOPA sites in E. coli, 6 sites were conserved throughout all six species, and 6 sites were conserved in five species (Supplementary_table 2). Among these 12 sites, two sites are from the SOD family and two sites are from GAPDH, including the two PB-DOPA sites that were also identified in HeLa mitochondria analysis.
Structural analysis of PB-DOPA sites in E. coli
Protein phosphorylation has been shown to preferably occur in the unstructured regions, while lysine acetylation is found in structured motifs 38, 39. We studied the secondary structure characteristics of PB-DOPA sites identified in the E. coli lysate, and compared them to the secondary structure characteristics of all Tyr sites in E. coli proteins. Our data showed that PB-DOPA is enriched on the helix structure (p=0.0012) and significantly less represented on the turn structure (p=0.012) comparing to all the tyrosine residues (Figure 5A). Hence, PB-DOPA modification, similar to lysine acetylation, is likely to be enriched in the structured sequences and less abundant in the unstructured regions.
Figure 5.

Bioinformatic analysis. Secondary structure analysis (A) sequence logo analysis of PB-DOPA sites (B) and enrichment analysis (C) in the biological process in proteins bearing PB-DOPA in E. coli.
We further performed sequence logo analysis for the PB-DOPA sites identified in the E. coli lysate (Figure 5B) 40, and compared the abundance of different amino acids on each flanking position of the PB-DOPA site with that of all Tyr residues in E. coli proteins from the database. We found that the positively charged amino acid Lys was enriched in several positions including +9, −5, −1 and +10, while negatively charged amino acid Glu was enriched in +2 and +3 positions.
Biological process enrichment analysis
To elucidate cellular pathways that may be associated with PB-DOPA, we performed enrichment analysis of the biological processes on the identified PB-DOPA proteins by comparing the Gene Ontology (GO) annotations of the PB-DOPA proteins and E. coli proteome (Table 1 and Supplementary_table 1). Our data revealed that proteins bearing PB-DOPA were dominantly enriched in carbohydrate metabolism such as glycolysis and glucogenesis (Figure 5C). Among the 27 biological processes with enrichment p value less than 0.01, 14 processes were involved with carbohydrate metabolism, and all the enriched processes were involved with energy metabolism in the cell.
DISCUSSION
Identification of the PTM substrate proteins and their PTM sites are typically the first steps toward dissection of the PTM pathways, and understanding their biological functions. The protein substrates of tyrosine hydroxylation cannot easily be identified by conventional isotopic labeling. Additionally, there are still no modification-specific pan antibodies, such as those for phosphotyrosine and lysine acetylation, to detect and confirm its PTM peptides. Accordingly, biology studies of tyrosine hydroxylation have previously proceeded very slowly.
To address this problem, we carried out the first proteomics screening to identify PB-DOPA proteins. The study described here identified 67 novel sites of PB-DOPA in 43 proteins in E. coli, and 9 novel PB-DOPA sites in 7 proteins from HeLa mitochondria. Our approach involves unrestrictive sequence alignment using PTMap software developed in-house. Unlike conventional database searching software, PTMap requires exclusive modification-site localization for all unrestrictive PTM identifications. Given that hydroxylation or oxidation can occur on various amino acids (http://www.unimod.org), this feature of PTMap offers unique advantage by removing ambiguous PTM identifications with oxidation on amino acids other than Tyr.
A total of 4,715 and 3,012 Tyr residues were identified from 12,184 non-redundant E. coli peptides and 8,151 HeLa mitochondrial peptides, respectively. These results allow us to estimate the relative abundance of PB-DOPA to be ~ 1.4% and ~ 0.3% in E. coli and HeLa mitochondria, respectively. The result was a surprise to us, as mitochondria is the major cellular organelle that generates radicals and other reactive oxygen species (ROS). The much lower abundance of PB-DOPA in mitochondria than in E. coli could suggest that either ROS may not be the major cause of PB-DOPA in E. coli, or the mechanisms to prevent oxidative damage in mammalian cells are much better developed than E. coli.
Our results suggest that PB-DOPA is likely involved in the regulation of SOD2 activity. The same PB-DOPA site was found in SOD2 protein from both E. coli and HeLa mitochondria. Structure analysis showed that PB-DOPA preferred structured protein segments to unstructured regions. More importantly, proteins bearing PB-DOPA were found to be highly enriched in the pathways involved in cellular energy metabolism and especially carbohydrate metabolism. Our data therefore indicate the possibility of intrinsic cellular mechanism that regulates the PB-DOPA pathway. Finally, to our knowledge, this is the first study demonstrating that the PB-DOPA is present in prokaryotic cells and eukaryotic mitochondria.
Hydroxylation of proline and lysine and their regulatory enzymes such as prolyl hydroxylases are known to contribute to a variety of physiolgical and pathophysiological processes, such as tumorigenesis, erythropoiesis, angiogenesis 41. Hydroxylation at the tyrosine residue changes its structure. It is anticipated that such change is likely to induce alteration of structure and functions for a portion of its substrates, similar to proline hydroxylation (e.g., HIFα). Furthermore the alteration of protein structure can induce the preference of degradation in biological pathway 11, 42.
Many questions remain in the field of tyrosine hydroxylation. Based on the information obtained in this study, PB-DOPA is an abundant protein post-translational modification. It is highly likely that multiple enzymes remain to be identified in the cells. Is the PB-DOPA status dynamically changed between cells in different organs, cell lines, developmental stages, and environmental conditions? What are the main factors that determine the levels of PB-DOPA? What are the major functions of PB-DOPA in cellular functions and pathological conditions? The PB-DOPA substrates identified in this study could drive experimental efforts toward functional characterization of the PB-DOPA proteins and the anatomy of the PB-DOPA pathways.
Supplementary Material
Acknowledgments
This work was supported by NIH grants to Y.Z. and P.S.
Footnotes
Supplemental Informations
This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Walsh CT, Garneau-Tsodikova S, Gatto GJ., Jr. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl. 2005;44(45):7342–72. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 2.Ozer A, Bruick RK. Non-heme dioxygenases: cellular sensors and regulators jelly rolled into one? Nat Chem Biol. 2007;3(3):144–53. doi: 10.1038/nchembio863. [DOI] [PubMed] [Google Scholar]
- 3.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 4.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 5.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14(1):34–44. [PMC free article] [PubMed] [Google Scholar]
- 6.Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, Kessler B, Nielsen ML, Schmitz C, Butler DS, Yates JR, 3rd, Delahunty CM, Hahn P, Lengeling A, Mann M, Proudfoot NJ, Schofield CJ, Bottger A. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science. 2009;325(5936):90–3. doi: 10.1126/science.1175865. [DOI] [PubMed] [Google Scholar]
- 7.Myllyharju J, Kivirikko KI. Characterization of the iron- and 2-oxoglutarate-binding sites of human prolyl 4-hydroxylase. EMBO J. 1997;16(6):1173–80. doi: 10.1093/emboj/16.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozeki H, Ito S, Wakamatsu K, Ishiguro I. Chemical characterization of pheomelanogenesis starting from dihydroxyphenylalanine or tyrosine and cysteine. Effects of tyrosinase and cysteine concentrations and reaction time. Biochim Biophys Acta. 1997;1336(3):539–48. doi: 10.1016/s0304-4165(97)00068-8. [DOI] [PubMed] [Google Scholar]
- 9.Claus H, Decker H. Bacterial tyrosinases. Syst Appl Microbiol. 2006;29(1):3–14. doi: 10.1016/j.syapm.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 10.Liu P, Mehn MP, Yan F, Zhao Z, Que L, Jr., Liu HW. Oxygenase activity in the self-hydroxylation of (s)-2-hydroxypropylphosphonic acid epoxidase involved in fosfomycin biosynthesis. J Am Chem Soc. 2004;126(33):10306–12. doi: 10.1021/ja0475050. [DOI] [PubMed] [Google Scholar]
- 11.Rodgers KJ, Hume PM, Dunlop RA, Dean RT. Biosynthesis and turnover of DOPA-containing proteins by human cells. Free Radic Biol Med. 2004;37(11):1756–64. doi: 10.1016/j.freeradbiomed.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Umeda A, Thibodeaux GN, Zhu J, Lee Y, Zhang ZJ. Site-specific protein cross-linking with genetically incorporated 3,4-dihydroxy-L-phenylalanine. Chembiochem. 2009;10(8):1302–4. doi: 10.1002/cbic.200900127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirshner N, Goodall M. The formation of adrenaline from noradrenaline. Biochim Biophys Acta. 1957;24(3):658–9. doi: 10.1016/0006-3002(57)90271-8. [DOI] [PubMed] [Google Scholar]
- 14.Kirshner N. Pathway of noradrenaline formation from DOPA. J Biol Chem. 1957;226(2):821–5. [PubMed] [Google Scholar]
- 15.Goole J, Amighi K. Levodopa delivery systems for the treatment of Parkinson’s disease: an overview. Int J Pharm. 2009;380(1-2):1–15. doi: 10.1016/j.ijpharm.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 16.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci U S A. 1964;51:786–94. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu W, Shi XL, Roeder RG. Synergistic activation of transcription by CBP and p53. Nature. 1997;387(6635):819–23. doi: 10.1038/42972. [DOI] [PubMed] [Google Scholar]
- 18.Kaiser C, James SR. Acetylation of insulin receptor substrate-1 is permissive for tyrosine phosphorylation. BMC Biol. 2004;2:23. doi: 10.1186/1741-7007-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen T, Yao TP. AcK-knowledge reversible acetylation. Sci STKE. 2004;(245):e42. doi: 10.1126/stke.2452004pe42. 2004. [DOI] [PubMed] [Google Scholar]
- 20.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23(4):607–18. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 21.Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, Delmonte T, Villeneuve A, Sladek R, Xu F, Mitchell GA, Morin C, Mann M, Hudson TJ, Robinson B, Rioux JD, Lander ES. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A. 2003;100(2):605–10. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Chen W, Cobb MH, Zhao Y. PTMap--a sequence alignment software for unrestricted, accurate, and full-spectrum identification of post-translational modification sites. Proc Natl Acad Sci U S A. 2009;106(3):761–6. doi: 10.1073/pnas.0811739106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Kwon SW, Kim SC, Zhao Y. Integrated approach for manual evaluation of peptides identified by searching protein sequence databases with tandem mass spectra. J Proteome Res. 2005;4(3):998–1005. doi: 10.1021/pr049754t. [DOI] [PubMed] [Google Scholar]
- 25.Levin JM, Robson B, Garnier J. An algorithm for secondary structure determination in proteins based on sequence similarity. FEBS Lett. 1986;205(2):303–8. doi: 10.1016/0014-5793(86)80917-6. [DOI] [PubMed] [Google Scholar]
- 26.Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18(1):207–8. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- 27.Chen CS, Korobkova E, Chen H, Zhu J, Jian X, Tao SC, He C, Zhu H. A proteome chip approach reveals new DNA damage recognition activities in Escherichia coli. Nat Methods. 2008;5(1):69–74. doi: 10.1038/NMETH1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng Z, Tang Y, Chen Y, Kim S, Liu H, Li SS, Gu W, Zhao Y. Molecular characterization of propionyllysines in non-histone proteins. Mol Cell Proteomics. 2009;8(1):45–52. doi: 10.1074/mcp.M800224-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang YC. Efficient precipitation and accurate quantitation of detergent-solubilized membrane proteins. Anal Biochem. 1992;205(1):22–6. doi: 10.1016/0003-2697(92)90573-p. [DOI] [PubMed] [Google Scholar]
- 30. Available: www.unimod.org. In.
- 31.Afonso V, Champy R, Mitrovic D, Collin P, Lomri A. Reactive oxygen species and superoxide dismutases: role in joint diseases. Joint Bone Spine. 2007;74(4):324–9. doi: 10.1016/j.jbspin.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Hunter T, Ikebukuro K, Bannister WH, Bannister JV, Hunter GJ. The conserved residue tyrosine 34 is essential for maximal activity of iron-superoxide dismutase from Escherichia coli. Biochemistry. 1997;36(16):4925–33. doi: 10.1021/bi9629541. [DOI] [PubMed] [Google Scholar]
- 33.Landis GN, Tower J. Superoxide dismutase evolution and life span regulation. Mech Ageing Dev. 2005;126(3):365–79. doi: 10.1016/j.mad.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 34.McCord JM, Edeas MA. SOD, oxidative stress and human pathologies: a brief history and a future vision. Biomed Pharmacother. 2005;59(4):139–42. doi: 10.1016/j.biopha.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 35.Miller AF, Sorkin DL, Padmakumar K. Anion binding properties of reduced and oxidized iron-containing superoxide dismutase reveal no requirement for tyrosine 34. Biochemistry. 2005;44(16):5969–81. doi: 10.1021/bi0476331. [DOI] [PubMed] [Google Scholar]
- 36.Castellano I, Cecere F, De Vendittis A, Cotugno R, Chambery A, Di Maro A, Michniewicz A, Parlato G, Masullo M, Avvedimento EV, De Vendittis E, Ruocco MR. Rat mitochondrial manganese superoxide dismutase: amino acid positions involved in covalent modifications, activity, and heat stability. Biopolymers. 2009;91(12):1215–26. doi: 10.1002/bip.21208. [DOI] [PubMed] [Google Scholar]
- 37.Sorkin DL, Miller AF. Spectroscopic measurement of a long-predicted active site pK in iron-superoxide dismutase from Escherichia coli. Biochemistry. 1997;36(16):4916–24. doi: 10.1021/bi963047z. [DOI] [PubMed] [Google Scholar]
- 38.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 39.Gnad F, de Godoy LM, Cox J, Neuhauser N, Ren S, Olsen JV, Mann M. High-accuracy identification and bioinformatic analysis of in vivo protein phosphorylation sites in yeast. Proteomics. 2009;9(20):4642–4652. doi: 10.1002/pmic.200900144. [DOI] [PubMed] [Google Scholar]
- 40.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fong GH, Takeda K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008;15(4):635–41. doi: 10.1038/cdd.2008.10. [DOI] [PubMed] [Google Scholar]
- 42.Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83(3-4):301–10. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.