

Abstract
Although the key neuropathology associated with diencephalic amnesia are lesions to the thalamus and/or mammillary bodies, functional deactivation of the hippocampus and associated cortical regions also appear to contribute to the memory dysfunction. For example, there is loss of forebrain cholinergic neurons and alterations in stimulated acetylcholine (ACh) levels in hippocampus and cortex in animal models of diencephalic amnesia associated with thiamine deficiency. In the present study, the pyrithiamine-induced thiamine deficiency (PTD) rat model was used to assess the functional relationships between thalamic pathology, behavioral impairment, ACh efflux and cholinergic innervation of the hippocampus and cortex. In PTD-treated rats, ACh efflux during behavioral testing was blunted to differing degrees in the hippocampus, medial frontal cortex and the retrospenial cortex. In addition, significant reductions in cholinergic fiber densities were observed in each of those regions. However, only hippocampal cholinergic fiber density correlated significantly with ACh efflux in the same region suggesting that the reduction in cortical ACh efflux in cases of diencephalic amnesia cannot be fully explained by a loss of cholinergic fiber innervation. This notion supports the emerging theory that the functional consequences of the distal effects of lesions go beyond simple deafferentation. Specifically, some frontal cortical regions exhibit hypersensitivity to deafferentation that is only detected during behavioral and/or physiological demand.
Keywords: Acetylcholine, Frontal cortex, Retrospenial cortex, Hippocampus, Spontaneous alternation, Rat, Amnesia
Introduction
Diencephalic amnesia caused by chronic malnourishment or infarct/lesion is associated with neuropathology in the thalamus and mammillary bodies. Patients with amnesia due to Wernicke-Korsakoff Syndrome (WKS) display lesions to those regions and accompanied cortical atrophy (Kopelman, Thompson, Guerrino & Marshall, 2009; Mair, 1994). Interestingly, WKS patients are impaired on tasks of declarative memory, despite relatively intact hippocampal cytoarchitecture (Gold & Squire, 2006; Mayes, Meudell, Mann & Pickering, 1988; Victor, Adams & Collins, 1971; Weiskrantz, 1982). In vivo studies have demonstrated that under conditions of behavioral or chemical stimulation, hippocampal and cortical regions are not fully activated in WKS patients (Caulo et al, 2005; Reed et al, 2003) or in the pyrithiamine-induced thiamine deficiency (PTD) rodent model of WKS (Pires et al, 2005; Roland & Savage, 2007; Savage, Chang & Gold, 2003). Such data support the theory that “functional lesions” of distal brain regions can contribute to the cognitive impairments seen after diencephalic damage (Aggleton, 2008).
The well-established rodent PTD model of WKS has been used to gain insight into brain regions critical for amnesia. This treament produces consistent bilateral lesions of the anterior, midline intralaminar, dorsolateral and posterior thalamic nuclei as well as the mammillary bodies, with more minor lesions in the pontine tegmentum, and periacqueductal grey (Langlais, Zhang & Savage, 1996; Mair, 1994; Witt, 1985). Although the extent of midline thalamic damage has been demonstrated to predict the severity learning and memory impairment in the PTD model (Mair et al, 1988; 1992), alteration in hippocampal and cortical functioning might also explain some of the dysfunction observed in the model and in WKS patients (Oscar-Berman et al, 2003; Reed et al, 2003). Our research group and others have found that after thiamine deficiency in rodents there are changes in hippocampal and cortical cholinergic function that correlate with behavioral impairment (Nakagawasai et al, 2000, Pires et al, 2001; 2005; Savage et al, 2003). The selective loss of basal forebrain cholinergic neurons (Pitkin & Savage, 2001, 2005; Savage, Roland & Klintsova. 2007; Roland and Savage 2009) may explain some of the changes in acetylcholine (ACh) release seen after thiamine deficiency. These neurons modulate hippocampal and cortical neural activity patterns (Hasselmo, 2006) and the loss of forebrain cholinergic neurons and their afferents could exacerbate the diencephalic amnestic syndrome.
To understand the role of cholingeric dysfunction in diencephalic amnesia, we conducted microdialysis during behavioral testing to measure ACh efflux in the hippocampus as well the medial frontal cortex (mFC) and retrospenial cortex (RSC) as these regions have been implicated in the neurobehavioral pathology associated with thiamine deficiency in humans (Oscar-Berman et al, 2004; Reed et al, 2003) and rodents (Kril & Homewood, 1993; Langlais & Zhang, 1997). In addition, acetylcholinesterase (AChE)-labeled axons in the hippocampus, mFC, and RSC were quantified using stereological methods. Using this unique combination of methodologies, we demonstrated significant correlations between behavioral performance, thalamic pathology, and ACh efflux in the hippocampus. Novel findings were that there was blunted ACh efflux in both cortical regions, but this impairment was not associated with a loss of cholinergic fibers. These results support the notion that functional consequences of the distal effects of lesions go beyond simple deafferentation.
Methods
Subjects
Forty male Sprague-Dawley rats (3–4 months; 350–400 g; Harlan Inc., Indianapolis, IN) were used as subjects in this study. They were group housed with unlimited access to water and Purina rodent chow in a colony room on a 12/12-hour light-dark cycle (onset at 7:00 am). All experiments were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1996). The experimental procedures were approved by the Binghamton University Committee on Animal Care and Use.
PTD Treatment
PTD treatment was similar to that previously described (Savage et al, 2007; Roland & Savage, 2009; Vetreno et al, 2008). Briefly, animals were randomly assigned to one of the following treatments: (i) pair-fed (PF, n=20), or (ii) pyrithiamine-induced thiamine deficiency (PTD, n=20). Subjects in the PTD treatment group were free-fed thiamine deficient chow (Teklad Diets, Madison, WI, USA) and received daily injections (0.25 mg/kg, i.p.) of pyrithiamine hydrobromide (Sigma-Aldrich, St. Louis, MO, USA). On days 14–16 of treatment, animals developed signs of local tonoclonic movement of the front and hind limbs, and generalized convulsions (seizures). Within 4 hr of seizure onset, PTD-treated animals were given an injection of thiamine hydrochloride (100 mg/kg, i.p., Sigma-Aldrich) every 8 hr until seizure activity disappeared and the rats regained an upright posture. The PF animals were fed an amount of thiamine deficient chow equivalent to the average amount consumed by the PTD group on the previous day of treatment and received daily injections of thiamine hydrochloride (0.4 mg/kg, i.p.). Animals that receive PF treatment did not demonstrate any changes in thiamine-dependent enzymes in the brain (Buttersworth & Heroux, 1989; Gigurere & Buttersworth, 1987). After treatment, all subjects were placed on regular chow and allowed 21 days to regain the weight lost during treatment.
Cannulae Surgery
Methods for cannulae implantation were as previously described (Anzalone et al, 2009; Savage et al., 2003; Roland & Savage, 2007). Three weeks after PTD or PF treatment, all subjects were prepared for implantation of two plastic guide cannulae for later in-vivo microdialysis (both CMA/11 mm, Carnegie Medicine Associates, Chelmsford, MA, USA). Prior to surgery, animals were anesthetized with an i.p. injection (0.1 ml/kg) of a ketamine (83 mg/kg)/xylazine (17 mg/kg) mixture. After subjects were non-responsive they were placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). All subjects had one cannula aimed at the hippocampus (N=40 [PF=20, PTD=20]) in one hemisphere. In the other hemisphere a cannula was placed in the cortex: Half of the subjects had the second cannula placed in the RSC (n=20 [PF=10 PTD=10]), while the remaining rats had the second cannula implanted into the mFC (n=20 [PF=10, PTD=10]). Hemispheric placement (L, R) of cannulae (hippocampus, cortex) was counterbalanced across subjects. The first cannula was lowered into either the left or right hippocampus (5.0 mm posterior to bregma, 5.0 mm lateral to the midline, and 4.2 mm DV) and the other guide cannula was lowered into either the RSC (6.3 mm AP, 1.0 mm lateral to the midline, and 1.25 mm DV) or the mFC (1.6 mm anterior to Bregma, 2.0 mm lateral to the midline, and 1.20 mm DV) according to the atlas of Paxinos and Watson (1986). After surgery, animals were individually housed and allowed a four day recovery period followed by five days of handling (5 min/day) prior to behavioral testing. All rats were fasted the night before behavioral testing.
In vivo Microdialysis and Behavioral Testing
Nine days following surgery two microdialysis probes (CMA/11, 3 mm for the hippocampus CMA/11, 2 mm for the RSC and mFC) were inserted into the guide cannulae. The probes were connected to 2 meters of plastic tubing and were perfused continuously (CMA/100 pump) at a rate of 2.0 μl/min with artificial CSF (in mM: 128 NaCl, 2.5 KCl, 1.3 CaCl, 2.1 MgCl, Na2HPO4, 2.0 NaH2PO4 and 1.0 glucose, brought to a pH of 7.4) containing the AChE inhibitor neostigmine (500 nM). Prior to maze testing, the microdialysis probes were inserted into the hippocampal and RSC or mFC guide cannulae and the animal was placed into a holding cage (41 cm × 30 cm × 35 cm) located in the testing room. After 60 min of stabilization, dialysis samples were collected into small plastic vials (Phenix Research Products, Candler, NC, USA) every 6 min (sample volume 12 μl) for a period of 18 min in the holding cage to determine basal levels of ACh in awake rats. During this initial baseline phase, the animals were free to move about the holding cage. After initial exploration, most animals sat in a corner and occasionally groomed. After 3 baseline samples (B1–B3) were collected, the rats were gently picked up and placed in the center of the novel maze. The subjects had no prior exposure to the plus maze used for behavioral testing. The apparatus was made of wood with clear Plexiglas sidewalls (12 cm high) and a painted black floor with the four arms of equal distance (55 cm). It was elevated 80 cm from the floor. The rats were allowed to traverse the maze freely for an 18 min period while dialysis samples continued to be collected every 6 min (Maze samples: M1 – M3) and the number and sequence of arms entered were recorded to determine alternation scores. An alternation was defined as an entrance into each of the 4 arms during a successive 4-choice sequence. The percent alteration score was equal to the ratio of: (actual alternations/possible alternations) × 100. Chance performance of this task is 9.3% ([4/4][3/4][2/4][1/4]=0.093). The maze testing room contained various extramaze cues (posters, doors, tables, etc). Upon completion of 18 min of maze testing, rats were transferred back to the holding cage for an additional 18 min and dialysis sample were collected (Post-baseline samples: A1 – A3). Each phase of maze testing (Baseline, Maze, Post-baseline) produced 3 dialysis samples that were frozen until the time of assay.
Dialysate samples were assayed for ACh using high performance liquid chromatography (HPLC) with electrochemical detection (Bio Analytic Systems, West Lafayette, IN, USA). The system included an ion-exchange microbore analytical column, a microbore ACh/Ch immobilized enzyme reactor containing acetylcholinesterase and choline oxidase, and a perioxidase wired working electrode. The detection limit of this system is 10 femtomoles. Acetylcholine peaks were quantified by comparison to peak heights of standard solutions (ACh [Sigma-Aldrich, St Louis, MO, USA] at 100 and 20nM).
Histology
After completion of behavioral testing, animals were anesthetized with 0.5 mg/kg, i.p. injection of Sleep-Away (26% sodium pentobarbital in 7.8% isopropyl alcohol and 20.7% propylene glycol solution; Fort Dodge, Iowa) and perfused transcardially using 0.9% saline solution and 4.0% phosphate-buffered paraformaldehyde (Electron Microscopy Services, Hatfield, PA). The brains were excised, post-fixed in a 10% formalin solution for at least 24 hr and then transferred to a 30% sucrose solution. Coronal sections from the brains were cut (60 μm thick) on a sliding microtome (Sm2000r; Leica Microsystems, Bannockburn, IL, USA) and later stained with cresyl violet for assessment of thalamic pathology and probe location.
Sections were also stained with an antibody for neuron-specific nuclear protein (NeuN) in order to visualize the lesion. Briefly, free floating sections were agitated in Tris buffered saline (TBS) and then rinsed in 0.3% hydrogen peroxide. After three TBS washes, slices were blocked and permebailized using a blocking solution containing 10% normal horse serum in TBS. Slices were then incubated in primary NeuN antibody (Monoclonal Mouse anti-NeuN antibody, 1: 500; Vector Labs, Burlingame, CA, USA ) for 48 hrs and then incubated again in the secondary antibody on the third day (biotinylated horse anti-mouse IgG, 1: 500; Vector Labs). After several TBS washes, the sections were incubated for 2 h in 1:200 avidin–biotin–horseradish peroxidase complex (ABC elite, Vector Laboratories) and visualized using a 0.001% diaminobenzidine in 0.0004% H2O2 solution (Sigma-Aldrich). The reaction was stopped by TBS rinses and sections were mounted and dried on gelatine-coated slides.
Acetylcholinesterase (AChE) Histochemistry
Localization of AChE-positive nerve fibers were visualized and assessed using the AChE Hedreen and Bacon modified Karnovsky-Roots method (Hedreen, et al., 1985). Briefly, brain sections from both PF- and PTD-treated animals were rinsed in a 0.1M sodium acetate buffer (pH 6.0) followed by incubation in a modified Karnovsky-Roots medium (0.1M sodium acetate buffer, acetylthiocholine iodide, sodium citrate, cupric sulfate, dH2O, K3Fe(CN)6, and ethopropazine). The tissue was then washed in a sodium acetate buffer and rinsed in a 4% ammonium sulfide solution. Following a wash in sodium nitrate buffer, the AChE-positive fibers were visualized in a solution containing 0.1% silver nitrate. Sections were then mounted, dehydrated through successive alcohol rinses (95%, 100%, 100%), and cover-slipped with Permount (Fisher Scientific, Fair Lawn, NJ, USA).
Unbiased AChE Stereology
A modified unbiased stereology procedure was conducted to quantify and assess AChE fiber density in the RSC, mFC and ventral hippocampus. A low powered image was captured via a digital camera (DVC-1310; DVC company, Austin, TX) with a 5X lens on a Zeiss microscope (Axioscope 2-plus, Carl Zeiss Microimaging, Thornwood, NY, USA) with an attached 3-axis motorized stage (Ludle, Hawthorne, NY). Histological landmarks were used to trace a contour around each region of interest using StereoInvestigator software (MicroBrightField, 8.0; Williston, VT, USA). Anatomical boundaries used to define the RSC included the outer dorsal border of the zonal layer of the superior colliculus that marked the ventral aspect of the RSC and the outer head of the forcep corpus callosum demarking its most lateral portions. The dorsal and ventromedial portion (the area of cortex located between the ventral portion of the forcep corpus callosum head and the dorsal border of the zonal layer of the superior colliculus) of the RSC were defined within an approximate 120° angle that stemmed from the outer head of the forcep corpus callosum to both the cortex bordering the midline and the outer dorsal surface of the zonal layer of the superior colliculus (approximately Intraural [IA]: 3.4–2.2 mm, Paxinos & Watson, 1986). The anatomical landmark demarcating the location of the mFC was the dorsal aspect of the forceps minor corpus callosum bordering the ventral floor of the mFC. In the most anterior slices (approximately IA: 11.70–10.70 mm) the medial and lateral mFC boundaries were defined within an approximate 55° angle from the dorsal forceps minor corpus callosum to the top of the slice, while more posterior sections were defined with a 30° angle (Approximately IA: 10.20–9.48 mm). In all hippocampal sections (approximately IA: 4.30–2.90 mm), the dorsal aspect of the ventral hippocampus was defined by a horizontal line starting from area Cornu Ammonis (CA)3 that borders the most lateral portion of the thalamus (ventral lateral geniculate nucleus) to area CA1 which neighbors the medial border of the alveus hippocampus. The CA1 and CA3 areas of the hippocampus defined the medial and lateral borders, respectively.
Acetylcholinesterase fibers were counted using an optical fractionator approach with a 100X objective. As can be seen in Figure 1, fibers that crossed the green borders of the counting frame were counted while fibers touching the red lines or fibers outside of the counting frame were not counted. The evaluation interval varied as a function of brain area and was as follows: With a random starting point, every 8th section was selected for mFC, every 6th section for the RSC, and every 5th section for the ventral hippocampus. The fractionator sampling consists of a section sampling fraction (ssf 1/5 or 1/6), an area sampling fraction representing a ratio between counting frame size and grid size (asf 10μm × 10μm/150μm × 150μm) and a height sampling fraction (hsf 20μm/60μm). The equation for determining AChE fiber density was: N=ΣQ ×1/ssf×1/asf×1/hsf, where Q is the number AChE fibers actually counted in the specimens, whereas N represents the total fiber estimation. An average section thickness after tissue processing was 27μm. The mean coefficient of error was less than 0.05.
Figure 1.

Methods for stereological quantification of acetylcholinesterase-labeled (AChE) fibers. X, Y (panel A) and Z dimensions (panel B) for sampling and counting frames used for quantification of AChE fibers. Representative photomicrograph of AChE-labeled fibers (panel C; 100x magnification) with superimposed counting frame.
Intraventricular Distances (IVD) Measure
The amount of midline thalamic tissue loss following PTD treatment can be estimated by measuring the IVD at the midline of the thalamus (Robinson & Mair, 1992; Roland & Savage, 2009b). The IVD (mm) was measured from the floor of the 3rd ventricle to the roof of the 3rd ventricle at the following approximate interaural (IA) locations: 7.20 mm (at the level where the stria medullaris runs longitudinal to the dorsal surface and the ventral lateral nucleus first appears) and 6.44 mm (at the first appearance of the lateral habenula) according to the atlas of Paxinos and Watson (1986). Quantitative measures were determined by the use of video images taken from a Nikon light microscope (Nikon Eclipse E400; Nikon Instruments, Melville, NY, USA) using a Scion 1394 camera JAVA module (Scion Corp; Fredrick, MD, USA) and analyzed using an image analyzer program (IMAGE-J, v.1.34, NIH, Bethesda, MD, USA) on a Macintosh G4 computer.
Results
Histology
We used NeuN immunohistochemistry to document the diencephalic pathology in PTD-treated rats as the expression of NeuN is observed in most neuronal cell types and not in glial cells. Representative thalamic and mammillary body brain sections of both PF (A, C, E) and PTD (B, D, F) treated animals immunostained against NeuN are shown in Figure 2. PTD animals undergo selective neuronal loss in various thalamic and mammillary body nuclei. Specifically, virtually complete neuronal loss was observed in the anteroventral ventrolateral (AVVL) thalamic nucleus with relative sparing of the dorsal anteroventral dorsomedial (AVDM) and anterodorsal nucleus (AD). Marked cell loss was also observed in several midline intralaminar nuclei including central medial nucleus (CM), paracentral nucleus (PC) and centrolateral (CL). This lesion also extended into the posterior (Po) thalamic nucleus. Although the majority of the medial dorsal (MD) complex was spared, there was some loss of neurons in the most ventral portion of MD. In addition, there was significant neuronal loss in the in the hypothalamus including the supramammilary nucleus (SuM) and the medial mammillary nucleus (MM). There was moderate neuronal loss in both the lateral mammillary (LM) and lateral medial mammillary (ML) nuclei.
Figure 2.
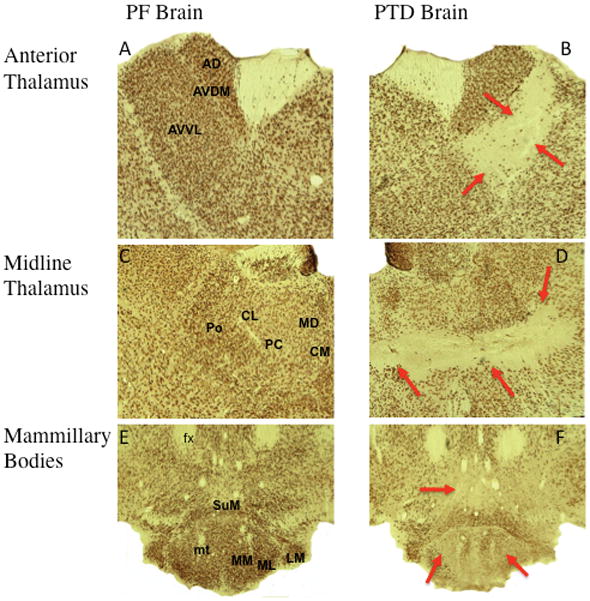
Neuronal specific nuclear protein (NeuN) stained slides comparing the thalamus and mammillary bodies of PF and PTD rats. The top row (A and B) show the state of anterior thalamic nuclei in both PF and PTD animals including the anteroventral ventrolateral (AVVL), anteroventral dorsal medial (AVDM) and anterodorsal nucleus (AD). The middle row of images (C and D) display representative images of midline and intralaminar thalamic structures including the following nuclei and fiber tracts: internal medullary laminae (IML), paracentral nucleus (PC), central medial nucleus (CM), interanteromedial nucleus (IAM), intermediodorsal nucleus (IMD), paraventricular nucleus (PV), central mediodorsal nucleus (MDC), medial and lateral mediodorsal nucleus (MDM/MDL). The bottom row (E and F) display representative samples of the mammillary bodies of both groups including the mammillothalamic tract (MT), mammillary peduncle (mp) medial mammillary nucleus (MM) posterior medial mammillary nucleus (MP), lateral medial mammillary nucleus (ML) lateral mammillary nucleus (LM) and the supramammillary nucleus (SuM).
PTD treatment resulted in significant changes in thalamus. Quantitative analysis of the thalamus showed a significant loss (approximately 30%) of thalamic mass as measured by IVD (Both F’s [1,30]> 10.34, p’s<.01) in PTD rats (2.02 mm ± 0.19 [SEM]) relative to PF rats (2.88 mm ± 0.15).
Cannulae placement was analyzed in all animals. Representative photomicrographs of acceptable cannulae placements are shown in Figure 3 for the hippocampus (A), mFC (B) and RCS (C). Eight subjects were not included in the analyses due to misplaced cannulae. The final number of subjects for each treatment condition was 16 PF rats (mFC/hippo=8, RSC/hippo=8) and 16 PTD (mFC/hippo=8, RSC/hippo=8).
Figure 3.

Histological verification of cannulae placement. Representative photomicrographs of Nissl-stained sections of cannula tracts in medial frontal cortex (mFC; A), retrospenial cortex (RSC, B), and hippocampus (C). Schematic representation of cannula placement of all animals implanted in the mFC (D), RSC (E) and hippocampus (F).
AChE Fiber Quantification
Acetylcholinesterase fiber densities were analyzed on a representative subpopulation of the subjects in the mFC (n=12 [PTD=6, PF=6]), RSC (n=12 [PTD=6, PF=6]) and hippocampus (n=12 [PTD=6, PF=6]) using a one-factor between-subjects analysis of variance (ANOVA: Group: PTD vs. PF) for each region. As shown in Figure 4, the estimated number of AChE positive fibers in both the RSC and mFC of PTD treated animals were significantly reduced compared to PF animals (mFC: F[1,10]= 47.84, p<.0001; RSC: F[1,10] = 14.65, p<.01). Specifically, AChE positive fibers were reduced by approximately 25% in the mFC and 18% in the RSC as a function of PTD treatment. Significant reductions (28%) of cholinergic fibers were also observed in the hippocampus of PTD animals compared to the PF animals (F[1,10] = 8.9, p<.05).
Figure 4.

Reduced estimated number of acetylcholinesterase labeled (AChE) fibers in PTD rats. Schematic representation of regions sampled in the mFC, RSC, and hippocampus (left panels in A–C, respectively) and approximate Interaural distances of sampled sections (middle panels in A–C, respectively). Comparison of estimated total number of AChE fibers in mFC, RSC, and hippocampus (right panels in A–C, respectively). Asterisks indicate significant difference (p<0.05).
Spontaneous Alternation
Behavioral performance on the spontaneous alternation task was analyzed with a 1-factor between subjects ANOVA (Group: PTD vs. PF). As shown in Figure 5, PF rats had higher alternation scores than the PTD rats (Group effect: F[1, 30] = 20.9, p<.0001). However, activity levels, as measured by the number of arm entries, were not different as a function of Group (F [1,30] <1): PTD rats entered 26.94± 2.33 (mean±SEM) arms, whereas PF rats entered 28.93± 2.97 arms during the 18-minute maze test period.
Figure 5.
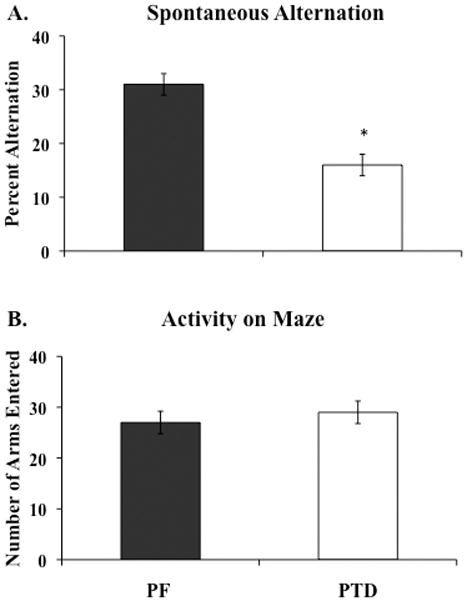
Reduced spontaneous alternation (Mean ± SEM) but not activity levels in PTD rats. Percent alternation scores (panel A) and number of arms entered (panel B) during the 18 min testing period in PF and PTD rats. Asterisks indicate significant difference (p<0.01).
Acetylcholine Efflux
Microdialysis/HPLC data were analyzed three ways: First, to examine differences in hippocampal ACh efflux, all rats were included in a mixed model ANOVA design that included one between-subjects factor (Group: PTD vs. PF) and two within subjects factors (Phase [baseline, maze, post-baseline], Sample time [one, two, three]). Second, given that half the animal had a cortical microdialysis cannula in the mFC and the remaining rats had a cortical microdialysis cannula in the RSC, separate one between-subjects (Group), two within-subjects factors (Phase, Sample Time) repeated measures ANOVAs were conducted for each cortical region. Third, two between-subjects (Group, Brain Region) repeated measures ANOVAs were calculated to assess differences in ACh release during the different phase periods as a function of brain region. Figure 6A–C displays the changes in ACh efflux, relative to baseline levels in each region, as a function of region and behavioral testing and sample time.
Figure 6.

Comparisons of acetylcholine (ACh) efflux profiles (Mean rise above baseline ± SEM) of the medial frontal cortex (mFC, A), retrospenial cortex (RSC, B) and hippocampus (C) of PF (circles) and PTD (squares) animals during baseline (B1, B2, B3), maze (M1, M2, M3) and post-baseline (A1, A2, A3) phases. Acetylcholine efflux significantly increased by 110% over baseline in the mFC and 80% in the hippocampus of PF animals, while PTD animals displayed no significant ACh rise in the mFC and a blunted ACh efflux of only 20% above baseline in the hippocampus (p’s<.01). ACh efflux in the RSC was only mildly impaired as PF rats displayed a 100% increase above basal ACh levels during maze traversal and PTD animals displayed a 60% increase relative to baseline (p=.056). The three graphs also illustrate the differences in ACh profiles between the hippocampus and cortex of intact PF animals.
Medial Frontal Cortex
Basal amounts of ACh in the mFC (91.9 ± 23.43 femtomoles) were not different as a function of Group (F [1,14]=3.32, p=.09). However, as seen in Figure 6-A, there was a significant Group × Phase interaction once behavioral tested began (F [2,28] = 3.78, p<.05): Higher levels of ACh release during maze traversal occurred in the PF rats compared to PTD rats (F [1, 14] =26.03, p<.001). Upon further analyses, PTD-treated animals showed a non-significant rise in behaviorally-evoked ACh efflux during maze exploration (F[1,7]=3.51, p>.09). In contrast, PF rats displayed a significant increase in ACh release during the maze testing (F[1 7]= 80.41, p<.0001), and there was a trend for higher ACh levels to persist after maze running (F [1,7]=4.96, p=.06).
In addition, a Phase × Sample time interaction (F[4,56]=4.76, p<.01) was also observed. Post-hoc analyses revealed that during maze exploration PF animals showed the greatest rise in ACh release during both M2 and M3 (M1 vs. M2: F[1, 7]=8.63, p<.05; M1 vs. M3: F [1, 7]=5.65, p<.05) while no differences in time were seen during post-baseline measures (F[2, 14]=0.44, p>.5). Thus, PF rats demonstrated peak elevations in ACh during the late phases of maze exploration that persisted into the post-baseline phase of behavioral performance. The PTD treatment attenuated this behaviorally-evoked ACh release in the mFC during both maze traversal and post-baseline measurements.
Retrosplenial Cortex
Assessment of baseline levels of ACh efflux (80.18±20.03 femtomoles) in the RSC of PTD and PF rats showed no significant differences (F[1,14]=2.87, p>.1). Retrosplenial ACh levels increased during behavioral testing, relative to baseline levels in the holding cage, for all rats (main effect of Phase: F[2, 28]=30.71, p<.0001). The Group × Phase interaction approached significance (F[2, 28]=3.19, p=.056). Figure 6-B shows that PTD-treated rats had a slightly lower level of ACh release in the RSC (expressed as percent increase above baseline values) during maze testing, relative to PF rats. Sample time interacted with Phase (F[4, 56]=4.46, p<.01) an effect due to differences in peak rises in RSC ACh efflux during different time points (Sample Time) during maze testing (M1 vs M2: F[1,7]=7.39, p<.05) and post-baseline (after) phases (A1 vs A2: F[1, 7]=5.97, p<.05; A1 vs A3: F[1, 7]=6.19, p<.05).
Hippocampus
Basal amounts of ACh in the hippocampus (78.85±15.6 femtomoles) were not different as a function of Group (F[1,30]<1). Hippocampal ACh levels increased during behavioral testing relative to baseline levels in the holding cage for all rats (main effect of Phase: F[2,60]=34.99, p<.0001; see Figure 6-C). However, when animals were behaviorally tested a significant Group × Phase interaction (F[2,60]=8.84, p<.001) emerged for the hippocampal ACh samples. Figure 6-C shows that PTD-treated rats had a significantly lower level of hippocampal ACh release (expressed as percent increase above baseline values) during maze testing, relative to PF rats (F[1,30] =19.48, p<.0001). Sample time did not interact with any other variables (all F’s<1).
Comparisons across brain regions
Across all three regions, PTD rats had blunted ACh efflux compared to PF rats (all F’s [1,14]>7.6, p<.05). The effect in all structures was a function of the facilitated ACh efflux due to behavioral testing (Group × Phase interaction, all F’s[2,28]>3.85, p<.05). However, there were also significant differences in the magnitude of ACh efflux as a function of Brain Region. Both cortical regions had higher behaviorally-induced ACh efflux than the hippocampus. Contrasting ACh efflux in the mFC to that of the hippocampus led to a significant Region × Phase × Sample time interaction (F[4,54]=3.31, p<.05). Specifically, post-hoc analyses showed that ACh efflux was higher in the mFC during both maze performance (F[1, 7]=9.65, p<.05) and post-baseline measurements (F[1, 7]=8.50, p<.05) when compared to the hippocampus in PF rats. No such effect occurred in PTD rats (F[1, 7] <1). When ACh efflux in the RSC was contrasted to that in the hippocampus a similar phenomenon was observed: There was Region × Phase interaction (F [2,28] = 5.13, p<.05). The effect was due to the post-baseline period after maze testing being higher in the RSC than the hippocampus (F [1, 7] = 5.37, p=.05) in PF rats.
Finally, no differences were observed in the magnitude of the behaviorally driven increases in ACh efflux between the two cortical regions (all F’s<1). Both cortical regions displayed a similar pattern of ACh efflux change: The greatest rise was in the later time periods of maze testing (M2 & M3) and the increase in ACh efflux persisted past behavioral testing into the post-baseline period (A1).
Correlations between thalamic tissue loss, fiber density, ACh efflux and spontaneous alternation
As shown in Figure 7, simple regression analyses revealed that alternation behavior was positively correlated with hippocampal (r =0.58, p<.05) and mFC (r =0.56, p<0.05) ACh efflux, but not RSC ACh efflux. However, as shown in Figure 8, the loss of thalamic mass was correlated with the loss of cholinergic fibers in each region (all r’s >0.63, p’s<.05). The highest correlation was seen between hippocampal fiber density and thalamic tissue mass (r=0.79, p<.01). Furthermore, hippocampal fiber density was positively correlated with hippocampal ACh efflux (r =0.76, p<.01). In contrast, cortical ACh efflux did not correlate with fiber density in either the mFC or RSC (see Figure 9).
Figure 7.
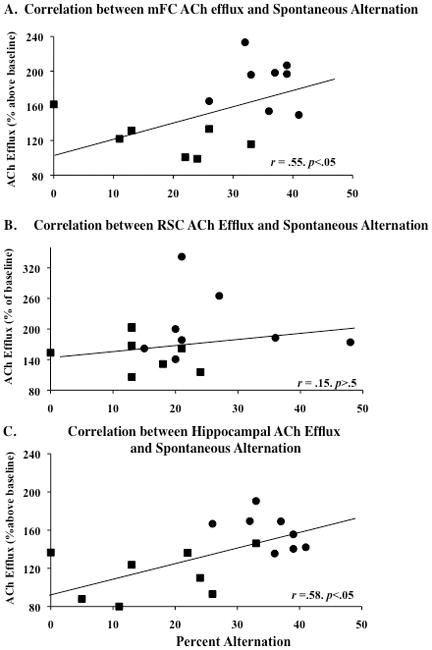
Positive correlations (p’s<.05) between spontaneous alternation performance and acetylcholine (ACh) efflux in the mFC and hippocampus but not RSC. Relationship between spontaneous alternation performance and ACh efflux in the mFC (A), RSC (B), and hippocampus (C) observed in PF (circles) and PTD (squares) groups. The trend lines on each graph show the combined regression analyses for the PTD (squares) and PF (circles) groups.
Figure 8.
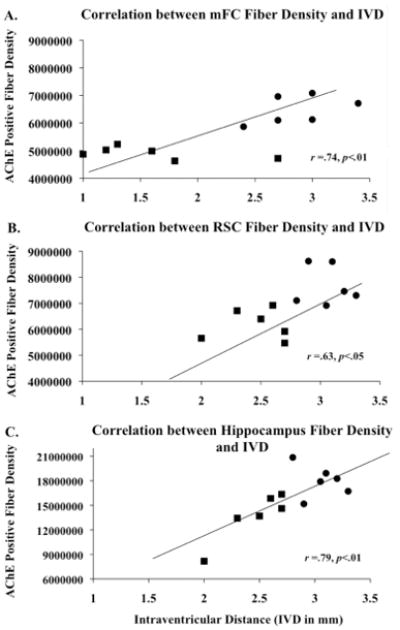
Positive correlations (p’s<.05) between thalamic mass/intraventricular distance (IVD) and acetylcholinesterase-labeled (AChE) fibers in the medial frontal cortex (mFC), retrospenial cortex (RSC), and hippocampus. Relationship between IVD and ACh fibers in the mFC (A), RSC (B), and hippocampus (C) observed in PF (circles) and PTD (squares) groups.
Figure 9.
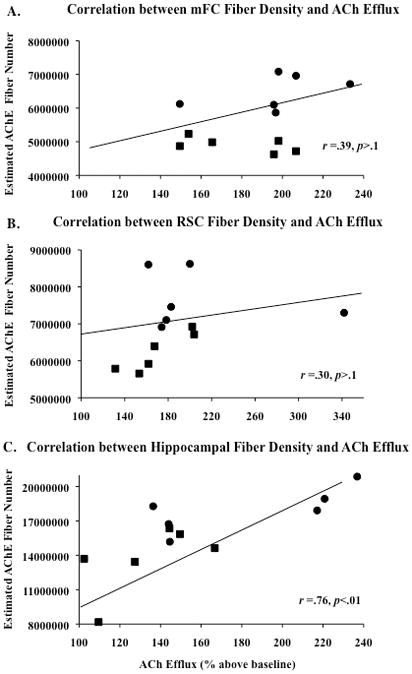
Correlations between estimated acetylcholinesterase-labeled (AChE) fiber number and ACh efflux in the he medial frontal cortex (mFC, A), retrospenial cortex (RSC, B) were not significant. However, there was a positive correlation between AChE positive fiber numbers and acetylcholine (ACh) efflux in the hippocampus (C: p<.01). Trend lines of each graph display the combined regression analyses for the PTD (squares) and PF (circles) groups.
Discussion
The present study is the first to demonstrate that behaviorally-evoked ACh efflux is not only blunted in the hippocampus, but is also reduced in two critical cortical regions in the PTD model. However, different cortical regions display varying degrees of cholinergic dysfunction. Although PTD treatment led to reductions in AChE fiber density in the hippocampus, mFC and the RSC as well as blunted ACh efflux, only hippocampal and mFC ACh efflux was positively correlated with spontaneous alternation behavior. In contrast, while RSC ACh efflux was blunted in PTD rats, this effect did not correlate with behavioral performance.
It should be noted that the midline thalamic damage is critical to the development of the amnestic syndrome seen in the PTD model and the human disorder of WKS (Mair, 1994). The reported loss of cortical (mFC and RSC) and hippocampal cholinergic fiber density significantly correlated with loss in thalamic mass (as measured by intraventricular distance [IVD]). Thus, in PTD-treated rats there was an association between loss of cholinergic fiber density and shrinkage of midline thalamic tissue. In addition, our previous work has also demonstrated a strong correlation (r=0.84) between a loss of cholinergic forebrain neurons and IVD; furthermore, both of those brain measures significantly correlated (r’s=0.66 and 0.65) with alternation performance (Roland & Savage, 2009b). These data match the literature demonstrating that the behavioral impairment in the PTD model is consistently associated with the degree of thalamic tissue loss (Langlais & Savage, 1995; Robinson & Mair, 1992) and that lesions to the midline thalamic nuclei produce behavioral impairment that mimics the PTD model (Burk & Mair, 1998; Mair & Lancourse, 1992; Savage et al, 1997). It remains unknown whether there are unique contributes of midline thalamic pathology, forebrain cholinergic cell loss and cholinergic fiber loss to impaired hippocampal or cortical ACh release and behavioral impairment. It is extremely difficult -- if not impossible -- to determine exact behavioral contributes of a single structure: Even selective lesions lead to tissue shrinkage and cell loss (Savage et al, 1998) or functional deactivation (Vann & Albasser, 2009; Jenkins et al, 2004) in distal brain regions including the cortex. A second consideration is whether the distal effects of lesions can be more than the degree of deafferentation. It appears that in the hippocampus and some cortical regions there is a greater loss plasticity than deafferentation after damage to subcortical structures, rendering the regions “functionally lesioned” (Aggleton, 2008).
Acetylcholine levels in the cortex and hippocampus are blunted during maze exploration in the PTD model-- but to varying degress of amplitude and temporal progression. For example, both the mFC and RSC displayed higher ACh efflux relative to the hippocampus and show asymptotic levels later during maze traversal. These findings are consistent with previously established data demonstrating higher levels of cortical ACh during various cognitive tasks (Acquas et al., 1996; Anzalone et al., 2009; Giovannini et al., 1998; Passetti et al., 2000; Pepeu & Giovannini, 2008; Thiel et al., 1998a). Similar to the present study, Giovannini and colleagues (1998) found that spontaneous alternation induced an approximate 300% increase in cortical ACh release whereas other studies have shown only a 130–180% increase in the hippocampus (McIntyre et al., 2002; Ragozzino et al., 1996, 1998; Roland et al, 2008; Savage et al., 2007). However, caution should be taken with this interpretation, as the different probe lengths and variation in AChE activity across the regions may contribute to regional differences in ACh efflux. With this is mind, all data in the present studies as a function of baseline for each region. The temporal change in ACh efflux between the cortex and hippocampus also replicates our recent findings in non-manipulated rats (Anzalone et al., 2009).
Although in the PTD model we have consistently demonstrated a moderate loss of cholinergic neurons (25–30%) in the medial septum/diagonal band (Pitkin & Savage, 2001, 2005; Savage et al, 2007; Roland & Savage, 2009b), this moderate loss of cholinergic cells alone cannot fully account for the extent of behavioral impairment observed as selective cholinergic cell loss alone does not produce a complete amnestic syndrome (for a review see Parent & Baxter, 2004). It is possiable that thiamine deficiency-induced damage to the thalamus and mammillary bodies affects both hippocampal and cortical cholinergic functioning. Acetylcholine efflux has been used a measurement of structural activation during cognitive processing (Gold, 2004). Anatomical evidence that these brain regions are strongly interconnected and behavioral findings suggest that the hippocampus, anterior thalamic nuclei and mammillary bodies depend on each other during the acquisition of some spatial tasks (Aggleton, 2008). We do know that lesion of the anterior thalamus disrupts behaviorally-evoked immediate early gene activation in the hippocampus and limbic cortical regions (Jenkins et al 2002). However, it has not been determined whether selective lesions of the diencephalic structures disrupt ACh release in the hippocampus or cortex during cognitive processing.
Recent data, including the current findings, provide some evidence that cholinergic cell loss, the accompanied loss of cholinergic fibers and blunted ACh release does contribute to the amnestic syndrome in the PTD model. We do know that selective stimulation of the medial septal or hippocampal regions by drugs that increase ACh levels in the PTD model (Roland, Levinson, Vetreno & Savage, 2010; Roland, Mark, Vetreno & Savage, 2008; Roland & Savage, 2009a) or by electrical stimulation of the midline thalamus (Mair & Hembrook, 2008) can lead to cognitive recovery or enhancement. Such results suggest that activation of some memory-related structures can enhance cognitive processing throughout multiple circuits. Thus, the results from the present study suggest that cortical -- in particular frontal cortical -- hypoactivation contributes to the amnesic state reported in the thiamine deficiency model of diencephalic amnesia.
Previous work has revealed widespread degeneration of cortical fibers and a minimal loss of hippocampal fibers in both the pre- and post-lesion phases of PTD treatment (Langlais & Zhang, 1997). To gain insight into the mechanisms of reduced functional ACh activation, the density of cholinergic fibers (AChE) into these regions was determined. There was a comparable modest -- but significant -- loss of cholinergic fibers innervating all three regions. However, only fiber loss in the hippocampus correlated with corresponding ACh efflux suggesting that the loss of medial septal/diagonal band cholinergic neurons that we have reported in recent papers contributes to the observed hippocampal ACh efflux dysfunction (Savage et al, 2007; Roland & Savage, 2009). Furthermore, we have previously documented a nonsignficant loss of cholinergic neurons in the nucleus basalis magnocellularis of PTD-treated rats (Savage et al, 2007). This non-significant loss of nucleus basalis magnocellularis cholinergic neurons that project to the cortex likely contributes to the lack of correlation between cholinergic fiber density and ACh efflux in the cortical regions. It is interesting that even though there was not a significant loss of cholinergic neurons projecting to the cortex -- there was a significant loss of cholinergic fibers. Thus, even a subtle loss of projection neurons can significantly impact fiber innervation of the target region. However, the loss of fibers did not fully explain the scale of reduction in behaviorally stimulated ACh release in PTD rats. The region that had the greatest reduction of behaviorally stimulated ACh release after PTD treatment, relative to PF rats, was the mFC (95% reduction), followed by the hippocampus (80%) and then the RSC (40%). Only fiber loss in the hippocampus correlated with both ACh efflux and behavior. Alternation performance also correlated with mFC ACh release, but not mFC fiber density. Thus, although there is evidence of specific AChE fiber loss in the current data set and more global fiber degeneration in the PTD model (Langlais & Zhang, 1997), the loss of fibers does not appear to fully explain the behavioral impairment. A previuos study did document that rats with the most severe thalamic lesions had the greatest loss of both cortical white and gray matter along with the maximal behavioral impairment (Langlais & Savage, 1995).
In the RSC, there were no correlations between AChE fibers, ACh release or behavior. This could be accounted for by two factors: First, damage to the RSC appears to more severely disrupt spatial performance when interoceptive cues are more important than external visual cues (Pothuizen, Aggleton & Vann, 2008). In the standard alternation task, both types of cues are available and down-regulation of the RSC could be partially compensated for by greater reliance on external cues (Pothuizen, Davies, Aggleton, & Vann, 2010). Second, cholinergic innervation of the RSC is more diverse than either the hippocampus or mFC. Cholinergic cells in the rat forebrain are organized into 4 subdivisions (Ch1-4) that have different innervation patterns: Ch1 (medial septal area) and Ch2 (diagonal band) innervate the hippocampus and surrounding cortical areas; Ch3 (Horizontal limb of the diagonal band of Broca) innervates the olfactory bulb, whereas Ch4 (nucleus basalis magnocellularis) innervates the neocortex and amygdala (Dougherty et al, 1991). Amaral and Kurz (1985) further noted three distinct pathways within the Ch1/Ch2 cholinergic projection system (dorsal, ventral, and intermediate). The dorsal group innervates the ventral hippocampus, whereas the ventral group innervates the dorsal hippocampus. In contrast, the intermediate group innervates the cingulate cortex—without providing hippocampal connections. Recent evidence suggests that the RSC receives greater innervation from Ch2 over Ch1 (Gonzalo-Ruiz & Morte, 2000). In addition, the RSC receives abundant cholinergic innervation from Ch4 (Big et al, 1982, Eckenstein et al, 1988; Gage et al, 1994; Mesulam et al, 1983). This more diverse cholinergic innervation may make the RSC less vulnerable, relative to the hippocampus or mFC, to thiamine deficiency.
Thus, a direct loss of cholinergic fibers in the hippocampus correlates with blunted hippocampal ACh and behavioral impairment in the PTD model. However, such relationships between cholinergic fiber loss and reductions in behaviorally-driven ACh efflux do not exist in the cortex. A previous study looking specifically at axon degeneration reported qualitively greater axon degeneration in the frontal cortex than in the RSC (Langlais & Zhang, 1997). Thus, although AChE fiber number did not correlate with ACh release, it is possiable that the generalized cortical fiber degeneration that occurs in the PTD model could account for the differences in cortical vulnerability to the cholinergic deficit.
A few other studies have looked at neurochemical dysfunction in the cortex after recovery from thiamine deficiency. Serotonin has been shown to be increased (Langlais, Mair, Anderson & McEntee, 1988), whereas norpinephrine was found to be decreased (Mair, Anderson, Langlais & McEntee, 1985) in homogenate tissue samples that included both the hippocampus and cortex. However, no correlations between neurochemical changes and behaviors were conducted in those studies. In addition, stimulated frontal cortical glutamate release is decreased in rats recovered by PTD treatment, but it did not correlate with behavioral impairment on the water maze (Carvalho et al, 2006). In contrast, as in the present study, a number of measures of cholinergic function after thiamine deficiency have been found to correlate with poor memory performance. Like the present study, significant correlations were found between cortical and hippocampal AChE activity and/or ACh release and measures of memory performance (Pires, Pereira, Oliveria-Silva, Franco & Ribeiro, 2005; Savage et al., 2003). Thus, to date we have the most information about the role of ACh dysfunction contrubuting to the behavioral impairments observed in the PTD model.
In summary, our results suggest that certain cortical regions may exhibit hypersensitivity to the deafferentation and reorganization of the brain after damage to the thalamus and hypothalamus. There is some loss of cholinergic innervation of the frontal cortex (25%), but the modest extent of this fiber loss cannot account for the complete lack of behaviorally-related increase in mFC ACh release. Other models of diencephalic amnesia have shown that behaviorally driven activation of cortical regions, as measured by immediate early genes, is severely impaired following lesions of the anterior thalamus (Jenkins et al., 2002; 2004) or mammillary bodies (Albasser & Vann, 2009). Such results suggest that cortical deafferentation likely plays a minor role in behaviorally driven hypoactivity of cortical regions in models of diencephalic amnesia. Rather, it appears that cholinergic cortical dysfunction associated with diencephalic amnesia is likely a consequence of a lack of cortical plasticity during cognitive processing that can not be attributed to simple deafferentation. Thus, the lack of cholinergic activation (ACh efflux) during behavioral testing in the frontal cortex in the PTD model fits the criteria for a “functionally-lesioned” structure (see Aggleton, 2008).
Acknowledgments
This research was funded by grant NINDS 054272 to LMS. The authors would like to thank Jess Blackwolf and Obeta Lavin for their assistance in histology.
Abbreviations
- ACh
Acetylcholine
- AChE
Acetylcholinesterase
- ANOVA
Analysis of Variance
- CA
Cornu Ammonis
- HPLC
High Performance Liquid Chromatography
- IVD
Intraventricular distance
- mFC
medial Frontal Cortex
- PTD
Pyrithiamine-induced Thiamine Deficiency
- PF
Pair-fed
- RSC
Retrospenial Cortex
References
- Acquas E, Wilson C, Fibiger HC. Conditioned and unconditioned stimuli increase frontal cortical and hippocampal acetylcholine release: Effects of novelty, habituation and fear. Learn Mem. 1996;16:21–27. doi: 10.1523/JNEUROSCI.16-09-03089.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggleton JP. Understanding anterograde amnesia: Disconnections and lesions. Q J Exp Psychol. 2008;61:1441–1471. doi: 10.1080/17470210802215335. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Kurz J. An analyses of the origins of the cholinergic and non-cholinergic septal projections to the hippocampal formation of the rat. J Comp Neurol. 1985;240:37–59. doi: 10.1002/cne.902400104. [DOI] [PubMed] [Google Scholar]
- Anzalone SA, Roland J, Vogt B, Savage LM. Acetylcholine efflux from retrosplenial areas and hippocampal sectors during maze exploration. Behav Brain Res. 2009;201:272–278. doi: 10.1016/j.bbr.2009.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Big IV, Woolf NJ, Butcher LL. Cholinergic projections from the basal forebrain to frontal, parietal, temporal, occipital, and cingulate corticies: A combined fluorescent tracer and acetylcholinesterase analysis. Brain Res. 1982;8:727–763. doi: 10.1016/0361-9230(82)90101-0. [DOI] [PubMed] [Google Scholar]
- Burk JA, Mair RG. Thalamic amnesia reconsidered: excitotoxic lesions of the intralaminar nuclei, but not the mediodorsal nucleus, disrupt place delayed matching-to-sample performance in rats (Rattus norvegicus) Behav Neurosci. 1998;112:54–67. [PubMed] [Google Scholar]
- Butterworth RF, Héroux M. Effect of pyrithiamine treatment and subsequent thiamine rehabilitation on regional cerebral amino acids and thiamine-dependent enzymes. J Neurochem. 1989;52:1079–1084. doi: 10.1111/j.1471-4159.1989.tb01850.x. [DOI] [PubMed] [Google Scholar]
- Caulo M, Van Hecke J, Toma L, Ferretti A, Tartaro A, Colosimo C, Romani GL, Uncini A. Functional MRI study of diencephalic amnesia in Wernicke-Korsakoff syndrome. Brain. 2005;128:1584–1594. doi: 10.1093/brain/awh496. [DOI] [PubMed] [Google Scholar]
- Carvalho FM, Pereira SR, Pires RG, Ferraz VP, Romano-Silva MA, Oliveira-Silva IF, Ribeiro AM. Thiamine deficiency decreases glutamate uptake in the prefrontal cortex and impairs spatial memory performance in a water maze test. Pharmacol Biochem Behav. 2006;83:481–489. doi: 10.1016/j.pbb.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Chang Q, Gold PE. Impaired and spared cholinergic functions in the hippocampus after lesions of the medial septum/vertical limb of the diagonal band with 192 IgG-saporin. Hippocampus. 2004;14:170–179. doi: 10.1002/hipo.10160. [DOI] [PubMed] [Google Scholar]
- Dougherty KD, Turchin PI, Walsh TJ. Septocingulate and septohippocampal cholinergic pathways: involvement in working/episodic memory. Brain Res. 1991;810:59–71. doi: 10.1016/s0006-8993(98)00870-1. [DOI] [PubMed] [Google Scholar]
- Eckenstein FP, Baughman RW, Quinn J. An anatomical study of cholinergic innervation in rat cerebral cortex. Neurosci. 1988;25:457–474. doi: 10.1016/0306-4522(88)90251-5. [DOI] [PubMed] [Google Scholar]
- Gage SL, Keim SR, Simon JR, Low WC. Cholinergic innervation of the retrosplenial cortex via the fornix pathway as determined by high affinity choline uptake, choline acetyltransferase activity, and muscarinic receptor binding in the rat. Neurochem Res. 1994;19:1379–1386. doi: 10.1007/BF00972466. [DOI] [PubMed] [Google Scholar]
- Giguère JF, Butterworth RF. Activities of thiamine-dependent enzymes in two experimental models of thiamine deficiency encephalopathy: 3. Transketolase Neurochem Res. 1987;12:305–310. doi: 10.1007/BF00972141. [DOI] [PubMed] [Google Scholar]
- Giovannini MG, Bartolini L, Kopf SR, Pepeu G. Acetylcholine release from the frontal cortex during exploratory activity. Brain Res. 1998;784:218–227. doi: 10.1016/s0006-8993(97)01161-x. [DOI] [PubMed] [Google Scholar]
- Gold PE. Coordination of multiple memory systems. Neurobiol Learn Mem. 2004;82:230–242. doi: 10.1016/j.nlm.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Gold JJ, Squire LR. The anatomy of amnesia: neurohistological analysis of three new cases. Learn Mem. 2006;13:699–710. doi: 10.1101/lm.357406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo-Ruiz A, Morte L. Localization of amino acids, neuropeptides and cholinergic markers in neurons of the septum-diagonal band complex projecting to the retrosplenial granular cortex of the rat. Brain Res Bull. 2000;52:499–510. doi: 10.1016/s0361-9230(00)00287-2. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME. The role of acetylcholine in learning and memory. Curr Opin Neurobiol. 2006;16:1–6. doi: 10.1016/j.conb.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedreen JC, Bacon SJ, Price DL. A modified histochemical technique to visualize acetylcholinesterase-containing axons. J Histochem Cytochem. 1985;33:134–140. doi: 10.1177/33.2.2578498. [DOI] [PubMed] [Google Scholar]
- Jenkins TA, Dias R, Amin E, Brown MW, Aggleton JP. Fos imaging reveals that lesions of the anterior thalamic nuclei produce widespread limbic hypoactivity in rats. J Neurosci. 2002;22:5230–5238. doi: 10.1523/JNEUROSCI.22-12-05230.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins TA, Vann SD, Amin E, Aggleton JP. Anterior thalamic lesions stop immediate early gene activation in selective laminae of the retrosplenial cortex: evidence of covert pathology in rats? Eur J Neurosci. 2004;12:3291–3304. doi: 10.1111/j.0953-816X.2004.03421.x. [DOI] [PubMed] [Google Scholar]
- Kopelman MD, Thomson AD, Guerrini I, Marshall EJ. The Korsakoff syndrome: clinical aspects, psychology and treatment. Alcohol. 2009;44:148–154. doi: 10.1093/alcalc/agn118. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Homewood J. Neuronal changes in the cerebral cortex of the rat following alcohol treatment and thiamine deficiency. J Neuropathol Exp Neurol. 1993;52:586–593. doi: 10.1097/00005072-199311000-00005. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Mair RG, Anderson CD, McEntee WJ. Long-lasting changes in regional brain amino acids and monoamines in recovered pyrithiamine treated rats. Neurochem Res. 1988;13:1199–1206. doi: 10.1007/BF00971639. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Savage LM. Thiamine deficiency in rats produces cognitive and memory deficits on spatial tasks that correlate with tissue loss in diencephalon, cortex and white matter. Behav Brain Res. 1995;68:75–89. doi: 10.1016/0166-4328(94)00162-9. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX, Savage LM. Neuropathology of thiamine deficiency: an update on the comparative analysis of human disorders and experimental models. Metab Brain Dis. 2006;11:19–37. doi: 10.1007/BF02080929. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX. Cortical and subcortical white matter damage without Wernicke’s encephalopathy after recovery from thiamine deficiency in the rat. Alcohol Clin Exp Res. 1997;21:434–443. doi: 10.1111/j.1530-0277.1997.tb03788.x. [DOI] [PubMed] [Google Scholar]
- Mair RG. On the role of thalamic pathology in diencephalic amnesia. Rev Neurosci. 1994;5:105–140. doi: 10.1515/revneuro.1994.5.2.105. [DOI] [PubMed] [Google Scholar]
- Mair RG, Anderson CD, Langlais PJ, McEntee WJ. Behavioral impairments, brain lesions and monoamine activity in the rat following a bout of thiamine deficiency. Behav Brain Res. 1988;27:223–239. doi: 10.1016/0166-4328(88)90119-2. [DOI] [PubMed] [Google Scholar]
- Mair RG, Hembrook JR. Memory enhancement with event-related stimulation of the rostral intralaminar thalamic nuclei. J Neurosci. 2008;28:14293–142300. doi: 10.1523/JNEUROSCI.3301-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair RG, Knoth RL, Rabchenuk SA, Langlais PJ. Impairments of olfactory, auditory and spatial serial reversal learning in rats recovered from pyrithiamine induced thiamine deficiency. Behav Neurosci. 1991;105:360–374. [PubMed] [Google Scholar]
- Mair RG, Lacourse DM. Radio-frequency lesions of the thalamus produce delayed-nonmatching-to-sample impairments comparable to pyrithiamine-induced encephalopathy in rats. Behav Neurosci. 1992;106:634–645. doi: 10.1037//0735-7044.106.4.634. [DOI] [PubMed] [Google Scholar]
- Mayes AR, Meudell PR, Mann D, Pickering A. Location of lesions in Korsakoff’s syndrome: neuropsychological and neuropathological data on two patients. Cortex. 1988;24:367–388. doi: 10.1016/s0010-9452(88)80001-7. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, Pal SN, Marriott LK, Gold PE. Competition between memory systems: acetylcholine release in the hippocampus correlates negatively with good performance on an amygdala-dependent task. J Neurosci. 2002;22:1171–1176. doi: 10.1523/JNEUROSCI.22-03-01171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Levey AI, Wainer BH. Cholinergic Innervation of cortex by the basal forebrain: Cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol. 1983;214:170–197. doi: 10.1002/cne.902140206. [DOI] [PubMed] [Google Scholar]
- Nakagawasai O, Tadano T, Hozumi S, Tan-No K, Niijima F, Kisara K. Immunohistochemical estimation of brain choline acetyltransferase and somatostatin related to the impairment of avoidance learning induced by thiamine deficiency. Brain Res Bul. 2000;52:189–196. doi: 10.1016/s0361-9230(00)00248-3. [DOI] [PubMed] [Google Scholar]
- Oscar-Berman M, Kirkley SM, Gansler DA, Couture A. Comparisons of Korsakoff and non-Korsakoff alcoholics in neuropsychological tests of prefrontal brain functioning. Alcohol Clin Exp Res. 2004;28:667–675. doi: 10.1097/01.alc.0000122761.09179.b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oscar-Berman M, Marinkovic K. Alcoholism and the brain: an overview. Alcohol Res Health. 2003;27:125–133. [PMC free article] [PubMed] [Google Scholar]
- Parent MB, Baxter MG. Septohippocampal acetylcholine: involved in but not neccesary for learning and memory? Learn Mem. 2004;11:9–20. doi: 10.1101/lm.69104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passetti F, Dalley JW, O’Connell MT, Everitt BJ, Robbins TW. Increased acetylcholine release in the rat medial prefrontal cortex during performance of a visual attentional task. Eur J Neurosci. 2000;12:3051–3058. doi: 10.1046/j.1460-9568.2000.00183.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego: Academic Press; 1986. [Google Scholar]
- Pepeu G, Giovannini G. Changes in acetylcholine extracellular levels during cognitive processes. Learn Mem. 2008;11:21–27. doi: 10.1101/lm.68104. [DOI] [PubMed] [Google Scholar]
- Pires RG, Pereira SR, Pittella JE, Franco GC, Ferreira CL, Fernandes PA, Ribeiro AM. The contribution of mild thiamine deficiency and ethanol consumption to central cholinergic parameter dysfunction and rats’ open-field performance impairment. Pharmacol Biochem Behav. 2001;70:227–235. doi: 10.1016/s0091-3057(01)00593-7. [DOI] [PubMed] [Google Scholar]
- Pires RG, Pereira S, Oliveira-Silva IF, Franco GC, Ribeiro AM. Cholinergic parameters and the retrieval of learned and re-learned spatial information: A study using a model of Wernicke-Korsakoff syndrome. Behav Brain Res. 2005;162:11–21. doi: 10.1016/j.bbr.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Pitkin SR, Savage LM. Aging potentiates the acute and chronic neurological symptoms of pyrithiamine-induced thiamine deficiency in the rodent. Behav Brain Res. 2001;119:167–177. doi: 10.1016/s0166-4328(00)00350-8. [DOI] [PubMed] [Google Scholar]
- Pitkin SR, Savage LM. Age-related vulnerability to diencephalic amnesia produced by thiamine deficiency: the role of time of insult. Behav Brain Res. 2004;148:93–105. doi: 10.1016/s0166-4328(03)00208-0. [DOI] [PubMed] [Google Scholar]
- Pothuizen HH, Aggleton JP, Vann SD. Do rats with retrosplenial cortex lesions lack direction? Eur J Neurosci. 2008;28:2486–2498. doi: 10.1111/j.1460-9568.2008.06550.x. [DOI] [PubMed] [Google Scholar]
- Pothuizen HH, Davies M, Aggleton JP, Vann SD. Effects of selective granular retrosplenial cortex lesions on spatial working memory in rats. Behav Brain Res. 2010;208:566–575. doi: 10.1016/j.bbr.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Ragozino ME, Pal SN, Unick K, Stefani MR, Gold PE. Modulation of hippocampal acetylcholine release and spontaneous alternation scores by intrahippocampal glucose injections. J Neurosci. 1998;18:1595–1601. doi: 10.1523/JNEUROSCI.18-04-01595.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragozzino ME, Unick KE, Gold PE. Hippocampal acetylcholine release during memory testing in rats: augmentation by glucose. Proc Natl Acad Sci. 1996;93:4693–4698. doi: 10.1073/pnas.93.10.4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LJ, Lasserson D, Marsden P, Stanhope N, Stevens T, Bello F, Kingsley D, Colchester A, Kopelman MD. FDG-PET findings in Wernicke-Korsakoff syndrome. Cortex. 2003;39:1027–1045. doi: 10.1016/s0010-9452(08)70876-1. [DOI] [PubMed] [Google Scholar]
- Robinson JK, Mair RG. MK-801 prevents brain lesions and delayed non-matching to sample deficits produced by pyrithiamine-induced encephalopathy in rats. Behav Neurosci. 1992;106:623–633. [PubMed] [Google Scholar]
- Roland JJ, Levinson M, Vetreno R, Savage LM. Differential effects of systemic and intraseptal administration of the acetylcholinesterase inhibitor tacrine on the recovery of spatial behavior in an animal model of diencephalic amnesia. Eur J Neurosci. 2010;629:31–39. doi: 10.1016/j.ejphar.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Mark K, Vetreno RP, Savage LM. Increasing hippocampal acetylcholine levels enhance behavioral performance in an animal model of diencephalic amnesia. Brain Res. 2008;1234:116–127. doi: 10.1016/j.brainres.2008.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Savage LM. Blunted hippocampal, but not striatal, acetylcholine efflux parallels learning impairment in diencephalic-lesioned rats. Neurobiol Learn Mem. 2007;87:123–132. doi: 10.1016/j.nlm.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Savage LM. Blocking GABA-A receptors in the medial septum enhances hippocampal acetylcholine release and behavior in a rat model of diencephalic amnesia. Phamacol Biochem Behav. 2009a;92:480–487. doi: 10.1016/j.pbb.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Savage LM. The Role of Cholinergic and GABAergic Medial Septal/Diagonal Band Cell Populations in the Emergence of Diencephalic Amnesia. Neurosci. 2009b;160:32–41. doi: 10.1016/j.neuroscience.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Castillo R, Langlais PJ. Effects of thalamic intralaminar nuclei and internal meduallary lamina on spatial memory and object discrimination. Behav Neurosci. 1998;112:1339–1352. doi: 10.1037//0735-7044.112.6.1339. [DOI] [PubMed] [Google Scholar]
- Savage LM, Sweet AJ, Castilo R, Langlais PJ. The Role of Internal Medullary Lamina Nuclei and Posterior Thalamic Nuclei in Learning, Memory, and Habituation in the Rat. Behav Brain Res. 1997;82:133–147. doi: 10.1016/s0166-4328(97)80983-7. [DOI] [PubMed] [Google Scholar]
- Savage LM, Chang Q, Gold PE. Interactions between diencephalic and hippocampal processing. Learn Mem. 2003;10:242–246. doi: 10.1101/lm.60003. [DOI] [PubMed] [Google Scholar]
- Savage LM, Roland J, Klintsova A. Selective hippocampal- but not forebrain amygdalar- cholinergic dysfunction in diencephalic amnesia. Brain Res. 2007;1139:210–219. doi: 10.1016/j.brainres.2006.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliebs R, Arendt T. The significance of the cholinergic system in the brain during aging and alzheimer’s disease. J Neural Transm. 2006;113:1625–1644. doi: 10.1007/s00702-006-0579-2. [DOI] [PubMed] [Google Scholar]
- Thiel CM, Huston JP, Schwarting RK. Cholinergic activation in frontal cortex and nucleus accumbens related to basic behavioral manipulations: Handling and the role of post-handling experience. Brain Res. 1998;812:121–132. doi: 10.1016/s0006-8993(98)00961-5. [DOI] [PubMed] [Google Scholar]
- Vann SD, Albasser MM. Hippocampal, retrosplenial, and prefrontal hypoactivity in a model of diencephalic amnesia: Evidence towards an interdependent subcortical-cortical memory network. Hippocampus. 2009;19:1090–102. doi: 10.1002/hipo.20574. [DOI] [PubMed] [Google Scholar]
- Vetreno RP, Anzalone SJ, Savage LM. Impaired, spared, and enhanced ACh efflux across the hippocampus and striatum in diencephalic amnesia is dependent on task demands. Neurobiol Learn Mem. 2008;90:237–244. doi: 10.1016/j.nlm.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff syndrome: A clinical and pathological study of 245 patients, 82 with post-mortem examinations. Contemp Neurol Ser. 1971;7:1–206. [PubMed] [Google Scholar]
- Weiskrantz L. Comparative Aspects of Studies of Amnesia. Phil Trans R Soc Lond B Biol Sci. 1982;298:97–109. doi: 10.1098/rstb.1982.0075. [DOI] [PubMed] [Google Scholar]
- Witt ED. Neuroanatomical consequences of thiamine deficiency: a comparative analysis. Alcohol. 1985;20:201–21. [PubMed] [Google Scholar]