

Abstract
Knr4, recently characterized as an intrinsically disordered Saccharomyces cerevisiae protein, participates in cell wall formation and cell cycle regulation. It is constituted of a functional central globular core flanked by a poorly structured N-terminal and large natively unfolded C-terminal domains. Up to now, about 30 different proteins have been reported to physically interact with Knr4. Here, we used an in vivo two-hybrid system approach and an in vitro surface plasmon resonance (BIAcore) technique to compare the interaction level of different Knr4 deletion variants with given protein partners. We demonstrate the indispensability of the N-terminal domain of Knr4 for the interactions. On the other hand, presence of the unstructured C-terminal domain has a negative effect on the interaction strength. In protein interactions networks, the most highly connected proteins or “hubs” are significantly enriched in unstructured regions, and among them the transient hub proteins contain the largest and most highly flexible regions. The results presented here of our analysis of Knr4 protein suggest that these large disordered regions are not always involved in promoting the protein–protein interactions of hub proteins, but in some cases, might rather inhibit them. We propose that this type of regions could prevent unspecific protein interactions, or ensure the correct timing of occurrence of transient interactions, which may be of crucial importance for different signaling and regulation processes.
Keywords: intrinsically unstructured (disordered), proteins, disordered protein regions, protein–protein interactions, two-hybrid system, surface plasmon resonance
Introduction
Knr4 is a Saccharomyces cerevisiae protein which localizes at sites of polarized cell growth and influences cell wall formation and cell cycle progression.1,2 An unusually large number (194) of other S. cerevisiae genes show synthetic lethal interactions with the KNR4/SMI1 gene,3 and at the protein level, about 30 different proteins have been reported to physically interact with Knr4.4–8 We recently showed that this protein is constituted of a central globular core flanked by a poorly structured N-terminal domain and a large natively unfolded C-terminal domain.9 Therefore, Knr4 belongs to the family of intrinsically disordered proteins (IDP) which, being characterized by the absence of stable secondary and/or tertiary structures and existing instead as very dynamic ensembles of conformations under physiological conditions in vitro, fulfill crucial biological functions (for reviews see, e.g., Refs. 10–17). Many members of this family are able to interact with multiple partners and are very often involved in cell-signaling and regulatory functions.18–22 Although the central, structured part of Knr4 protein seems to ensure most of the functions of the protein, we showed that the N-terminal region of Knr4 is indispensable for cell viability when PKC1 pathway is deficient.9 Interaction of Knr4 through this domain with other proteins thus seems crucial for the functioning of a parallel pathway that maintains cell integrity in the absence of a functional PKC1 pathway. However, the role of the large Knr4 C-terminal domain with its highly disordered structure so far remains undefined. Here we have investigated the influence of both N-terminal and C-terminal domains on protein–protein interactions between Knr4 and its binding partners, in vivo using the yeast two-hybrid system and in vitro by surface plasmon resonance (BIAcore). We show that these domains display unusual interactions properties that are in fact common toward all different Knr4 partners.
Results
Global screen for specific partners of Knr4 terminal domains by two-hybrid system approach
As a result of several studies on protein interactions, where the entire Knr4 protein was used as the bait, about 30 protein partners of Knr4 were identified.4–8 In this work, we carried out different large scale two-hybrid screenings, using deletion variants of Knr4 lacking either N- or C-termini as baits. Our aim was to identify Knr4 binding partners specifically interacting with either N- or C-terminal domains, to better understand the functions of these two protein parts.
For this purpose, four genome-wide two-hybrid screenings were performed with different fragments of Knr4 (1–505, 1–340, 80–505, and 80–340) fused with the Gal4 DNA-binding domain (BD) [Fig. 1(A)]. These baits were individually crossed using a robotic programmable apparatus (Biomek 2000 from Beckman) with the arrayed library of Gal4 activating domain (AD)-fused proteins4 representing almost the entire genome of S. cerevisiae. Resulting diploids were replicated on synthetic medium lacking adenine to screen for two-hybrid interactions.23 We noticed that on the 3rd day of growth on this media, many large colonies appeared when Knr4 lacking C-terminus (1–340) was used as bait. On the contrary, in the two screenings with N-terminus lacking constructs (80–340 and 80–505), the colony size of potential positive clones was smaller than in the screen with the full-length Knr4-BD fusion. To eliminate false positives, a total of 103 potential protein partners selected in all four screenings were then re-examined in two steps for their ability to interact with the different Knr4 fragments. First, we used the strategy of the original screening, where the corresponding haploid strains from the library were crossed with the haploids of the opposite mating type transformed by the same four BD-Knr4 fusions and a control plasmid expressing only the DNA BD of Gal4. This step was followed by the growth detection of resulting diploids in the interaction-selective media. Ten candidates showing detectable growth compared with the negative control (diploid strain containing pOBD2 and pOAD plamsids) were kept after this step. The second verification round consisted in retransforming the plasmids extracted from the 10 selected library strains into the haploids carrying Knr4-BD-expressing plasmids. Similarly to the first verification, growth of the resulting double transformants was tested on interaction-selective media. After this step, the interactions with Knr4-BD fragments were confirmed only for four proteins: a very strong one was found for Tid3 and weaker ones for Rvs167, Asm4, and Bud3 proteins. Importantly, the strength of interaction of each partner with Knr4 was dependent on the Knr4 fragment used in combination. The strongest interaction was always observed for the Knr4 1–340 fragment, slightly weaker for the full-length protein (1–505), and significantly weaker interactions were observed for the two fragments lacking the protein N-terminus (80–505 and 80–340 fragments). This observation is in agreement with the differences in colony sizes of the diploids grown on synthetic medium lacking adenine that we saw during the initial screenings. Thus, these data suggest that the absence of C-terminal part could increase the interactivity of Knr4 protein, whereas removal of N-terminus would weaken the interactions.
Figure 1.

Interaction between different Knr4 parts and Tid3 in the two-hybrid system. A: Schematic representation of the two-hybrid constructions expressing different parts of Knr4 fused to the Gal4 DNA binding domain. B: Transformants of pJ69-4A strain each carrying pOAD-Tid3 plasmid and pOBD2 plasmid with or without Knr4 deleted variants were grown on SD-Trp-Leu solid media at 30°C for 1 day. Next day, replica was made on two plates: new SD-Trp-Leu and on SD-Ade. Presented photograph was taken after 72 h of growth. C: β-galactosidase activity measured in the cell extracts from the strains expressing AD-Tid3 fusion protein (Tid3 fused to the activating domain of Gal4) and different Knr4 deleted variants fused to the BD of Gal4. The results (triplicates) were normalized to protein concentrations and expressed in nanomoles of O-nitrophenyl-β-d-galactopyranoside converted/min/mg proteins. The upper bar corresponds to the negative control strain with only activating (AD) and DNA binding (BD) domains of Gal4 expressed.
These four new physical partners of Knr4 come in addition to the ones that we and other labs previously isolated.4–8 These partners fall into different functional categories, but most of them are related to cell morphogenesis either directly (actin and dynactin cytoskeleton elements, polarisome components) or through stress related signaling pathway (mostly, the CWI pathway and transcription factors), which is fully coherent with the biological function of Knr4 that we previously published.2,9 However, the purpose of this work was not to further study the function of the full protein in the cell, but to investigate the specific roles of its disordered domains.
Refined in silico characterization of the disordered nature of Knr4 protein
We established earlier that Knr4 protein is divided into three domains: a central globular core flanked by a poorly structured N-terminal region and a large natively unfolded C-terminal domain.9 Because we observed in the present study the crucial importance of the two terminal parts of Knr4 protein for its interaction capacity, we performed a new in-depth in silico structure analysis to explain these properties. To analyze in details the intrinsically disordered nature of Knr4, we first carried out the composition profiling of this protein as described by Vacic et al.24 The tendency of a given protein to be intrinsically disordered is determined by a set of specific features of its amino acid sequence and composition.11,25 For example, intrinsically disordered proteins are significantly depleted in bulky hydrophobic (I, L, and V) and aromatic amino acid residues (W, Y, and F), which would normally form the hydrophobic core of a folded globular protein, and also possess low content of C and N residues. These residues, I, L, V, W, F, Y, C, and N, were proposed to be called order-promoting amino acids. On the other hand, intrinsically disordered proteins and regions were shown to be substantially enriched in disorder-promoting amino acids: E, K, R, G, Q, S, P, and A.
The results of the analysis that we conducted on Knr4 sequence are shown in Figure 2(A). At first glance, the composition profile of the full-length Knr4 differed significantly from that of a set of well-characterized intrinsically disordered proteins from the DisProt database.27 However, when this analysis was restricted to the N- and C-terminal fragments of this protein (Knr41–79 and Knr4341–505 domains, respectively), the composition profiling produced plots typical for intrinsically disordered proteins, with noticeable depletion in major order-promoting residues (excepted for the phenylalanine residues which seem to be rather abundant in Knr41–79 domain) and enrichment in major disorder-promoting residues [Fig. 2(A)]. On the other hand, the central core of the protein, the Knr480–340 domain, displayed a composition profile that was expected for an ordered protein [Fig. 2(A)]. Figure 2(A) also shows that the Knr4341–505 domain is more disordered than Knr41–79, because it contains essentially less major order-promoting residues (especially, W, F, Y, C, and L) and noticeably more major disordered-promoting residues (such as K, E, and Q) than any other protein analyzed in this study.
Figure 2.

Computational evaluation of the intrinsic disorder propensity of Knr4 and its N- and C-terminal domains. A: Composition profiling of Knr41–505 (red bars), Knr41–79 (green bars), Knr480–340 (yellow bars), and Knr4341–505 (blue bars) compared with a set of ordered proteins from PDB. Data for a set of disordered proteins from DisProt are shown by black bars. The bar for a given amino acid represents the fractional difference in composition between a given protein (or set of proteins) and the set of ordered proteins. The fractional difference is calculated as (CX − Cordered)/Cordered, where CX is the amount of the amino acid in a given protein/set, and Corder is the corresponding amount in the set of ordered proteins. Residues are ordered by their disorder propensity. Negative values indicate residues less abundant in the given protein/set than in the set of ordered proteins, positive indicates residues more abundant in the given protein/set than in the set of ordered proteins. B: Distribution of the PONDR®-FIT score over the sequences of Knr4. In PONDR plot, segments with scores above 0.5 correspond to disordered regions, whereas those below 0.5 correspond to ordered regions. Red and blue bars represent the respective positions of the alpha-helical and beta-structural elements predicted by the NPS (network protein sequence analysis) consensus secondary structure server,26 which runs the input sequence against several different secondary structure prediction tools and generates a consensus secondary structure out of them.
To get stronger evidences on the disordered structure of the N- and C-terminal domains of Knr4, these parts of the protein were analyzed using a recently developed disorder predictor PONDR®-FIT.28 This bioinformatics tool represents a meta-predictor, which combines the outputs of several disorder predictors, such as PONDR-VLXT,29 PONDR-VSL2,30 PONDR-VL3,31 FoldIndex,32 IUPred,33 and Top-IDP.34 The prediction performance of this meta-predictor is 2–3% higher than the performance of the best of its individual components (some of which are among the most accurate disorder predictors currently available). The results of the Knr4 analysis are shown in Figure 2(B), which clearly illustrates that both the N-terminal (residues 1–80) and the C-terminal domains (residues 340–505) are predicted to be highly disordered because their PONDR-FIT curves are located mostly above the 0.5 threshold. This conclusion was further confirmed by the prediction of secondary structure propensity in Knr4 protein. In fact, Figure 2(B) shows that both N-terminal and C-terminal domains of this protein contain very limited amount of predicted α-helices and β-strands.
It has been emphasized that predictions of short ordered regions in otherwise highly disordered proteins might correspond to protein regions that mediate interaction with other proteins or DNA, so called molecular recognition elements or molecular recognition fragments (MoREs or MoRFs, respectively).24,35–38 Figure 2(B) shows that the intrinsically disordered N- and C-terminal domains of Knr4 contain several potential binding sites identified as characteristic deeps in the PONDR-FIT curve. In the N-terminal domain, the only deep centered at residue ∼26 is due to the presence of several aromatic residues (Y21, Y24, and F32). So, it is likely that this region is responsible for the observed in vivo N-terminus interaction indispensability. In the C-terminal domain, there are four noticeable deeps centered at residues ∼315, ∼360, ∼400, and ∼450. The major C-terminal deep is determined by the hydrophobic 313LVF315 motif, the second and third deeps are due to the hydrophobic 361YV362 and 401VKIV404 patches, whereas the last deep does not have continuous hydrophobic motifs, being characterized by few hydrophobic residues sparse through the sequence. Earlier, it has been emphasized that the presence of multiple aromatic side chains within a highly disordered structure can confer crucial biological function to an IDP.39
Roles of the Knr4 protein unstructured terminal regions in protein–protein interactions
Detailed interaction analysis using the two-hybrid system
To meticulously characterize the role of different Knr4 domains in the interactions with its protein partners, we created a set of new plasmids bearing additional “bait” constructions from Knr4. In particular, we wanted to verify the in silico predicted “interacting” motif within N-terminal part (around AA 26) of the protein. The constructed plasmids expressed either the Knr4 central part (AA 80–340) with or without the flanking regions, or only the terminal regions of the protein fused to the Gal4 DNA binding domain (BD) [Fig. 1(A)]. To test for the strength of interactions of different Knr4 fragments, we used pJ69-4a strains already carrying plasmids with different partners of Knr4 fused to Gal4 AD and transformed them with the plasmids described above expressing fusions of Gal4 BD with different parts of Knr4. In the resulting double transformants, the Gal4-dependent expression of all three markers (ADE2, HIS3, and LacZ) was analyzed [Fig. 1(B,C)]. These experiments were conducted with the different Knr4 protein partners found in the screening described above: Tid3, Asm4, Rvs167, and Bud3, and similar interaction profiles with Knr4 fragments were obtained for all of them (data not shown). The results observed with Tid3, which displayed the strongest interaction with Knr4 of these four new partners, are presented on Figure 1(B,C).
To test for the general aspect of these interactions properties, we decided to conduct similar experiments with other previously identified Knr4 partners. We choose Tys1, a known in vivo protein partner of Knr4 isolated in our lab in a previous global two-hybrid screen, because the interaction between these two proteins is strong and has an established biological significance.5 Tys1 interaction with Knr4 in the two-hybrid system was reinvestigated in details, and its interaction profile with Knr4 fragments is shown on Figure 3. Although the interaction strength measured is higher for Tys1 as expected, the interaction profile with the different Knr4 fragments is similar to the one observed for Tid3 shown in Figure 1(B,C).
Figure 3.

Interaction between different Knr4 parts and Tys1 in the two-hybrid system. A: Transformants of pJ69-4A strain each carrying pOAD-Tid3 plasmid and pOBD2 plasmid with or without Knr4 deleted variants were grown on SD-Trp-Leu solid media at 30°C for 1 day. Next day, replica was made on two plates: new SD-Trp-Leu and on SD-Ade. The presented photograph was taken after 72 h of growth. B: β-galactosidase activity measured in the cell extracts from the strains expressing AD-Tid3 fusion protein (Tid3 fused to the activating domain of Gal4) and different Knr4 deleted variants fused to the BD of Gal4. The results (triplicates) were normalized to protein concentrations and expressed in nanomoles of O-nitrophenyl-β-d-galactopyranoside converted/min/mg proteins. The upper bar corresponds to the negative control strain with only activating (AD) and DNA binding (BD) domains of Gal4 expressed.
The results of these tests clearly indicated that the first 40 amino acids of the Knr4 protein were absolutely needed for its ability to interact with its binding partners. Furthermore, looking either at the ADE2 marker expression [Fig. 1(B)] or at the β-galactosidase activity [Fig. 3(A)], we were able to detect a quite strong interaction even for the Knr4 N-terminal domain alone (construction BD 1–80), although its strength seems to depend on the partner and on the reporter gene considered. On the other hand, the comparison of the interaction strength of different pairs of constructs, for example, comparison of results for the fusion carrying 1–505 with that carrying 1–340, or 80–505 fusion with 80–340 fusion, or fusions 40–505 and 40–340 [Figs. 1(B) and 3(A)] revealed that the presence of the disordered C-terminus in the corresponding Knr4 fragments had a clear diminishing effect on the interaction capacity of the protein. Interestingly, even basal β-galactosidase activity measured in the control strain expressing only BD of Gal4 was higher than the β-galactosidase activity measured for the BD-Knr4 340–505 fusion [Figs. 1(C) and 3(B)]. This observation can be explained by the existence of strong negative effects of the C-terminal region of Knr4 on the occasional weak interactions between the Gal4 BD and either the AD fusions or possibly proteins of the yeast transcriptional machinery itself.
In vitro surface plasmon resonance test for protein–protein interaction specificity
We strongly believe that the evidence of the general aspect of the interaction properties is stronger if it is demonstrated using both different partners and different methods. Thus, to confirm our results about the role of the two unstructured Knr4 termini in protein interaction obtained in vivo, we chose an in vitro approach, surface plasmon resonance (SPR). In this analysis system (BIAcore), one of the interacting molecules is immobilized onto the surface of a sensor chip and a potential interaction partner is then injected in solution and flows over the sensor surface. The binding of an interaction partner to the immobilized molecules results in a change in SPR signal that is detected in real time and presented as a sensorgram. In this experiment, we investigated in vitro interaction of Knr4 and its two truncated variants with Tys1, the best known in vivo protein partner of Knr4 isolated in our lab.5 Purified Knr4 fragments 1–340, 1–505(full protein), or 80–505 were fixed on a CM5 BIAcore chip, then solutions containing different concentrations of purified Tys1 protein were flowed over the chip and the binding kinetics were measured. Representative sensorgrams shown in Figure 4 illustrate that Tys1 protein binds with a much higher efficiency on the Knr41–340 lacking the C-terminal domain than on the full size protein. On the opposite, only a very weak binding is observed on the Knr480–505 lacking the N-terminal domain. In addition, the dissociation constant calculated for Knr41–340 (1.15 × 10−8M) is significantly lower than the ones for the two other fragments (4.22 × 10−7M and 6.66 × 10−7M). Sensorgrams also show that the SPR signal increases significantly with the concentration of Tys1 in the flowing solution.
Figure 4.
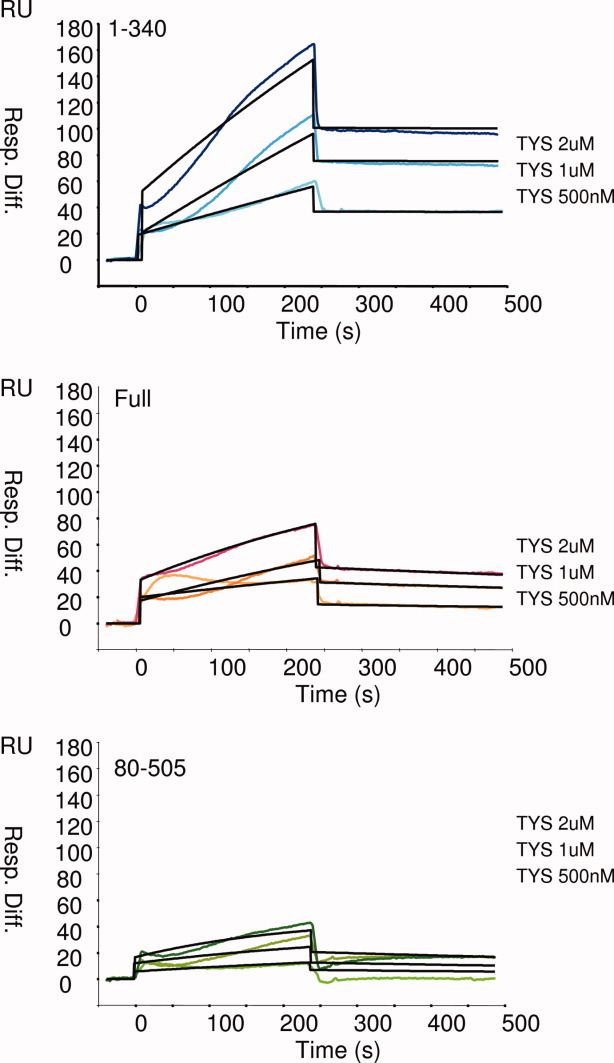
Effect of the N- and C-terminal domains of Knr4 protein on its in vitro protein binding capacity. Purified Knr4 protein fragments 1–340, 1–505, and 80–340 were fixed on three channels of a CM5 BIAcore sensorchip. Solutions containing different concentrations (500 nM, 1 μM, and 2 μM) of purified Tys1 protein were flowed over the chip and the binding kinetics were measured and quantified versus the chip forth channel used as a negative control. Sensorgrams are expressed in resonance units (RU) as a function of time in seconds. Resonance units on the Y axis represent Tys1 binding to Knr4 fragments.
Discussion
Numerous recent studies revealed the existence of a correlation between the presence of intrinsically disordered regions in proteins and their ability to interact with a large number of partners.16,40,41 The presence of disordered regions in such proteins and the existence of the binding-induced disorder-to-order transitions were proposed to allow specific but reversible interactions, which are very important for the proteins involved in all kind of cellular regulation processes.12,14,16,18–22,42–45 In many cases, the conclusion on the advantage provided by intrinsically disordered regions for promiscuous behavior of the corresponding protein was based on the computational analysis of the protein interactions and expression databases, combined with the protein structure predictions. In this work, the promiscuity of the potential yeast hub protein Knr4, which is shown to be composed of three differently structured domains, was investigated using a set of experimental tools complementary to the computational approaches. The strength of the yeast two-hybrid interactions of this protein with its binding partners was shown to be dependent on the presence of the Knr4 N- and C-terminal domains. The N-terminus appears to be essential for the Knr4 interactions, first because, being fused alone to the DNA binding domain of Gal4, this fragment assured the in vivo interaction. Second, the removal of this fragment from the protein resulted in a noticeable decrease in the interaction strength. Detailed computational analysis of this N-terminal domain sequence revealed the presence of a characteristic deep in the PONDR-FIT curve, centered at residue 26, which is likely to be responsible for the protein–protein interactions observed.
Given the presence of two deeps at positions 361 and 401 and the strongly disordered character of the C-terminal domain of the protein, a positive involvement of this protein part in the interactions could also be expected. However, on the contrary, we observed an inhibitory effect of the whole C-terminal part on protein–protein interactions. Some weak interactions were even found in vivo for the constructs lacking the N-terminal region when the C-terminal domain was deleted too. In addition, we noticed that the fusion of this disordered C-terminal domain to the Gal4 binding domain abolished the residual background interaction observed between this domain and the Gal4 activating domain fusion proteins (or possibly the yeast transcriptional machinery itself). Furthermore, in vitro SPR analyses clearly demonstrated the negative influence of the unstructured C-terminus of Knr4 protein on its binding capacity. Altogether, these data indicate that this large intrinsically disordered C-terminal part of the protein is not directly involved in the protein–protein interaction, but rather serves as a negative regulator of these interactions. It is possible that to fulfill some of its functions Knr4 needs to interact very strongly with other proteins. The C-terminal domain could then be specifically cleaved to allow these interactions to take place at the proper time or cell location. Indeed, the several in vivo phosphorylation sites as well as the PEST sequence found this domain (data not shown) could in these cases participate in ensuring the appropriate degradation of this part of the protein.
Intrinsically unstructured (or disordered) proteins (IUP or IDP) and regions are very abundant among the most highly connected proteins or “hubs” in protein interactions networks.18,19,41,46 Among them, the transient or “date” or “sociable” hubs, which interact with different partners at different times and locations, are statistically enriched in large and highly flexible regions compared with the “party” hubs which form stable complexes with their partners.47,48 It has been proposed that these large disordered regions are implicated in transient binding interactions by favoring disorder to order transition, which would render the interactions highly reversible as maintaining their high specificity. The enrichment of intrinsic disorder in date hubs may facilitate transient interactions, which might be required for date hubs to interact with different partners at different times.49 Furthermore, the abundance of IDPs in the cell is precisely regulated at the levels of transcription, transcript clearance, translation, and proteolytic degradation.42,50 This is coherent with the functions of IDPs, because fidelity in signal transduction may require the presence of most signaling pathways elements in appropriate amounts and only as long as they are needed. Indeed, variations in the abundance of IDPs in the cell are associated with perturbed cellular signaling that may lead to pathological conditions such as cancer.51 The fact that disordered regions are prone to make promiscuous molecular interactions when their concentration is increased is likely the cause of this dosage sensitivity.52
On the basis of these recent findings and on the presented analysis of the yeast Knr4, we propose that the large disordered and highly flexible regions of transient hub proteins are not always involved in promoting their protein–protein interactions, but in some cases, might rather inhibit them. Theoretically, they could sterically prevent unspecific protein–protein interactions, or ensure the occurrence only at the correct timing of the transient interactions of “date” or “sociable” hub proteins. These mechanisms might be of crucial importance for different signaling and regulation processes.
Material and Methods
Bacteria and yeast strains, media, and growth conditions
Escherichia coli BL21 codon plus cells (Novagen, Madison, USA) used for proteins expression were cultivated in LB supplemented with ampicillin (150 mg/L) and chloramphenicol (40 mg/L) at 37°C. S. cerevisiae strains used for this work were PJ69-4α and PJ69-4a (leu2-3,112 ura3-52 trp1-901 his3-200 gal4Δ gal80Δ GAL-ADE2 lys2::GAL1-HIS3 met2:: GAL7-LacZ53). Yeast cultures were routinely done at 30°C on a rotational shaker (220 rpm) in either a standard YEPD (10 g/L yeast extract, 20 g/L bactopeptone, 20 g/L glucose) medium or in SD medium (1.7 g/L yeast nitrogen base without amino acids and ammonium, 5 g/L ammonium sulfate, and 20 g/L glucose) supplemented with the auxotrophic requirements. For solid media, 20 g/L of agar was added.
Oligonucleotides and plasmids constructions
Primers used for the construction of bait plasmids are listed in Table I. DNA fragments of KNR4 were obtained by PCR amplification from genomic DNA. To amplify deleted variants of Knr4, different combinations of forward and reverse primers shown in the Table I were used. Amplified fragments were cloned into BamH1/Pst1 of pOBD2 plasmid. For the expression of GST-fusion proteins and purification of three deleted Knr4 variants needed for the BIAcore test, we used the protocol and the plasmids described by Durand et al.9 Tys1-GST fusion protein was expressed from the plasmid described in Ref. 5.
Tabel 1.
Primers Used in This Study
| Name of the primer (restriction site) | Sequence |
|---|---|
| Knr4-1 forward BamH1 | GGATCCTGGATCTATTCAAAAGAAAAGTTAAA |
| Knr4-80 forward BamH1 | GGATCCCCACGGAGTCAAACGATGGTGTCTCTGAA |
| Knr4-40 forward BamH1 | GGATCCACAGCAACAATGGTCAGGTAAATCC |
| Knr4-340 forward BamH1 | GGATCCTGCAAGAAAACTTGAGATCACAA |
| Knr4-505 reverse Pst1 | CTGCAGTCATAAAGCTATATTTTCAAATTCTTC |
| Knr4-340 reverse Pst1 | CTGCAGTCATTGATACTTGATCCACGTTCTTC |
Two-hybrid large scale screen
The screen for interacting proteins was done using a genome-wide array consisting of ∼6000 yeast haploid strains, each containing a plasmid (pOAD) with an ORF fused to the Gal4 AD. The bait plasmids pOBD2 carrying the different Knr4 fragments were transformed into yeast strain PJ69-4a. These transformants were then crossed to the yeast array (16 plates, each with 384 positions) composed of the PJ69-4α strains transformed with pOAD plasmids carrying the AD-fused ORFs. All replications and inoculations were carried out using the 384-pin replicator of a Biomek 2000 Laboratory Automation Workstation, with movements programmed using the BioWorks Version software (Beckman Coulter). After mating, PJ69-4 diploids were selected by plating onto SD without leucine and tryptophan. Screening for protein–protein interactions was done by pinning these diploids onto SD lacking leucine, tryptophan, and adenine. Positive colonies, which grew on this selection medium, were identified by their position in the array. Growth was scored after 4, 7, 10, 16, and 20 days at 30°C.
Surface plasmon resonance (BIAcore) analysis
Materials
Biomolecular interaction analyses were carried out in HBS-buffer (150 mM NaCl, 0.05% (v/v) Surfactant P20, 10 mM Hepes, pH 7.4 or pH 5.5) using the BIAcore® 3000 (Biacore AB, Uppsala, Sweden). Recombinant Tys1-GSTp, Knr4, and its different fragments were expressed and purified as described.5,9 Protein concentration was determined using NanoDrop 1000 (Nyxor Biotech).
Immobilization
Knr4 protein fragments were immobilized on a CM5 sensorchip (BIAcore) using the Amine Coupling Kit (BIAcore). The surface of the sensorchip was activated with 70 μL EDC/NHS (100 mMN-ethyl-N′-(dimethyl-aminopropyl)-carbodimide-hydrochloride, 400 mMN-hydroxysuccinimide) using a flow rate of 10 μL/min. Protein fragments, 20–40 μg/mL, were diluted in 10 mM sodium acetate pH 4.0. Subsequently, the sensorchip was deactivated with 70 μL of 1M ethanolamine hydrochloride pH 8.5 (flow rate: 10 μL/min) and HBS flowed for 5 min.
BIAcore analysis
Binding analyses were performed with multiple injections of different protein concentration solutions over the immobilized surfaces at 25°C. All samples were diluted in HBS-EP buffer (Hepes 10 mM, NaCl 150 mM, EDTA 3 mM, polysorbate 0.005%) and were injected over the sensor surface for 4 min at a flow rate of 20 μL/min. All diluted samples were injected at the same time over the four channels (flow cells). Flow cell 1 was used to obtain control sensorgrams showing nonspecific binding to the sensorchip surface. Control sensorgrams were subtracted from sensorgrams obtained with immobilized fusion proteins to yield true binding responses. Kinetics constants (kon, koff, and KD) were calculated using BIAevaluation 4.0.1 software.
In silico analysis
Compositional profiling
To gain insight into the relationships between sequence and disorder, the amino acid compositions of Knr4 and its fragments were compared using an approach developed for the analysis of intrinsically disordered proteins.11,24 To this end, the fractional difference in composition between given protein or a protein set (Knr4, its fragments and a set of intrinsically disordered proteins,27 and a set of ordered proteins24 was calculated for each amino acid residues. The fractional difference was calculated as (CX − Cordered)/Cordered, where CX is the content of a given amino acid in a given protein (or protein set), and Cordered is the corresponding content in a set of ordered proteins and plotted for each amino acid. This analysis was performed using Composition Profiler, a tool that automates this task and graphically summarizes the results,24 which is available at http://www.cprofiler.org/.
Predictions of intrinsic disorder
PONDR®-FIT28 bioinformatics tool represents a meta-predictor which is a consensus artificial neural network prediction method combining the outputs of several disorder predictors, such as PONDR-VLXT,29 PONDR-VSL2,30 PONDR-VL3,31 FoldIndex,32 IUPred,33 and Top-IDP.34 These individual predictors were selected because they use different predictive approaches, emphasize different features of the sequence, and all give acceptable accuracies as individual predictors.28 The PONDR®-FIT meta-predictor represents a completely new combination of predictors not used in the development of any previous meta-predictors, such as metaPrDOS54 and MD.55 PONDR®-FIT was shown to be comparable with the other meta-predictors with significantly improved accuracy in the aggregate as compared with its individual component predictors. In fact, by eightfold cross-validation PONDR®-FIT was found to improve the prediction accuracy over a range of 3–20% with an average of 11% compared with the single predictors, depending on the datasets being used.28
Acknowledgments
We are indebted to Pr Claudio De Virgilio for generously making his laboratory facilities available and for the supervision of the Two-Hybrid global screen experiments.
References
- 1.Martin H, Dagkessamanskaia A, Satchanska G, Dallies N, Francois J. KNR4, a suppressor of Saccharomyces cerevisiae cwh mutants, is involved in the transcriptional control of chitin synthase genes. Microbiology. 1999;145:249–258. doi: 10.1099/13500872-145-1-249. [DOI] [PubMed] [Google Scholar]
- 2.Martin-Yken H, Dagkessamanskaia A, Basmaji F, Lagorce A, Francois J. The interaction of Slt2 MAP kinase with Knr4 is necessary for signalling through the cell wall integrity pathway in Saccharomyces cerevisiae. Mol Microbiol. 2003;49:23–35. doi: 10.1046/j.1365-2958.2003.03541.x. [DOI] [PubMed] [Google Scholar]
- 3.Lesage G, Bussey H. Cell wall assembly in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2006;70:317–343. doi: 10.1128/MMBR.00038-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, Qureshi-Emili A, Li Y, Godwi B, Conover D, Kalbfleisch T, Vijayadamodar G, Yang M, Johnston M, Fields S, Rothberg JM. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 5.Dagkessamanskaia A, Martin-Yken H, Basmaji F, Briza P, Francois J. Interaction of Knr4 protein, a protein involved in cell wall synthesis, with tyrosine tRNA synthetase encoded by TYS1 in Saccharomyces cerevisiae. FEMS Microbiol Lett. 2001;200:53–58. doi: 10.1111/j.1574-6968.2001.tb10692.x. [DOI] [PubMed] [Google Scholar]
- 6.Basmaji F, Martin-Yken H, Durand F, Dagkessamanskaia A, Pichereaux C, Rossignol M, Francois J. The ‘interactome’ of the Knr4/Smi1, a protein implicated in coordinating cell wall synthesis with bud emergence in Saccharomyces cerevisiae. Mol Genet Genomics. 2006;275:217–230. doi: 10.1007/s00438-005-0082-8. [DOI] [PubMed] [Google Scholar]
- 7.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrín-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St Onge P, Ghanny S, Lam MH, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O'Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 8.Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, Malitskaya Y, Vogel J, Bussey H, Michnick SW. An in vivo map of the yeast protein interactome. Science. 2008;320:1465–1470. doi: 10.1126/science.1153878. [DOI] [PubMed] [Google Scholar]
- 9.Durand F, Dagkessamanskaia A, Martin-Yken H, Graille M, Van Tilbeurgh H, Uversky VN, Francois JM. Structure-function analysis of Knr4/Smi1, a newly member of intrinsically disordered proteins family, indispensable in the absence of a functional PKC1-SLT2 pathway in Saccharomyces cerevisiae. Yeast. 2008;25:563–576. doi: 10.1002/yea.1608. [DOI] [PubMed] [Google Scholar]
- 10.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 11.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 12.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 13.Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 14.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 15.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunker AK, Silman I, Uversky VN, Sussman JL. Function and structure of inherently disordered proteins. Curr Opin Struct Biol. 2008;18:756–764. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Dunker AK, Oldfield CJ, Meng J, Romero P, Yang JY, Chen JW, Vacic V, Obradovic Z, Uversky VN. The unfoldomics decade: an update on intrinsically disordered proteins. BMC Genomics. 2008;9(Suppl 1):S1. doi: 10.1186/1471-2164-9-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 19.Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18:343–384. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 20.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–1898. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6:1899–1916. doi: 10.1021/pr060393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cagney G, Uetz P. High-throughput screening for protein-protein interactions using yeast two-hybrid arrays. Curr Protoc Protein Sci Chapter 19. 2001 doi: 10.1002/0471140864.ps1906s24. Unit 19.6. [DOI] [PubMed] [Google Scholar]
- 24.Vacic V, Oldfield CJ, Mohan A, Radivojac P, Cortese MS, Uversky VN, Dunker AK. Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res. 2007;6:2351–2366. doi: 10.1021/pr0701411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92:1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deleage G, Blanchet C, Geourjon C. Protein structure prediction. Implications for the biologist. Biochimie. 1997;79:681–686. doi: 10.1016/s0300-9084(97)83524-9. [DOI] [PubMed] [Google Scholar]
- 27.Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, Obradovic Z, Dunker AK. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007;35:D786–D793. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 2010;1804:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequencecomplexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 30.Obradovic Z, Peng K, Vucetic S, Radivojac P, Dunker AK. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins. 2005;61:176–182. doi: 10.1002/prot.20735. [DOI] [PubMed] [Google Scholar]
- 31.Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK. Predicting intrinsic disorder from amino acid sequence. Proteins. 2003;53:566–572. doi: 10.1002/prot.10532. [DOI] [PubMed] [Google Scholar]
- 32.Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 33.Dosztanyi Z, Csizmok V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol. 2005;347:827–839. doi: 10.1016/j.jmb.2005.01.071. [DOI] [PubMed] [Google Scholar]
- 34.Campen A, Williams RM, Brown CJ, Meng J, Uversky VN, Dunker AK. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept Lett. 2008;15:956–963. doi: 10.2174/092986608785849164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Callaghan AJ, Aurikko JP, Ilag LL, Gunter Grossmann J, Chandran V, Kuhnel K, Poljak L, Carpousis AJ, Robinson CV, Symmons MF, Luisi BF. Studies of the RNA degradosome-organizing domain of the Escherichia coli ribonuclease RNase E. J Mol Biol. 2004;340:965–979. doi: 10.1016/j.jmb.2004.05.046. [DOI] [PubMed] [Google Scholar]
- 36.Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 37.Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of molecular recognition features (MoRFs) J Mol Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 38.Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, Dunker AK. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry. 2007;46:13468–13477. doi: 10.1021/bi7012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng KP, Potikyan G, Savene RO, Denny CT, Uversky VN, Lee KA. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proc Natl Acad Sci USA. 2007;104:479–484. doi: 10.1073/pnas.0607007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han JD, Bertin N, Hao T, Goldberg DS, Berriz GF, Zhang LV, Dupuy D, Walhout AJ, Cusick ME, Roth FP, Vidal M. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature. 2004;430:88–93. doi: 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- 41.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 42.Gsponer J, Futschik ME, Teichmann SA, Babu MM. Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science. 2008;322:1365–1368. doi: 10.1126/science.1163581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wright PE, Dyson HJ. Intrinsically, unstructured proteins: reassessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 44.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 45.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patil A, Nakamura H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006;580:2041–2045. doi: 10.1016/j.febslet.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 47.Ekman D, Light S, Bjorklund AK, Elofsson A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol. 2006;7:R45. doi: 10.1186/gb-2006-7-6-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Higurashi M, Ishida T, Kinoshita K. Identification of transient hub proteins and the possible structural basis for their multiple interactions. Protein Sci. 2008;17:72–78. doi: 10.1110/ps.073196308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh GP, Ganapathi M, Dash D. Role of intrinsic disorder in transient interactions of hub proteins. Proteins. 2007;66:761–765. doi: 10.1002/prot.21281. [DOI] [PubMed] [Google Scholar]
- 50.Uversky VN, Dunker AK. Biochemistry. Controlled chaos. Science. 2008;322:1340–1341. doi: 10.1126/science.1167453. [DOI] [PubMed] [Google Scholar]
- 51.Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, Jakel H, Kullmann M, Kriwacki RW, Hengst L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–280. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 52.Vavouri T, Semple JI, Garcia-Verdugo R, Lehner B. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell. 2009;138:198–208. doi: 10.1016/j.cell.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 53.James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishida T, Kinoshita K. Prediction of disordered regions in proteins based on the meta approach. Bioinformatics. 2008;24:1344–1348. doi: 10.1093/bioinformatics/btn195. [DOI] [PubMed] [Google Scholar]
- 55.Schlessinger A, Punta M, Yachdav G, Kajan L, Rost B. Improved disorder prediction by combination of orthogonal approaches. PLoS One. 2009;4:e4433. doi: 10.1371/journal.pone.0004433. [DOI] [PMC free article] [PubMed] [Google Scholar]