

Abstract
Surface lysine methylation (SLM) is a technique for improving the rate of success of protein crystallization by chemically methylating lysine residues. The exact mechanism by which SLM enhances crystallization is still not clear. To study these mechanisms, and to analyze the conditions where SLM will provide the optimal benefits for rescuing failed crystallization experiments, we compared 40 protein structures containing N,N-dimethyl-lysine (dmLys) to a nonredundant set of 18,972 nonmethylated structures from the PDB. By measuring the relative frequency of intermolecular contacts (where contacts are defined as interactions between the residues in proximity with a distance of 3.5 Å or less) of basic residues in the methylated versus nonmethylated sets, dmLys-Glu contacts are seen more frequently than Lys-Glu contacts. Based on observation of the 10 proteins with both native and methylated structures, we propose that the increased rate of contact for dmLys-Glu is due to both a slight increase in the number of amine-carboxyl H-bonds and to the formation of methyl C–H···O interactions. By comparing the relative contact frequencies of dmLys with other residues, the mechanism by which methylation of lysines improves the formation of crystal contacts appears to be similar to that of Lys to Arg mutation. Moreover, analysis of methylated structures with the surface entropy reduction (SER) prediction server suggests that in many cases SLM of predicted SER sites may contribute to improved crystallization. Thus, tools that analyze protein sequences and mark residues for SER mutation may identify proteins with good candidate sites for SLM.
Keywords: intermolecular contacts, lysine methylation, protein crystallization, surface entropy reduction, data mining
Introduction
X-ray crystallography is the most important tool available in the arsenal for in-depth structural assignment of biological macromolecules such as proteins, RNA, and DNA. However, obtaining well-diffracting crystals of macromolecules is still a major bottleneck in X-ray crystallographic studies.1 The first challenge is to obtain crystals of macromolecules and second is to optimize conditions to obtain diffraction quality crystals. Numerous approaches have been developed to improve the rate and quality of protein crystallization. Some of these techniques are based on various theories of crystallization, while others are efficient methods for randomly sampling the many different external factors that affect crystallization. The majority of these methods focus on altering the physical and/or chemical parameters of the crystallization experiment.2,3 However, as the properties of the protein molecules strongly influence the crystallization process, an alternate approach is to modify the macromolecule rather than the external conditions of the crystallization experiment. These approaches include limited proteolysis,4 selective mutagenesis of surface residues,5,6 and modification of certain side chain residues, such as lysine methylation.7,8
Artificial fusion or affinity tags are often introduced into protein sequences for purposes of solubility, purification or other applications.9 Often these tags are disordered or adopt multiple conformations, which decrease the probability of crystal formation. In general, these tags are removed prior to crystallization using enzymatic cleavage techniques, however, in some instances an ordered affinity tag has provided additional biological information10 or improved crystallization.11 Two proteolytic techniques may be used to remove solvent-exposed and presumably conformationally flexible fragments of a protein and may improve the structural order of the macromolecular surface, and thus potentially improving the probability of getting crystals.12 In limited proteolysis, proteins are treated with a protease, and stable protein fragments are isolated. The fragments may be analyzed to prepare a new cloning construct expressing only that fragment, or purified directly after cleavage. In the case of in situ proteolysis, samples are treated with dilute mixtures of proteases immediately before crystallization. In situ proteolysis is faster and simpler, but also may introduce heterogeneity into the crystallization experiment. The major drawback of both techniques is the possibility of cleavage of biologically important fragments of the protein. Nevertheless, the use of both limited13-15 and in situ proteolysis16,17 is reported to significantly increase the success rate of protein structure determination. Selective deletions of C- or N-terminal tags18 or flexible omega-loops19 have also proven successful for improving protein crystallization.
Another innovative approach is surface entropy reduction (SER), where specific mutagenesis of certain amino acid residues improves the order of the surface of a protein and hence can increase the propensity for crystallization. In SER, specific surface residues like lysine or glutamate with long side chains, and thus high conformational entropy, are mutated to residues with much lower entropy, such as alanine.5 The surface entropy reduction prediction (SERp) server uses bioinformatics methods to identify clusters of residues that (1) contain numerous potentially highly-disordered side chains (residues like lysine and glutamate), (2) are not predicted to affect secondary structure, and (3) are not included in conserved sequence regions.20,21 Crystallization aided by SERp guided mutagenesis has been reported to yield a high success rate,5 indicating importance of analyzing the surface features of the protein towards crystal formation. Application of all these methods nonetheless does not guarantee that protein will crystallize.
The surface lysine methylation (SLM) technique,22,23 which modifies the protein surface by chemical means, has proven successful. A large-scale study has been conducted to evaluate the SLM success rate.8 Some of the major advantages of this method are speed and simplicity; it is unnecessary to mutate the gene and express mutated protein.24 A variety of chemical modifications may be made in a highly efficient and residue-selective way and the process takes hours to a day versus days or a week for mutation. Chemical modification of native protein is a well-established process for understanding the functional role of proteins. SLM has been reported to succeed in many cases,7,25 emphasizing the simplicity and effectiveness of the technique. However, as methylated protein is produced via a chemical process, it is often difficult or impossible to selectively methylate certain surface lysines. In addition, partial methylation introduces heterogeneity not present in the point mutation approach, which may cause difficulties in crystallization.
The protocol for reductive methylation uses formaldehyde as the methylating agent in the presence of a dimethylamine-trifluoroborane complex (ABC), which serves as a reducing agent. The reaction takes place in a low ionic strength buffer which reduces protein denaturation, though partial precipitation of macromolecule is still observed in some cases.8 The degree of completion of the reaction is determined by mass spectrometry analysis of samples before and after the methylation process. All exposed primary amine groups, namely Nɛ atoms of lysines, and the N-terminus of the protein can be converted to mono- or dimethylated amines. Under these reaction conditions lysine amine quarternization is not observed. Monomethylated lysines have been observed but are typically rare, since the secondary monomethylated amine is more reactive than the primary amine of the parent, and hence only dimethylated lysines are observed when an excess of reagents is used. If the diffraction resolution of a given protein crystal is poor, then the probability of assigning an incorrect number of methyl groups to a modified lysine residue is a distinct possibility. In the current investigation, we focused our analysis only on N,N-dimethyl-lysines.
Surface modification of polar flexible side chain residues of a protein may increase the hydrophobic surface in aqueous media, which may in turn allow for favorable protein–protein hydrophobic contacts leading to better organization and packing of the crystal. Any understanding and quantification of protein–protein hydrophobic contacts through polar residue modification may help in the development of a predictive model for proteins, which have a high-polar surface area and are difficult to crystallize. In the present work, we compare the surface contacts of methylated and nonmethylated proteins, both by statistical analysis of the PDB and by direct comparison of proteins for which both nonmethylated and methylated structures are available.
Results and Discussion
A modification of native protein by methylation is expected to produce a structure isomorphous to the unmodified protein, presumably due to the fact that the modification occurs mostly at the surface of the protein after it is fully expressed and folded correctly. In the cases of the 10 (to date) proteins with both nonmethylated and methylated structures in the PDB, all 10 pairs of structures are isomorphous, with pair wise Cα RMSDs ranging from 0.2 to 1.4 Å (for the lowest resolution structure) and only minor changes seen in the conformation of flexible loops. Although the overall folds of each of the 10 proteins are not significantly altered by the lysine modification, other physicochemical properties, such as the degree of basicity of nitrogen, are more affected. Of the 10 cases, where crystal structures were solved for both native and dimethyl-lysine-containing protein, in eight the crystals of the dimethylated proteins scattered better and produced higher resolution data. The properties of methylated lysines in proteins have been investigated by NMR. The pKa of dimethylated lysine in calmodulin was measured to be 9.29–10.23, which is slightly lower than the value measured for lysine 9.84–10.71.26,27
Effect of methylation of lysine on basicity
The change in the pKa of lysine upon methylation depends on the local environment.28 Replacement of amine protons with methyl groups increases the amine Lewis basicity.29 In general, upon lysine methylation the protein pI usually decreases by ∼1 for pI values higher than ∼8, but only a slight decrease is observed for lower pI values.7 The decrease in pKa for dimethylated lysine is consistent with a decrease in protein isoelectric point after methylation. However, the pI distributions of a nonredundant set of all PDB structures versus the set of methylated structures (Fig. 1) appear to be similar. This agrees with the prior observation that no correlation has been shown between either the pH or probability of crystallization and the protein pI value.8,30
Figure 1.
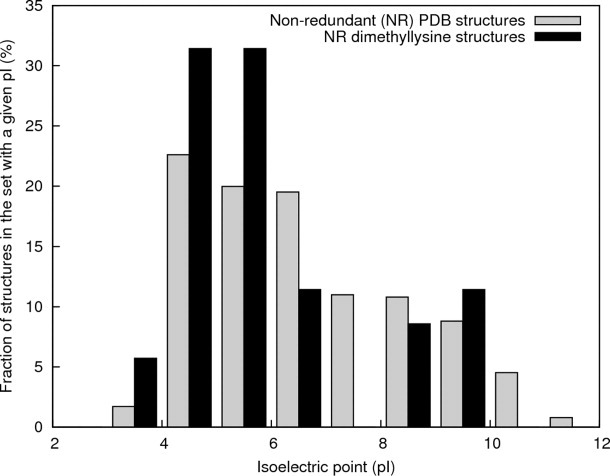
Histograms of calculated isoelectric points for the native counterparts of the methylated proteins in PDB structures containing methylated lysines (black bars) and for a set of nonredundant X-ray diffraction PDB structures (gray bars).
Protein surface stabilization by intramolecular interactions
Introduction of methyl groups affects electron density on the nitrogen and may make it possible to form new contacts. To study this phenomenon, we performed statistical analysis of the neighborhood of the basic residues Arg, Lys, and dmLys in the PDB (Table I), by calculating the number of interresidue interactions of the specified basic residue with another residue type, and dividing that number by the number of specific basic residues. (This average number of interresidue interactions for a given basic residue will subsequently be called the “contact rate.”) The contact rates for dmLys with Glu, Ile, Leu, Phe, and Ser are significantly greater than the corresponding contact rates for Lys (Table I and Fig. 2). Interestingly, the differences in contact frequencies of these five residues for Arg versus dmLys were not statistically significant (Fig. 2). At first glance, an increase in the contact rate of Lys with Leu, Ile and Phe upon methylation may be explained by the increase in volume, which allows dmLys to get closer to other residues and form favorable interactions. Adding methyl groups effectively increases the interaction radius of lysine by 1–1.2 Å. However, the increases in contact frequencies with hydrophobic residues are less clear given that molecular dynamics studies seem to indicate that dmLys has a larger hydration sphere than Lys.31 Similarly, reasons for increased propensity toward glutamate must be more complex, as a similar increase in the contact rate upon methylation is not observed for aspartate.
Tabel I.
Comparison of Surface Contact Frequencies
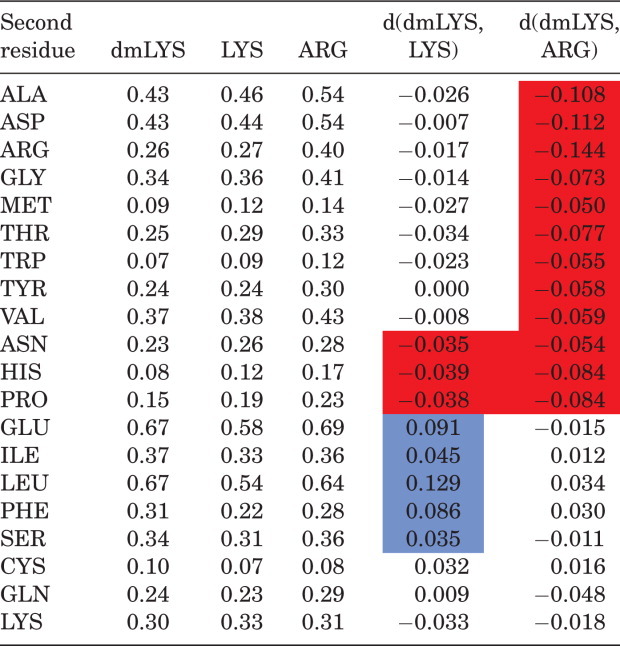 |
The first three columns give the average number of interresidue interactions per methylatedlysine (dmLYS), Lysine (LYS), and Arginine (ARG) to a specific second amino acid residue. Differences between the average numbers of interactions between dmLYS and LYS are specified by d(dmLYS,LYS) and between dmLYS and ARG by d(dmLYS, ARG). The statistically significant (p ≤ 0.05) differences for d(dmLYS,ARG) and d(dmLYS,LYS) are marked in red and blue, for decrease and increase, respectively.
Figure 2.
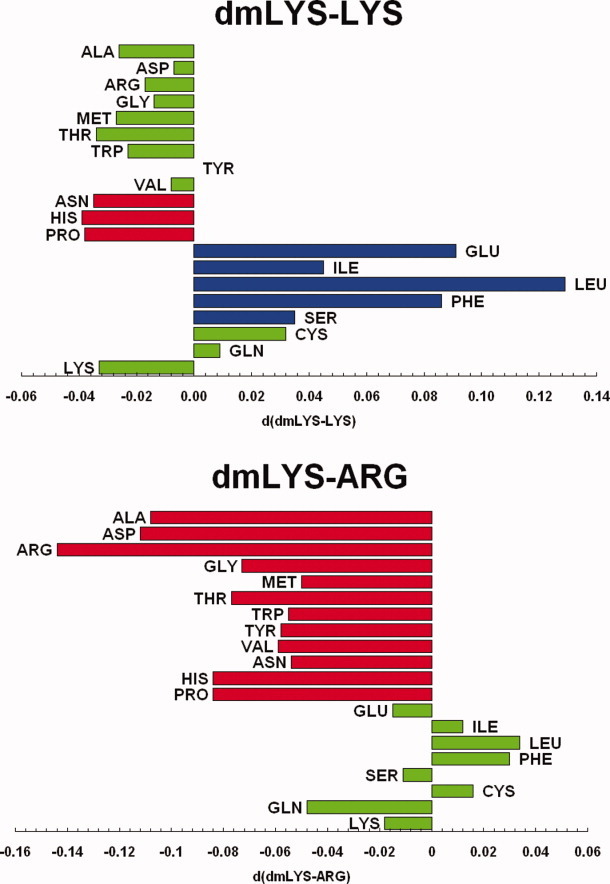
Comparison of surface contact frequencies of specific basic residue, Arg, Lys, or N,N-dimethyl-lysine (dmLys), contacts to a given type of residue, normalized by the total number of basic residues, described by the pair wise differences for dmLys-Lys and dmLys-Arg (green, not statistically significant; red and blue, statistically significant decrease and increase, respectively).
The contact rates of Lys-Glu and dmLys-Glu contacts were also analyzed for the set proteins with both methylated and nonmethylated structures in the PDB. In most of the cases, where more dmLys-Glu contacts were observed in the methylated structure than Lys-Glu contacts in the nonmethylated one, the methylated structure was solved at a higher resolution. For example, the methylated (PDB id 2FCL) and nonmethylated (PDB id 2EWR) structures of the putative nucleotidyl transferase TM1012 both contain 15 lysine residues (all of which are annotated as methylated in the case of 2FCL). 2EWR has 7 Lys-Glu contacts and diffracted to 1.6 Å, while 2FCL has 11 dmLys-Glu contacts and diffracted to 1.2 Å. Additionally, when an increase of Lys(dmLys)-Glu contacts was observed upon methylation, the increase in the number of Lys contacts in methylated protein was always greater then the increase in the number of amine-carboxyl hydrogen bonds. In some cases the methylated structure had only 1 or 2 additional amine-carboxyl H-bonds versus the unmodified protein structure. For example, in the previously discussed case of 2EWR versus 2FCL, the number of H-bonds increased from 1 to 3 upon methylation. For the remaining methylated–nonmethylated structure pairs, no additional H-bonds were observed. The relative increase in the number of dmLys-Glu interactions as compared to the overall increase regular amine-carboxyl H-bond interactions prompted us to calculate the overall frequency of H-bonds on the protein surface. The average H-bond frequency for the set of methylated proteins (0.07) and the nonredundant PDB set (0.08) turned out to be very similar, suggesting a minimal contribution of these effects on crystallization.
As methylation results in relatively few new amine-carboxyl H-bonds, other weak interactions should also be considered, such as cohesive C–H···O interactions, which were recently suggested to influence crystallization.32 An analysis of the local protein surfaces of the pairs of methylated–nonmethylated structure (both native and methylated lysine proteins) indicated that the methylated lysine residues induce distinct structural rearrangements in the nearby environment (Fig. 3). In one case, the methylated lysine side chain adopts a different conformation than the corresponding lysine in the nonmethylated structure to form a new hydrogen bond. In another case, Lys residues that were initially distant from a neighboring Glu residue moved toward it upon methylation forming two new cohesive C–H···O interactions. It has been suggested32 that methyl-carboxyl interactions facilitate crystallization by increasing the surface rigidity, but no comparative study has been done to confirm this hypothesis.
Figure 3.

Examples of local changes after methylation. Residues from methylated structures are shown in yellow, while unmodified structures are marked in grey. Dashed lines mark hydrogen bonds, while blue arrows show the change in the position of the side chain. A: Formation of hydrogen bond. In the unmodified molecule the distance between the Lys9 side chain nitrogen atom and the oxygen atom from the carboxyl group of Glu37 changes from 4.0 Å (PDB deposit: 1Z27) to 2.7 Å after methylation (PDB deposit: 1Z1Y). B: Different conformation of dmLys (PDB deposit: 2FCL) in comparison with Lys residue (PDB deposit: 2EWR) strengthens the hydrogen bond formed by Glu57 (the donor–acceptor distance changes from 3.0 to 2.7 Å).
The significant increase in the contact rate of dmLys to Glu, as compared to the difference of the contact rate of dmLys with Asp, may be due to the lengths of the side chains of Asp versus Glu. The length of the Glu side chain is longer, and more similar to that of dmLys and Lys. Thus, Glu may extend further and be more capable of forming stable interactions with less conformational strain than Asp. Furthermore, molecular dynamics studies suggest that the most favorable calculated H-bond distance involving a partially polarized carbon atom in a protonated amine is slightly longer than the H-bond distance observed for a protonated primary amine.31
Interestingly, the contact rates for dmLys to Glu, Ile, Leu, Phe, and Ser (i.e., those residues that showed a significant increase in contact frequency with Lys upon methylation) are similar to the corresponding contact rates observed for Arg (Table I and Fig. 2). This may be due to the structural similarity of Arg and dmLys (both side chains are similar in length and topology, and thus may have similar entropic and packing consequences) and the ability to form X–H···O interactions, where X = C for dmLys and X = N for Arg.
Two distinctly separate mechanisms are proposed for the observed enhanced dmLys-Glu interactions. First, the extended chain length of dmLys, as compared to native lysine, may result in new C–H···O/N interactions. Second, while the methylation of lysine affects its basicity and hence the ability of lysine to form hydrogen bonds, regardless very few new hydrogen bonds formed by lysine were observed upon methylation. These two mechanisms may operate either independently or synergistically to allow better crystal packing and hence better diffraction.
Previously, the role of methylation was usually explained by a decrease of protein solubility like in case of the scorpion toxin 1R1G, which failed to crystallize in natural form and yielded crystals only after lysine methylation33 or using racemic crystallization techniques.34 Nevertheless, C–H···O/N interactions are also present in these crystals and can be taken into account for their success of crystallization.
Crystal and intermolecular contacts formed by methylated lysines
The propensity of methylated lysine residues to form crystal contacts has been investigated as well. Lysine residues are rarely found in crystal contacts.35 As a result, in the SER approach, clustered surface lysines are selectively mutated to alanines or other low-entropy residues to improve the probability of forming well-diffracting crystals.5 Alanine residues are seen less frequently than other residues in specific surface contacts, but as its side chain is rigid, alanine is the most logical mutation for reducing conformation entropy. It was reported that methylated lysines are found to form crystal contacts in the structures of methylated proteins.7
To quantify the role of methylated lysines in the formation of crystal lattices, only the dmLys or Lys residues involved in crystal contacts were identified in a set of methylated structures and in a set of nonredundant PDB structures, respectively. For these residues, contact rates were calculated as before, save that only dmLys or Lys residues in crystal contacts were analyzed for contact partners. The contact rates for dmLys residues that form crystal contacts in methylated structures do not differ in a statistically significant way from the corresponding contact rates in the nonmethylated set (data not shown).
All of the sequences of methylated proteins in the PDB were also analyzed with the SERp server in order to identify lysine-containing high entropy clusters. The clusters proposed for mutation by the server were examined for their role in crystal contact formation. In about 60% of the considered cases, the methylated lysines in these clusters were perfectly ordered and formed crystal contacts (Table II). Most of the lysines designated for mutagenesis were also involved in stabilizing surface contacts following mainly cohesive C–H···O bond formation mechanism. dmLys residues have a different ability to form interactions compared with Ala, due to an extended side chain that exhibits complex rotameric behavior. The differences in the interactions formed by alanine and high entropy lysine residues are some of the rationales for the success of the SERp method. Nonetheless, substituting a methylated lysine or alanine for lysine can not be compared, since both phenomena offer different sets of properties, to an extent the same goal is achieved in some cases, surface entropy is reduced through hydrogen bonds over the protein surface, and an increased ability to form hydrogen bonds facilitates crystal contact formation.
Tabel II.
Lysine-Containing High-Entropy Clusters
| PDB id | Range | Sequence | Score | Contact |
|---|---|---|---|---|
| 132L | 31–33 | AAKFE | 1.68 | Crystal |
| 95–97 | AKK | 2.43 | Surface | |
| 2FCL (5.69) | 78–79 | KK | 5.27 | Surface |
| 86–87 | EK | 4.15 | Crystal | |
| 2FTZ | 14–16 | KKE | 4.91 | Surface |
| 3EGL | 73–74 | KE | 4.92 | Crystal + surface |
| 219–223 | EAAKQ | 3.51 | Surface | |
| 2I6G | 12–13 | EK | 4.04 | Crystal |
| 1LLN | 26–29 | KDKK | 6.81 | Surface |
| 114–115 | EK | 5.41 | Surface | |
| 43–46 | EQPK | 4.61 | Surface | |
| 2F4I (5.24) | 144–146 | KEE | 4.61 | Surface |
| 1Z1Y | 172–174 | KEK | 7.00 | Crystal + surface |
| 131–133 | EKK | 4.59 | Crystal + surface | |
| 48–50 | KKE | 4.22 | Crystal + surface | |
| 1R1G | 26 | K | 2.17 | Crystal + surface |
| 21 | K | 2.05 | Crystal | |
| 2QHQ (4.45) | 11 | K | 2.51 | Crystal |
| 1VIX (4.23) | 168–169 | EK | 3.69 | Surface |
| 1IV8 (7.11) | 108–111 | KKSK | 6.43 | Crystal + surface |
| 614–617 | KSKK | 6.18 | Crystal + surface | |
| 2P6V | 610–611 | KQ | 3.00 | Crystal |
| 3E3X | 48–51 | KEGK | 4.45 | Crystal + surface |
| 117–119 | KEE | 4.37 | Surface | |
| 316–317 | KK | 3.46 | Surface | |
| 3GS9 | 283–284 | EK | 5.26 | Surface |
| 324–325 | KK | 4.44 | Surface | |
| 138–139 | EK | 4.25 | Crystal | |
| 2ZPM | 52–52 | K | 2.41 | Crystal + surface |
| 2O3F (2.52) | 18–18 | K | 2.04 | Crystal |
| 2Q7X (5.28) | 261–262 | EK | 5.22 | Surface |
| 25–26 | EK | 4.90 | Surface | |
| 2VD8 (3.78) | 197–198 | KE | 2.90 | Crystal + surface |
| 2ETV | 197–199 | EAK | 4.58 | Surface |
| 305–308 | EEK | 4.33 | Surface | |
| 114–116 | QEK | 3.53 | Surface | |
| 2P4P | 53–54 | KK | 5.45 | Crystal + surface |
| 62–62 | K | 2.89 | Surface | |
| 1V1K (8.33) | 157–159 | EQK | 5.42 | Surface |
| 170–173 | QAKE | 5.31 | Crystal | |
| 3C8G | 82–82 | K | 2.28 | Crystal |
| 3EPQ (5.06) | 119–121 | EAK | 4.81 | Surface |
| 236–238 | QQKQ | 4.47 | Surface | |
| 226–227 | KK | 4.09 | Crystal + surface | |
| 2VIX | 70–71 | EK | 4.32 | Crystal + surface |
| 168–169 | EK | 3.69 | Surface | |
| 93–94 | EK | 3.44 | Crystal + surface |
SERp-server-predicted high-entropy clusters which contain methylated lysines and formed intermolecular contacts in the methylated protein structures. The contacts are labeled either as crystal contacts or surface interactions. For the remaining methylated proteins, no contacts were formed by dmLys in the high-entropy clusters identified. Values in parentheses next to the PDB id shows the score for the highest entropy lysine containing cluster found by the server if no contacts were found for lysines within it.
Crystal contact engineering involving substitution of lysine by arginine, a residue more frequently observed to form crystal contacts, has been already described in the literature.36,37 This method was demonstrated to be a successful alternative to the SER method: instead of decreasing the size of the entropic barrier, a residue is replaced with another with similar conformation entropy but an increased probability to form noncovalent interactions. Thus, this approach is more similar to SLM, as methylated lysine exhibits better noncovalent bonding ability than lysine but does not decrease its conformational entropy. Therefore, modification of Lys to dmLys may be seen as similar to replacing Lys with Arg. Most interactions with an arginine side chain occur approximately in the plane of the guanidinium moiety. However, interactions with a dimethyl group could happen in all directions, covering a greater interaction volume than Arg, although individual interactions may not be as strong as those with a guanidinium group. In this way, SLM may alter protein properties with no need for selective mutagenesis. However, SLM sacrifices the selectivity of the process as it affects the whole protein surface, and not some small local surface patches.
Thermodynamics of the methylation effect
The surface conformational entropy energy barrier of a single lysine residue at room temperature has been estimated as ∼2 kcal mol−1.38 Molecular modeling studies have estimated that dmLys has higher side-chain entropy than Lys by about 3.5 J mol−1 K−1, which by itself would create a greater barrier to crystal packing.31 The difference in entropic contribution to ground state energy between Arg and Lys (or dmLys) is unclear and debatable, because there is no consensus which residue has higher entropy in folded proteins.39,40 To allow crystallization, this barrier must be overcome by the entropy increase through the release of water molecules which were previously bound to residues engaged in protein–protein interfaces, and the stabilizing energetic effect of crystal contact interactions.41 As dmLys on the surface of a protein is predicted to form a larger solvation sphere, the entropic gain from dmLys upon crystallization can be estimated to be nearly 4 J mol−1 K−1 greater than the corresponding gain from Lys.31 This overcomes the penalty due to the higher conformational entropy for dmLys. The average energy of the discussed C–H···O contacts never exceeds a few kcal mol−1,42 which is approximately equal to the entropic barrier of the interacting residues. It is possible that dmLys residues form more stable noncovalent bonds because of their energetic effect described above.
Materials and Methods
Dataset
To calculate the properties of dmLys residues containing proteins with methylation introduced by chemical means, 40 structures in the Protein Data Bank (PDB) released before June 2009 were identified. To the best of our knowledge, this set contains all such proteins available in the PDB. As reference for comparative study of surface neighborhood and pI distribution, a nonredundant set of 18,971 proteins and protein complexes with structures solved by X-ray diffraction, as released in the PDB on or before June 2009 was used. This nonredundant set was constructed using the CD-Hit program, with clustering at 90% sequence identity.43
Analysis of neighborhood properties of the protein surface
Two residues were considered to be in contact when the distance between any two atoms from these residues was smaller than 3.5 Å. Such analysis is biased towards weaker interactions, because most strong H bonds would still be included if the cutoff distance was decreased to 3.2 Å. To validate that this bias would not significantly affect the statistics, we also performed analysis for 3.2 Å cutoff (see Supporting Information). If the part of the residue was missing in the PDB file, it was considered disordered, but contacts of its ordered part were still taken into account without further weighting or normalization. If double conformation was available, contacts of both conformations were taken into account without further weighting or normalization. The average number of residue A-to-residue B contacts (the “contact rate” of residue A to residue B) was calculated by summing the number of residues B in contact with each residue A, and dividing that number by the number of residues A. Only Lys, dmLys and Arg residues on the protein surface were considered. Surface residues in the reference set were identified using the program Surface Racer.44 The residue was considered to be on the surface if its solvent-accessible surface area was larger than 0 Å2. Because of the nature of the methylation process, all dmLys residues were assumed to be on the surface.
A hydrogen bond between Glu and Lys/dmLys residues was identified if (1) the Oδ1/Oδ2 atom of Glu and Nɛ atom of Lys were closer than 3.5 Å, and (2) this pair of atoms were the closest pair of atoms from these two residues. As every contact between the residues contributes to potential energy, the number of contacts was not otherwise normalized according to surface, volume, or number of atoms within the residue.
Interresidue contacts, which include both intramolecular and intermolecular contacts, were calculated using the SQL queries to the NEIGHBORHOOD relational database,45 which contains structural information about all residues, atoms, and contacts between residues and atoms.
Isoelectric point calculations
Isoelectric points (pI) were calculated from protein sequence information using a reimplementation of the algorithm used by the IEP program in the EMBOSS molecular biology software suite.46 By using the frequencies and theoretical pKa of each residue capable of donating protons, the weighted average of the effect of each type of amino acid on the overall charge is used to calculate the pH at which the charge is electrically neutral. The sequences were first filtered to remove polyhistidine fragments typically introduced at the termini of protein sequences to act as specific metal affinity tags. Specifically, fragments of 6 or more His residues in a sequence were identified. If a polyhistidine fragment lay 10 residues or closer to either the N- or C-terminus, the polyhistidine fragment and the residues between it and the terminus were excluded from the pI calculation. For the methylated proteins, isoelectric point calculations were performed in the same way for their nonmethylated counterparts.
Crystal contacts analysis
For purpose of automated crystal contact analysis a set of 3644 PDB entries, containing only proteins with a single chain in the asymmetric unit was chosen. Complexes with DNA or RNA were excluded from consideration. The entries were analyzed with the program CONTACT from the CCP4 suite,47 to calculate intermolecular protein-protein interactions. For the methylated set, both the CONTACT program and the NEIGHBORHOOD database were used to identify intermolecular contacts, between and inside asymmetric units, respectively. The distance cutoff for crystal contact was the same as previously used for surface contacts, that is, 3.5 Å.
Surface entropy reduction predictions
For the prediction of clusters with high conformational entropy in the set of 40 methylated proteins, the SERp server was used,21 with the default settings.
Statistical data analysis
Contact frequencies can be considered as proportional data and as such several statistical tests can be used to evaluate statistical significance of differences between pair of corresponding contact frequencies.48 In this report we consider a difference between two contact frequencies to be statistically significant according both to the t-test and z-test with the cut-off P-value of 0.05.
Summary
Simplicity and low labor and cost requirements makes SLM a very attractive rescue approach for proteins resistant to traditional methods of crystallization, and it is an easier alternative to mutagenesis-based methods, which are time consuming. The method could be also treated as a possible replacement for selective mutagenesis of lysines to arginines5 by chemical means, due to the similarity of arginine to methylated lysine in topology and surface contact formation ability. Tuning the properties of the side chain of lysine, specifically the steric shielding and electron density of the amine nitrogen, leads to alteration of numerous factors, which may affect the crystallization process significantly. The key factors that appeared to improve protein crystallization are the ability of methylated lysine to form new noncovalent interactions (mainly weak C–H···O/N bonds) and reduced surface entropy due to their formation.
Acknowledgments
The authors thank Zbigniew Dauter and the members of the Midwest Center for Structural Genomics for help and discussions.
Glossary
Abbreviations:
- dmLys
N,N-dimethyl-lysine
- PDB
Protein Data Bank
- SER
surface entropy reduction
- SERp
surface entropy reduction prediction
- SLM
surface lysine methylation.
References
- 1.O'toole N, Grabowski M, Otwinowski Z, Minor W, Cygler M. The structural genomics experimental pipeline: insights from global target lists. Proteins: Struct Funct Bioinformatics. 2004;56:201–210. doi: 10.1002/prot.20060. [DOI] [PubMed] [Google Scholar]
- 2.Giegé R, Ducruix A. An introduction to the crystallogenesis of biological macromolecules. In. In: Ducruix A, Giegé R, editors. Crystallization of nucleic acids and proteins: a practical approach. Oxford: Oxford University Press; 1999. pp. 1–13. [Google Scholar]
- 3.Mcpherson A. Crystallization of biological macromolecules. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press.; 1999. [Google Scholar]
- 4.Wilson WD, Foster JF. Conformation-dependent limited proteolysis of bovine plasma albumin by an enzyme present in commercial albumin preparations. Biochemistry. 1971;10:1772–1780. doi: 10.1021/bi00786a007. [DOI] [PubMed] [Google Scholar]
- 5.Derewenda ZS, Vekilov PG. Entropy and surface engineering in protein crystallization. Acta Crystallogr Sect D Biol Crystallogr. 2006;62:116–124. doi: 10.1107/S0907444905035237. [DOI] [PubMed] [Google Scholar]
- 6.Yamada H, Tamada T, Kosaka M, Miyata K, Fujiki S, Tano M, Moriya M, Yamanishi M, Honjo E, Tada H, Ino T, Yamaguchi H, Futami J, Seno M, Nomoto T, Hirata T, Yoshimura M, Kuroki R. ‘Crystal lattice engineering,’ an approach to engineer protein crystal contacts by creating intermolecular symmetry: crystallization and structure determination of a mutant human RNase 1 with a hydrophobic interface of leucines. Protein Sci. 2007;16:1389–1397. doi: 10.1110/ps.072851407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walter TS, Meier C, Assenberg R, Au KF, Ren J, Verma A, Nettleship JE, Owens RJ, Stuart DI, Grimes JM. Lysine methylation as a routine rescue strategy for protein crystallization. Structure. 2006;14:1617–1622. doi: 10.1016/j.str.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim Y, Quartey P, Li H, Volkart L, Hatzos C, Chang C, Nocek B, Cuff M, Osipiuk J, Tan K, Fan Y, Bigelow L, Maltseva N, Wu R, Borovilos M, Duggan E, Zhou M, Binkowski TA, Zhang RG, Joachimiak A. Large-scale evaluation of protein reductive methylation for improving protein crystallization. Nat Methods. 2008;5:853–854. doi: 10.1038/nmeth1008-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammarstrom M, Hellgren N, Van Den Berg S, Berglund H, Hard T. Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli. Protein Sci. 2002;11:313–321. doi: 10.1110/ps.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirillova O, Chruszcz M, Shumilin IA, Skarina T, Gorodichtchenskaia E, Cymborowski M, Savchenko A, Edwards A, Minor W. An extremely SAD case: structure of a putative redox-enzyme maturation protein from Archaeoglobus fulgidus at 3.4 A resolution. Acta Crystallogr Sect D Biol Crystallogr. 2007;63:348–354. doi: 10.1107/S0907444906055065. [DOI] [PubMed] [Google Scholar]
- 11.Zhang RG, Andersson CE, Skarina T, Evdokimova E, Edwards AM, Joachimiak A, Savchenko A, Mowbray SL. The 2.2 A resolution structure of RpiB/AlsB from Escherichia coli illustrates a new approach to the ribose-5-phosphate isomerase reaction. J Mol Biol. 2003;332:1083–1094. doi: 10.1016/j.jmb.2003.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mazza C, Segref A, Mattaj IW, Cusack S. Co-crystallization of the human nuclear cap-binding complex with a m7GpppG cap analogue using protein engineering. Acta Crystallogr Sect D Biol Crystallogr. 2002;58:2194–2197. doi: 10.1107/s0907444902015445. [DOI] [PubMed] [Google Scholar]
- 13.Gao X, Bain K, Bonanno JB, Buchanan M, Henderson D, Lorimer D, Marsh C, Reynes JA, Sauder JM, Schwinn K, Thai C, Burley SK. High-throughput limited proteolysis/mass spectrometry for protein domain elucidation. J Struct Funct Genomics. 2005;6:129–134. doi: 10.1007/s10969-005-1918-5. [DOI] [PubMed] [Google Scholar]
- 14.Koth CM, Orlicky SM, Larson SM, Edwards AM. Use of limited proteolysis to identify protein domains suitable for structural analysis. Methods Enzymol. 2003;368:77–84. doi: 10.1016/S0076-6879(03)68005-5. [DOI] [PubMed] [Google Scholar]
- 15.Lowe DM, Aitken A, Bradley C, Darby GK, Larder BA, Powell KL, Purifoy DJ, Tisdale M, Stammers DK. HIV-1 reverse transcriptase: crystallization and analysis of domain structure by limited proteolysis. Biochemistry. 1988;27:8884–8889. doi: 10.1021/bi00425a002. [DOI] [PubMed] [Google Scholar]
- 16.Dong A, Xu X, Edwards AM, Chang C, Chruszcz M, Cuff M, Cymborowski M, Di Leo R, Egorova O, Evdokimova E, Filippova E, Gu J, Guthrie J, Ignatchenko A, Joachimiak A, Klostermann N, Kim Y, Korniyenko Y, Minor W, Que Q, Savchenko A, Skarina T, Tan K, Yakunin A, Yee A, Yim V, Zhang R, Zheng H, Akutsu M, Arrowsmith C, Avvakumov GV, Bochkarev A, Dahlgren LG, Dhe-Paganon S, Dimov S, Dombrovski L, Finerty P, Jr, Flodin S, Flores A, Gräslund S, Hammerström M, Herman MD, Hong BS, Hui R, Johansson I, Liu Y, Nilsson M, Nedyalkova L, Nordlund P, Nyman T, Min J, Ouyang H, Park HW, Qi C, Rabeh W, Shen L, Shen Y, Sukumard D, Tempel W, Tong Y, Tresagues L, Vedadi M, Walker JR, Weigelt J, Welin M, Wu H, Xiao T, Zeng H, Zhu H. In situ proteolysis for protein crystallization and structure determination. Nat Methods. 2007;4:1019–1021. doi: 10.1038/nmeth1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wernimont A, Edwards A. In situ proteolysis to generate crystals for structure determination: an update. PLoS ONE. 2009;4:e5094. doi: 10.1371/journal.pone.0005094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pai EF, Kabsch W, Krengel U, Holmes KC, John J, Wittinghofer A. Structure of the guanine-nucleotide-binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nature. 1989;341:209–214. doi: 10.1038/341209a0. [DOI] [PubMed] [Google Scholar]
- 19.Dale GE, Kostrewa D, Gsell B, Stieger M, D'arcy A. Crystal engineering: deletion mutagenesis of the 24 kDa fragment of the DNA gyrase B subunit from Staphylococcus aureus. Acta Crystallogr Sect D Biol Crystallogr. 1999;55:1626–1629. doi: 10.1107/s0907444999008227. [DOI] [PubMed] [Google Scholar]
- 20.Cooper DR, Boczek T, Grelewska K, Pinkowska M, Sikorska M, Zawadzki M, Derewenda Z. Protein crystallization by surface entropy reduction: optimization of the SER strategy. Acta Crystallogr Sect D Biol Crystallogr. 2007;63:636–645. doi: 10.1107/S0907444907010931. [DOI] [PubMed] [Google Scholar]
- 21.Goldschmidt L, Cooper DR, Derewenda ZS, Eisenberg D. Toward rational protein crystallization: a web server for the design of crystallizable protein variants. Protein Sci. 2007;16:1569–1576. doi: 10.1110/ps.072914007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rypniewski WR, Holden HM, Rayment I. Structural consequences of reductive methylation of lysine residues in hen egg white lysozyme: an X-ray analysis at 1.8-A resolution. Biochemistry. 1993;32:9851–9858. doi: 10.1021/bi00088a041. [DOI] [PubMed] [Google Scholar]
- 23.Rayment I. Reductive alkylation of lysine residues to alter crystallization properties of proteins. Methods Enzymol. 1997;276:171–179. [PubMed] [Google Scholar]
- 24.Kim Y, Quartey P, Lezondra L, Hatzos C, Zhou M, Maltseva N, Li H, Wu R, Joachimiak A. Use of reductive methylation of proteins to increase crystallization efficiency at the Midwest Center for Structural Genomics. Acta Crystallogr Sect A Found Crystallogr. 2005;61:c437.. [Google Scholar]
- 25.Kobayashi M, Kubota M, Matsuura Y. Crystallization and improvement of crystal quality for X-ray diffraction of maltooligosyl trehalose synthase by reductive methylation of lysine residues. Acta Crystallogr Sect D Biol Crystallogr. 1999;55:931–933. doi: 10.1107/s0907444999002115. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M, Vogel HJ. Determination of the side chain pKa values of the lysine residues in calmodulin. J Biol Chem. 1993;268:22420–22428. [PubMed] [Google Scholar]
- 27.Zhang M, Thulin E, Vogel HJ. Reductive methylation and pKa determination of the lysine side chains in calbindin D9k. J Protein Chem. 1994;13:527–535. doi: 10.1007/BF01901534. [DOI] [PubMed] [Google Scholar]
- 28.Zhang M, Yuan T, Vogel HJ. A peptide analog of the calmodulin-binding domain of myosin light chain kinase adopts an alpha-helical structure in aqueous trifluoroethanol. Protein Sci. 1993;2:1931–1937. doi: 10.1002/pro.5560021114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith MB, March J. March's advanced organic chemistry. New York: Wiley.; 2001. [Google Scholar]
- 30.Canaves JM, Page R, Wilson IA, Stevens RC. Protein biophysical properties that correlate with crystallization success in Thermotoga maritima: maximum clustering strategy for structural genomics. J Mol Biol. 2004;344:977–991. doi: 10.1016/j.jmb.2004.09.076. [DOI] [PubMed] [Google Scholar]
- 31.Fan Y, Joachimiak A. Enhanced crystal packing due to solvent reorganization through reductive methylation of lysine residues in oxidoreductase from Streptococcus pneumoniae. J Struct Funct Genomics. doi: 10.1007/s10969-010-9079-6. (in press). DOI: 10.1007/s10969-010-9079-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaw N, Cheng C, Tempel W, Chang J, Ng J, Wang XY, Perrett S, Rose J, Rao Z, Wang BC, Liu ZJ. (NZ)CH...O contacts assist crystallization of a ParB-like nuclease. BMC Struct Biol. 2007;7:46. doi: 10.1186/1472-6807-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szyk A, Lu W, Xu C, Lubkowski J. Structure of the scorpion toxin BmBKTtx1 solved from single wavelength anomalous scattering of sulfur. J Struct Biol. 2004;145:289–294. doi: 10.1016/j.jsb.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 34.Mandal K, Pentelute BL, Tereshko V, Kossiakoff AA, Kent SB. X-ray structure of native scorpion toxin BmBKTx1 by racemic protein crystallography using direct methods. J Am Chem Soc. 2009;131:1362–1363. doi: 10.1021/ja8077973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucl Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anstrom DM, Colip L, Moshofsky B, Hatcher E, Remington SJ. Systematic replacement of lysine with glutamine and alanine in Escherichia coli malate synthase G: effect on crystallization. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:1069–1074. doi: 10.1107/S1744309105036559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Czepas J, Devedjiev Y, Krowarsch D, Derewenda U, Otlewski J, Derewenda ZS. The impact of Lys-->Arg surface mutations on the crystallization of the globular domain of RhoGDI. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:275–280. doi: 10.1107/S0907444903026271. [DOI] [PubMed] [Google Scholar]
- 38.Avbelj F, Fele L. Role of main-chain electrostatics, hydrophobic effect and side-chain conformational entropy in determining the secondary structure of proteins. J Mol Biol. 1998;279:665–684. doi: 10.1006/jmbi.1998.1792. [DOI] [PubMed] [Google Scholar]
- 39.Berezovsky IN, Chen WW, Choi PJ, Shakhnovich EI. Entropic stabilization of proteins and its proteomic consequences. PLoS Comput Biol. 2005;1:e47. doi: 10.1371/journal.pcbi.0010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu X, Kuhlman B. Protein design simulations suggest that side-chain conformational entropy is not a strong determinant of amino acid environmental preferences. Proteins: Struct Funct Bioinformatics. 2006;62:739–748. doi: 10.1002/prot.20786. [DOI] [PubMed] [Google Scholar]
- 41.Velikov KP, Christova CG, Dullens RP, Van Blaaderen A. Layer-by-layer growth of binary colloidal crystals. Science. 2002;296:106–109. doi: 10.1126/science.1067141. [DOI] [PubMed] [Google Scholar]
- 42.Desiraju G, Steiner T. The weak hydrogen bond in structural chemistry and biology. Oxford: Oxford University Press.; 1999. [Google Scholar]
- 43.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 44.Tsodikov OV, Record J MT, Sergeev YV. A novel computer program for fast exact calculation of accessible and molecular surface areas and average surface curvature. J Comput Chem. 2002;23:600–609. doi: 10.1002/jcc.10061. [DOI] [PubMed] [Google Scholar]
- 45.Zheng H, Chruszcz M, Lasota P, Lebioda L, Minor W. Data mining of metal ion environments present in protein structures. J Inorg Biochem. 2008;102:1765–1776. doi: 10.1016/j.jinorgbio.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 47.Collaborative Computational Project. The CCP4 suite: programs for protein crystallography. Acta Crystallogr Sect D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 48.Cleophas TJ, Zwinderman AH, Cleophas TF, Cleophas EP. Statistics applied to clinical trials. New York City: Springer; 2009. [Google Scholar]