

Abstract
The role of specific cleavage of transcription repressor proteins by proteases and how this may be related to the emerging theme of dinucleotides as cellular signaling molecules is poorly characterized. The transcription repressor NmrA of Aspergillus nidulans discriminates between oxidized and reduced dinucleotides, however, dinucleotide binding has no effect on its interaction with the zinc finger in the transcription activator AreA. Protease activity in A. nidulans was assayed using NmrA as the substrate, and was absent in mycelium grown under nitrogen sufficient conditions but abundant in mycelium starved of nitrogen. One of the proteases was purified and identified as the protein Q5BAR4 encoded by the gene AN2366.2. Fluorescence confocal microscopy showed that the nuclear levels of NmrA were reduced approximately 38% when mycelium was grown on nitrate compared to ammonium and absent when starved of nitrogen. Proteolysis of NmrA occurred in an ordered manner by preferential digestion within a C-terminal surface exposed loop and subsequent digestion at other sites. NmrA digested at the C-terminal site was unable to bind to the AreA zinc finger. These data reveal a potential new layer of control of nitrogen metabolite repression by the ordered proteolytic cleavage of NmrA. NmrA digested at the C-terminal site retained the ability to bind NAD+ and showed a resistance to further digestion that was enhanced by the presence of NAD+. This is the first time that an effect of dinucleotide binding to NmrA has been demonstrated.
Keywords: proteolysis, nitrogen metabolite repression, confocal microscopy, microcalorimetry, NmrA
Introduction
Aspergillus nidulans can utilize a wide range of nitrogen sources, but when given ammonium or glutamine in a mixture with a nonpreferred nitrogen source the enzymes and permeases necessary for the use of the nonpreferred source are not produced – this phenomenon is called nitrogen metabolite repression (NMR).1 The molecular mechanism for the signal transduction pathway controlling NMR is complex, involving the action of the transcription repressor and activator proteins NmrA and AreA, the control of mRNA stability mediated through the 3′ untranslated region of the areA-mRNA and AreA-dependent remodeling of chromatin domains.2-6 The activity of AreA is modulated post-translationally by interaction with the repressor NmrA.7 The levels of NmrA are depleted in mycelium exposed to nitrogen starvation conditions and the production of nmrA-specific mRNA is controlled by the bZIP transcription factor MeaB and is inversely related to that of areA-specific mRNA.8 For example, areA-specific mRNA is elevated in mycelium growing on nonpreferred nitrogen sources such as nitrate and nmrA-specific mRNA is lowered under the same conditions. The production of AreA and NmrA is therefore tightly controlled at the transcriptional level and the activity of AreA is proposed to be determined by its levels relative to that of NmrA.8
We have previously reported the structures of the unliganded wild-type NmrA protein as well as complexes with NAD+, NADP+ and the zinc finger of AreA.9-11 Structural comparisons revealed that NmrA has similarity to the SDR (short-chain dehydrogenase-reductase) superfamily,12 with the closest relationship to UDP-galactose-4-epimerase (UDP-GE). NmrA defines the NmrA-like structural family whose members include the Human protein HSCARG.13 HSCARG is essential for epithelial cell viability, binds NADPH much more strongly than NADP+ and may link changes in the NADP+: NADPH ratio to modulation of the production of the important cell signaling molecule nitric oxide.14-16 Microcalorimetry experiments showed that NmrA binds NAD+ and NADP+ with similar affinity but has a greatly reduced affinity for NADH and NADPH. The structure of NmrA in a complex with NADP+ has shown how repositioning a His 37 sidechain allows the different conformations of NAD+ and NADP+ to be accommodated.10 NmrA and a zinc finger (Zf)-containing 213 amino acid C-terminal fragment of AreA interact with a 1:1 stoichiometry and an apparent KD of 0.26 μM10 but this interaction is not affected by the presence of dinucleotides. In contrast, the Gal80p repressor of Saccharomyces cerevisiae that is involved in galactose metabolism apparently has its properties modulated by the differential binding of NAD+ and NADP+.17,18 Structural studies have shown that NmrA binds directly to the zinc finger of AreA mainly through interactions with alpha helices 1, 6, and 11, and amino acid substitutions in alpha helix 6 (E193Q/D195N or Q202E/F204Y) reduce the affinity of this interaction.9,11 Comparison with a nuclear magnetic resonance solution structure of the AreA Zf in a complex with a GATA-containing double stranded oligonucleotide19,20 shows parts of helices α-6 and α-11 of NmrA are positioned close to the GATA motif in the DNA, mimicking the major groove of the DNA.9
We show here for the first time that A. nidulans has three protease activities that digest NmrA(Mr 38.8 kDa) in an ordered manner to generate an N-terminal protease resistant 32 kDa fragment and that in vitro the presence of oxidized dinucleotides substantially enhances the resistance of this N-terminal fragment to further digestion by these proteases. The enhancement of resistance to proteolysis is the first time a potentially biologically significant property has been ascribed to the binding of dinucleotides to NmrA. We also show by confocal microscopy that the concentration of wild-type NmrA in the nucleus is at its minimum when the concentrations of the proteases are at their maximum. Taken together the data we present here reveal a potential new layer of control of NMR mediated through the ordered proteolytic cleavage of NmrA.
Results
NmrA levels are modulated in vivo in response to growth on nitrate or nitrogen starvation
We wished to determine the relative nuclear concentrations of wild-type NmrA when mycelium is grown on a repressing nitrogen source (ammonium) versus inducing (nitrate) or nitrogen starvation conditions using confocal microscopy. To do this polyclonal rabbit antibodies were raised against NmrA and affinity purified by chromatography on a column that had purified NmrA covalently bound to NHS-Sepharose (see Methods). NmrA was not detectable in cell free extracts of A. nidulans in preliminary western blot experiments using these affinity purified antibodies implying that the in vivo concentration of NmrA is low. To determine the relative concentration of NmrA in the nucleus under different growth regimes, we used fluorescently labeled anti-IgG antibodies in conjunction with confocal microscopy. The wild type and a mutant strain deleted for the nmrA gene were grown on cover slips and mycelium probed for the presence and location of NmrA as described in Methods. We used the nmrA deletion strain as a control to define the background level of the fluorescence signal when probing for NmrA under the different growth regimes. Mycelium was grown for 16 h in minimal medium with ammonium or nitrate as the nitrogen source, and for the nitrogen starvation experiments was initially grown with ammonium before being transferred to nitrogen-free medium for 4 h. Typical images probed for the presence of NmrA (red), tubulin (green), and DNA (blue) are shown in Figure 1, control experiments in which the primary antibody was omitted gave no measurable signal. For each growth condition mean fluorescence values were derived from measurements taken from mycelium grown on three individual slides prepared on separate days. For each slide 20 measurements were taken at random within the cytoplasm and single measurements were made within 20 individual nuclei.
Figure 1.
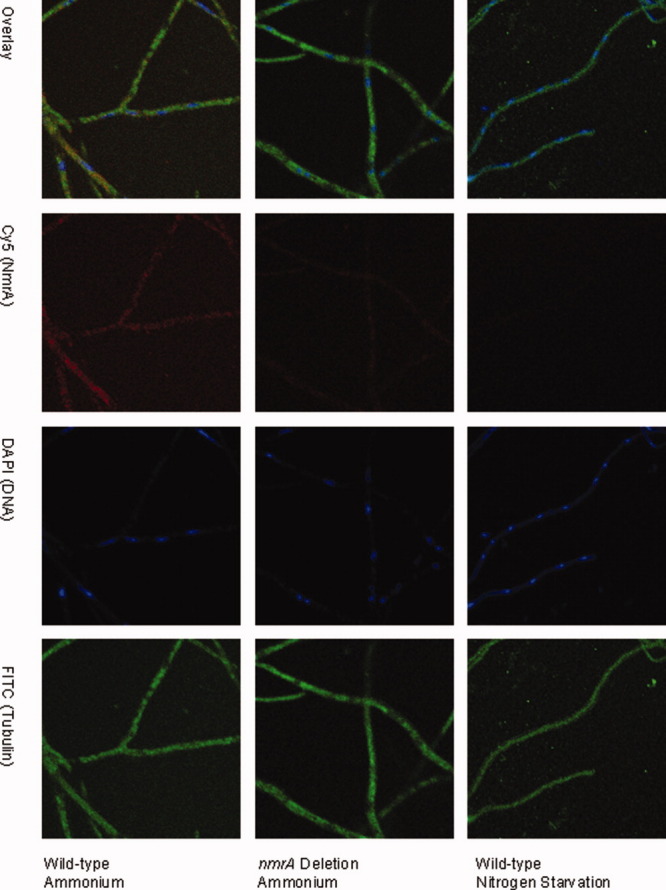
Confocal microscopy analysis of NmrA levels in wild type and nmrA deletion strains. wild type and nmrA deletion strains of A. nidulans were grown on cover slips on minimal medium containing 10 mM ammonium, 0.05% w/v glucose and 0.5% w/v quinic acid and for nitrogen starvation conditions transferred to liquid nitrogen free minimal medium. Mycelia were subsequently probed for the presence of NmrA by confocal fluorescence microscopy. Immunostaining was carried out as described in Methods with NmrA staining red, tubulin green (to delineate the boundaries of the hyphae), and DNA cyan (to define the boundaries of the nuclei). The top row shows all three images overlaid, the second row shows the fluorescence due to the presence of NmrA, the third row shows the fluorescence due to DNA staining in the nucleus and the bottom row shows the fluorescence due to staining tubulin.
T-test P-values of <0.0001 for comparisons between the total levels of NmrA in the wild type versus nmrA deletion strains were obtained when mycelium was grown on ammonium or nitrate, indicating a significant difference at the 95% confidence limit. When nitrogen starvation conditions were compared, the levels of fluorescence in the wild-type strain were greatly diminished giving a T-test P-value of 0.093 indicating no significant difference at the 95% confidence limit from the nmrA deletion strain. These latter data give confidence that the fluorescence signal measured in the wild-type strain reflects the true level of NmrA present under each of the growth conditions investigated. In the wild-type strain the T-test P-value for the comparison between the levels of nuclear NmrA in the ammonium versus nitrate grown mycelium was <0.0001 indicating a significant difference. Comparisons of the mean fluorescence measurements showed that there was a 38% reduction in the average nuclear level of NmrA in nitrate grown mycelium compared to ammonium grown (Fig. 2). These data also show that when wild-type mycelium is switched from growth on ammonium to nitrogen starvation conditions for 4 h, the fluorescence signal is reduced to the background level seen in the nmrA deletion strain. The levels of nmrA-specific mRNA are increased when mycelium is grown under repressing conditions and are decreased but not eliminated under nitrogen starvation conditions.8 These observations imply that the absence of NmrA in the nuclei of mycelium starved of nitrogen may in part be due to the action of proteases.
Figure 2.
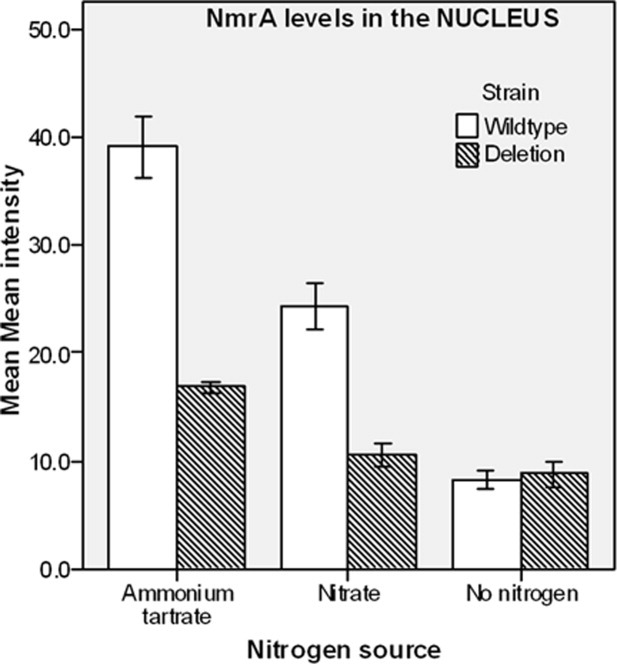
Statistical analysis of the nuclear levels of NmrA in different nitrogen regimes. wild type and nmrA deletion strains were grown under three conditions using medium containing 0.05% w/v glucose and 0.5% w/v quinic acid and (1) 10 mM ammonium tartrate or (2) 10 mM sodium nitrate as a nitrogen source, or (3) grown on ammonium tartrate and transferred to nitrogen free liquid media for 4 h for the starvation conditions. The bar chart shows the levels of NmrA in mycelium of wild type and nmrA deletion strains grown under the different nitrogen regimes. The y axis shows the mean mean fluorescence intensity and the x axis shows the nitrogen status of the growth medium. Each bar represents the average of 60 mean fluorescence intensity readings as detected by Leica software, and the error bars show the standard error of the mean.
NmrA is cleaved by three A. nidulans endoproteases
Cell-free extracts of A. nidulans mycelium starved of nitrogen were screened for protease activity using purified NmrA as the substrate. This screen demonstrated that NmrA was digested into discrete fragments with major products of Mr around 32kDa and, after prolonged digestion for around 2 h, 25kDa. In control experiments using cell-free extracts of A. nidulans grown under conditions that do not promote the production of proteolytic activity (see below), NmrA was resistant to prolonged incubation (approximately 20 h). Cell-free extracts containing protease activity were fractionated by chromatography as described in Methods, and screened for proteolytic activity in the presence and absence of the serine protease inhibitor benzamidine using purifed NmrA as the substrate. This procedure demonstrated the presence of three separable peaks of protease activity that produced NmrA fragments of slightly different size (Fig. 3). One protease activity, designated PNMA (for protease NMA), generated the digestion product FNM1A (for fragment NM1A) of Mr approximately 33 kDa. PNMA was benzamidine insensitive and washed through the column without binding. A second, benzamidine sensitive activity designated PNMB, (for protease NMB), generated the digestion product FNM1B (for fragment NM1B) of Mr approximately 32 kDa and was eluted from the column by around 0.5 M NaCl. A third, benzamidine insensitive, activity designated PNMC (for protease NMC) was eluted just before PNMB and generated an NmrA fragment indistinguishable in SDS-PAGE analysis from FNM1B. Additionally, all three proteases generated a third less intensely staining NmrA fragment of approximate Mr 25 kda in the standard proteolysis assay (see Methods). As column fractions containing PNMB were the most active, the generation of the 25 kDa fragment was investigated by subjecting NmrA to extended digestion (a time course of 4 h) with PNMB. Figure 4 shows the result of this digestion and demonstrates that after 4 h the 25 kDa proteolytic fragment is a major proteolytic product and was designated FNM2 (for fragment NM2).
Figure 3.
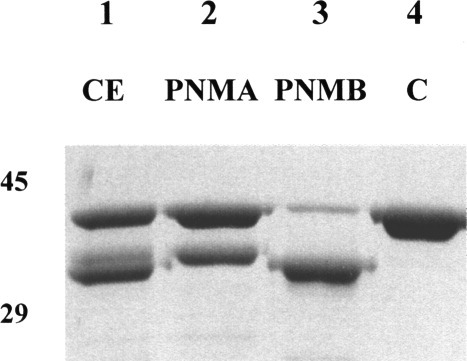
The specificity of PNMA and PNMB. 2 μg of NmrA was incubated at 25°C for 30 mins in 50 mM potassium phosphate pH 7.2, 1 mM DTT, with 1 μg of cell-free A. nidulans extract (lane 1), 10 μL PNMA from the highest activity fraction following DEAE HI Trap FF and superdex chromatography (lane 2) or 10 μL PNMB from the highest activity fraction following DEAE HI Trap FF and superdex chromatography (lane 3), and lane 4 contains a nondigested NmrA control. The digestion products were separated by SDS-PAGE using a 10% acrylamide separating gel and the positions of 45 and 29 kDa markers are shown.
Figure 4.
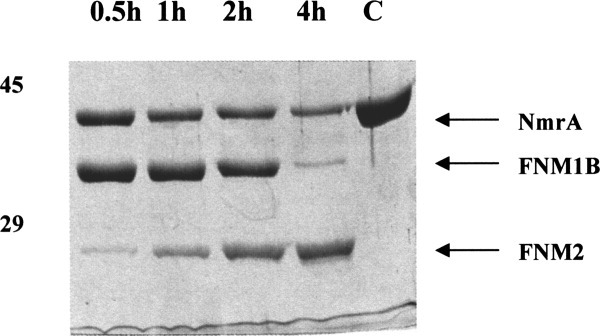
Time course digestion with PNMB. 2 μg of NmrA was incubated at 25°C in 50 mM potassium phosphate pH 7.2, 1 mM DTT, with approximately 5 units of PNMB purified by DEAE HI Trap FF and superdex chromatography. Samples were taken at intervals of 0.5 h (lane 1), 1 h (lane 2), 2 h (lane 3), and 4 h (lane 4), lane 5 is a nondigested NmrA control. The digestion products were separated by SDS-PAGE using a 10% acrylamide separating gel and the positions of 45 and 29 kDa markers are shown. The digestion products FNM1B and FNM2 are indicated by the arrows.
NmrA undergoes ordered proteolysis
The site of proteolysis in NmrA for the production of the fragments FNM1A and FNM1B was determined by a combination of Edman sequencing and Mr determination using LC-MS (see Methods). The protease PNMA produced an NmrA fragment of 33,404.2 Da (designated FNM1A), and PNMB produced a fragment of Mr 32,134.6 Da (designated FNM1B). As a control, the Mr of bovine carbonic anhydrase II was determined by this technique to be 29,023.7 Da. The measured value of the control Mr showed Dalton level accuracy implying the same level of accuracy with the NmrA peptide Mr determinations. All attempts to elute the fragment FNM2 (generated by digestion with PNMB) from gel slices were unsuccessful, consequently we were unable to determine the Mr of this proteolytic fragment.
To use the Mr information for mapping the cleavage sites within NmrA, the N-terminal amino acid sequence of each FNM1A and FNM1B and that of the stock NmrA substrate was determined (see Methods) and in each case the sequence AQQKKTIA was recovered. These results demonstrated that (a) the recombinant NmrA stock lacked an N-terminal methionine and (b) that each of the digested NmrA peptides recovered started at the same N-terminus and so must have different C-termini. Having determined that the stock of NmrA had no N-terminal methionine we determined its Mr by MALDI-TOF to be 38,663.8 Da which compares favorably with the predicted Mr of 38,662.6 Da and shows that the recombinant protein was not modified in any other way. However the Mr of a sample of the same stock of NmrA that had been subject to SDS-PAGE prior to LC-MS was found to be 38,737.5 Da. These measurements show that NmrA becomes covalently modified with an approximately 74 Da adduct (Table I) as a result of the SDS-PAGE or the subsequent gel extraction before LC-MS.
Tabel I.
The Measured and Predicted Mrs of NmrA and its Proteolytic Products
| Sample | Measured Mr (Da) after SDS-PAGE and gel extraction | Measured Mr (Da) prior to SDS-PAGE and gel extraction | Predicted Mr (Da) from the amino acid sequence | Difference (Da) from measured Mr of FNM1A | Difference (Da) from measured Mr of FNM1B |
|---|---|---|---|---|---|
| FNM1A | 33,402.2 | NA | — | — | — |
| FNM1B | 32,134.6 | 32,059.7 | 32059.8 | — | — |
| NmrA 2–352 | 38,737.5 | 38,663.8 | 38,662.6 | — | — |
| NmrA 2–291 | NA | NA | 32,059.7 | — | −74.9 Da |
| NmrA 2–292 | NA | NA | 32,116.8 | — | −17.8 Da |
| NmrA 2–293 | NA | NA | 32,230.0 | — | +95.4 Da |
| NmrA 2–306 | NA | NA | 33,330.2 | −74.0 Da | — |
| NmrA 2–307 | NA | NA | 33,458.4 | +54.2 Da | — |
The measured Mrs of NmrA lacking the N-terminal methionine (NmrA 2–352) and the FNM1B proteolytic fragment determined before and after SDS-PAGE and gel elution are shown with the predicted Mr derived from the amino acid sequence. A difference of +74.9 Da is evident in each case. Also shown are the measured Mrs of the NmrA proteolytic products FNM1A and FNM1B after gel extraction along with the predicted Mrs of C-terminally deleted NmrA possible candidate sequences.
Knowing the N-terminus of the proteolytic products FNM1A and FNM1B and their accurate Mrs allowed the deduction that the cleavage site of each protease lies within the C-terminal surface exposed nonordered region of NmrA (residues 281–309). The LC-MS technique provides accuracy at the Dalton level, however, there are no predicted cleavage products from NmrA that match the measured Mr values with this level of accuracy (Table I). Given that NmrA is covalently modified as a result of SDS-PAGE and subsequent gel extraction for LC-MS then, providing digestion by PNMA or PNMB does not remove the site of modification, it is reasonable to infer that the proteolytic fragments FNM1A and FNM1B will be similarly modified. The peptides 2–291 and 2–306 differ from the measured Mrs of FNM1B and FNM1A by −74.0 and −74.9 Da, respectively, implying that they, like nondigested NmrA, have been modified with an approximately 74 Da adduct as a result of SDS-PAGE and gel elution. In the case of the FNM1B fragment this hypothesis was tested directly by repeating the Mr determination with products of proteolysis that had not been subject to SDS-PAGE. In this case, the major digestion product had a measured Mr of 32059.65 kDa – a value that differs by 0.1 Da from the predicted Mr of an NmrA fragment consisting of amino acids 2–291. Therefore the most likely cleavage site to generate peptide FNM1A is between amino acids M306 and Q307, and to generate peptide FNM1B is between K291 and G292.
The FNM2 fragment of approximately 25 kDa formed by the digestion of NmrA with a crude cell-free extract of A. nidulans mycelium (therefore containing PNMA, B and C activities) was gel purified and LC-MS experiments produced an estimated Mr of 25,252.93 Da. Use of the Edman reaction determined the N-terminal amino acid sequence as AQQKKTIA implying it was generated by cleavage within the surface exposed α helix 8. The measured Mr of this NmrA fragment differs by 3.7 Da from the predicted Mr generated by digestion between residues A231 and F232. However, it is possible that this fragment is modified by the 74Da adduct described above, and if 74 Da is subtracted from the measured Mr the difference from the Mr of a fragment generated by digestion between residues A230 and A231 is 1 Da. It is likely therefore that this second proteolytic fragment is generated by digestion between A230 and A231. However this site of digestion is incompatible with the specificity of trypsin-like proteases such as PNMB (see below) and is therefore likely to be due to digestion by PNMA or C. Inspection of the amino acid sequence in this area shows that there is a potential PNMB cleavage site (R234/A235) 4 amino acids C-terminal to A230. Thus in a mixture of the PNMA, B and C, the FNM2 product of PNMB is a substrate for PNMA and/or C.
The in vitro generated data above show that NmrA is digested in an ordered fashion, with rapid cleavage occurring within the C-terminal unordered surface region generating fragments FNM1A and FNM1B. As FNM1A is a substrate for the protease PNMB, in crude cell-free extract FNM1A is converted to FNM1B. Similarly the FNM2 fragment that is generated at a slower rate by PNMB digestion within α helix 8 is a substrate for PNMA and C. The further digestion of FNM2 by PNMA, B and C does not generate stable intermediates implying that this fragment is disordered and that digestion is occurring at multiple sites simultaneously. Therefore the proteolytic fragment FNM1A/B appears to be inherently resistant to proteolysis and requires either an increase in digestion time or an increase in the concentration of protease of 10-fold or more to achieve quantitative conversion to lower Mr fragments.
Identification of PNMB
To try and identify the proteases that produce the fragments FNM1A and B proteolytically active fractions following Superdex chromatography were analyzed by SDS-PAGE following TCA precipitation (see Methods). For the fractions generating FNM1A, multiple weakly staining bands were observed. However in the fractions producing FNM1B a single strongly staining protein was observed. The identity of this protein was determined by a combination of peptide mass spectrometry and LCMS-MS. The peptides generated by tryptic digest were analyzed by two independent means, Mascot identity database searching and BLAST homology searching. Following de novo sequencing the Mascot analysis showed that several digest peptides matched significantly with the hypothetical protein Q5BAR4_EMENI from A. nidulans (Mascot score = 252; threshold score = 25; probability of a random match = 1 in 1021). An independent analysis method using the program DeNovoX was also used to attempt to sequence the peptides de novo (that is without prior reference to any databases) from their MS/MS spectra. The results of this analysis were manually inspected and the best 25–30 peptide MS/MS sequences were extracted to a list for BLAST homology searching in both directions. The results of the BLAST search showed that the only significant matching protein from searching the entire Swiss_Prot database identified the same hypothetical sequence as with Mascot. This match also provided extra sequence coverage while giving added confidence in the protein identification by an independent means of analysis. Q5BAR4_EMENI is a hypothetical protein that belongs to the Tryp_SPC (trypsin-like serine proteases) superfamily and is encoded by the gene AN2366.2. The predicted sites of cleavage by PNMB (see above), and the sensitivity to benzamidine are compatible with the specificity of trypsin-like proteases. This analysis was repeated with a further two separate purifications and generated the same results suggesting that the protease identified may correspond to PNMB and be responsible for the proteolysis of NmrA in vivo. BLAST searches of nonredundant protein databases revealed that Q5BAR4 is most closely similar to serine proteases in Diptera (45% identity and 67% similarity to Anopheles gambiae) but no matches to fungal homologues were identified in the top 100 hits. A more selective BLAST search of fungal genomes in the NCBI database revealed potential homologues in Phaeosporia, Sclerotinia, Gibberella, Nectria, Botryotinia, Verticillium, and Pyrenophora species. It is interesting to note that potential cleavage sites for PNMB are conserved in NmrA homologues in filamentous fungi including Aspergillus, Penicillium, Talaromyces, Gibberella, and Neurospora.
Production of the PNMA, B and C protease activities is associated with nitrogen limitation
The hypersensitivity of the two cleavage sites located within the unordered surface loop of NmrA provides a mechanism whereby its biological activity can be rapidly modulated in response to small increases in the concentration of the PNMA, B or C protease activities. To determine if the production of these protease activities is influenced by the nature of the nitrogen source, mycelium was grown for 18 h in liquid minimal medium with glucose as the carbon source and ammonium, nitrate, hypoxanthine, or proline at 10 mM as nitrogen source, and cell-free extracts assayed for protease activity. Protease activity was seen in all cases indicating that production of the proteases was not dependent on growth in one particular nitrogen source (data not shown). To look at the effect of nitrogen limitation, mycelium was grown in liquid minimal medium for 19 h with ammonium or nitrate at concentrations of 2.5, 5.0, or 40 mM, and cell-free extracts assayed for protease activity. Protease activity in cell-free extracts was associated with growth in the lower concentrations of nitrogen and was absent (even after prolonged overnight incubation) in cell-free extracts of mycelium grown at the highest nitrogen concentrations. Quinic acid is an abundant biomass in the natural environment of A. nidulans that can be used as a (noncatabolite repressing) carbon source.22 When quinic acid was used as carbon source in conjunction with 10 mm ammonium or nitrate, proteases were only detected in mycelium transferred to nitrogen free medium for 4 h. The reason for the apparent difference in protease production between glucose and quinic acid grown mycelium is likely to be due to a slower growth rate with quinic acid leading to a slower depletion of the nitrogen source during the overnight growth phase. This observation implies that the 38% diminution in nuclear NmrA levels seen in nitrate versus ammonium grown mycelium in the confocal microscopy experiments is not due to proteolysis. It is more likely to be a consequence of changes in nmrA-specific mRNA levels as these have been shown to vary with nitrogen source (ammonium versus alanine).8 Examples of the proteolytic activity associated with cell-free extracts prepared from mycelium grown on glucose, 5 and 40 mM ammonium and nitrate are, respectively, shown in Figure 5. This pattern of production of PNMA, B and C provides an in vivo mechanism whereby the lowering of the nitrogen concentration to a critical point, as a consequence of growth, can cause the in vivo pool of NmrA to be inactivated. As the critical nitrogen concentration is reached, a low concentration of protease is produced causing an increasing fraction of the NmrA pool to be rapidly digested by preferential cleavage at the two hypersensitive sites in the C-terminal region. As the concentration of nitrogen falls further below the critical point, higher protease concentrations are produced and the NmrA pool will be degraded by cleavage at other less-sensitive sites. The result of this diminution in NmrA concentration would be to free an increasing pool of transcriptionally active AreA and thereby potentiate the use of new nitrogen sources as they become available.
Figure 5.
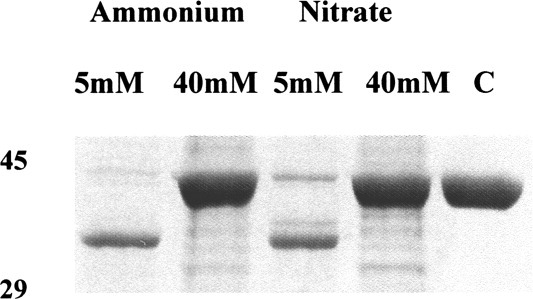
The effect of nitrogen limitation on the production of PNMA and PNMB. Wild-type A. nidulans mycelia were grown with glucose (0.4% w/v) as carbon source and ammonium tartrate or sodium nitrate as nitrogen sources at concentrations of 5 and 40 mM. Mycelia were harvested after 19 h growth and 1 μg of cell-free extract from each harvest was incubated with 2 μg NmrA as described in the Legend to Figure 3. Lane 1 = glucose plus 5 mM ammonium, Lane 2 = glucose plus 40 mM ammonium, Lane 3 = glucose plus 5 mM nitrate, lane 4 = glucose plus 40 mM nitrate, Lane 5 = nondigested NmrA control. The digestion products were separated by SDS-PAGE using a 10% acrylamide separating gel and the positions of 45 and 29 kDa markers are shown.
The resistance of FNM1 to further proteolysis is enhanced by binding oxidized dinucleotides
NmrA was digested singly with Superdex-purified PNMA and B and DEAE FF-purified PNMC in the presence and absence of 0.5 mM NAD+ and the appearance and further digestion of the FNM1 proteolytic fragments were monitored over a 6 h digestion (data not shown). In each case, the presence of NAD+ markedly protected the FNM1 fragments from the extensive digestion seen in the absence of dinucleotides or the presence of NADH. This effect was also seen when NADP+ was substituted for NAD+ but was more variable and on average less pronounced (data not shown). No protection was afforded by the addition of NAD(P)H. This analysis was repeated with three independent purifications of PNMB and typical results are shown in Figure 6. To test the hypothesis that it was the binding of oxidized dinucleotides to NmrA that was responsible for this affect the experiments were repeated with mutant NmrA that has a reduced ability to bind NAD+. The NmrA protein encoded by plasmid pMUT170 has the changes N12G/A18G (designated NmrAN12G/A18G) and a 13-fold increase in the KD for the binding of NAD+.11 NmrAN12G/A18G was purified in bulk and the digestion experiments repeated using Superdex-purified PNMA and B – typical results are shown in Figure 6. Inspection of Figure 6 shows that the disappearance of FNM1B from NmrAN12G/A18G digested by PNMB was not substantially modulated by the presence of NAD+. This observation supports the hypothesis that the protective effect of NAD+ is through binding to NmrA and FNM1B rather than by binding to and inhibiting the protease.
Figure 6.
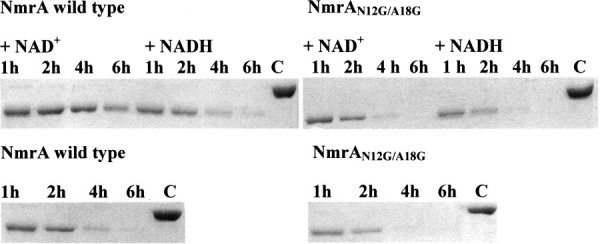
Digestion of wild type and mutant NmrA by PNMB in the presence and absence of NAD+ and NADH. 2 μg of NmrA or the mutant form NmrAN12G/A18G was digested with approximately 10 units of Superdex-purified PNMB in the presence and absence of 0.5 mM NAD+ or NADH for 1, 2, 4, or 6 h in a final volume of 10 μL in 50 mM potassium phosphate pH 7.2, 1 mM DTT. The lane labeled ‘C’ is nondigested NmrA. The digestion products were separated by SDS-PAGE using a 10% acrylamide separating gel.
Proteolytic digestion within its C-terminal unordered loop abrogates the ability of NmrA to bind the AreA Zf
The data in Figure 6 imply that the FNM1B proteolytic fragment retains sufficient structural integrity to bind NAD+, and we wished to know if FNM1B was able to bind to the AreA Zf. We have previously shown by ITC that NmrA binds endothermically to the C-terminal 213 amino acids of AreA (AreA664–876) with a 1:1 stoichiometry and a KD of approximately 0.26 μM.10 Crystallographic studies showed that NmrA specifically interacts with the AreA Zf contained within AreA amino acids 663–727, however these experiments gave no indication of the strength of binding. Therefore preliminary ITC experiments were carried out to quantify the interaction of NmrA with two overlapping amino acid sequences that encompassed the AreA Zf. The interaction of NmrA with AreA amino acids 637–727 (AreA637–727) and 663–727 (AreA663–727) was measured by ITC and a typical result is shown in Figure 7(A). Table II summarizes the thermodynamic parameters associated with the interactions. Inspection of Table II shows that absence of the C-terminal 149 amino acids (contained within AreA664–876) of AreA has a modest increase of around 4-fold in the KD for the interaction of NmrA with AreA663–727.
Figure 7.
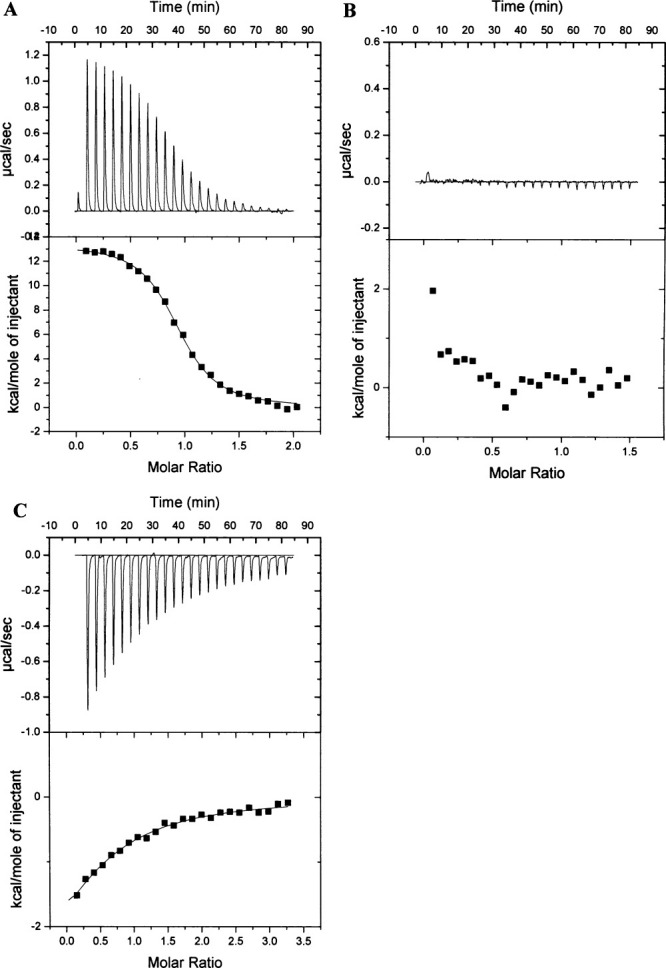
Typical ITC data for the endothermic binding of NmrA to the AreA Zinc Finger and the exothermic binding of NAD+ to FNM1B+C-term at 25 (C. (A): (Upper panel): Heat uptake upon injection (1 x 2 μL and 24 x 10 μL) of NmrA (300 μM) into the calorimetric cell (1.4 mL) containing AreA663–727 (28 μM). Heat pulses in the absence of AreA663–727 (not shown) were negligible. (Lower panel): Integrated heat pulses, normalized per mole of injectant, giving a differential binding curve that is adequately described by a single-site binding model; (B): (Upper panel): Heat uptake upon injection (1 x 2 μL and 24 x 10 μL) of FNM1B+C-term (200 μM) into the calorimetric cell (1.4 mL) containing AreA663–727 (25 μM). Heat pulses in the absence of AreA663–727 (not shown) were negligible. (Lower panel): Integrated heat pulses, normalized per mole of injectant; (C): (Upper panel): Heat uptake upon injection (1 x 2 μL and 24 x 10 μL) of NAD+ (2 mM) into the calorimetric cell (1.4 mL) containing FNM1B+C-term (112 μM). Heat pulses in the absence of FNM1B+C-term (not shown) were negligible. (Lower panel): Integrated heat pulses, normalized per mole of injectant, giving a differential binding curve that is adequately described by a single-site binding model.
Tabel II.
Thermodynamic Parameters for Binding of NmrA to the Area Zinc Finger and the Binding of NAD+ to FNM1B+C-term Determined by ITC at 250°C
| Ligand in syringe | Ligand in cell | n | KD(app) μM | Average KD(app) μM | ΔHobs kcal mol−1 | ΔS0 cal K−1 mol−1 | c |
|---|---|---|---|---|---|---|---|
| 0.30 mM NmrA | 28μM AreA637–727 | 0.9 (0.1) | 1.17 | 1.28 ± 0.26 | 13.6 (0.4) | 72.7 | 22 |
| 0.30 mM NmrA | 28μM AreA637–727 | 1.0 (0.1) | 1.10 | 13.5 (0.2) | 72.6 | 26 | |
| 0.30 mM NmrA | 28μM AreA637–727 | 1.1 (0.1) | 1.58 | 13.6 (0.2) | 72.2 | 19 | |
| 0.27 mM NmrA | 30 μM AreA663–727 | 1.0 (0.1) | 0.93 | 0.89 ± 0.08 | 13.5 (0.2) | 73.07 | 31 |
| 0.27 mM NmrA | 30μM AreA663–727 | 1.0 (0.1) | 0.79 | 14.0 (0.1) | 75.10 | 37 | |
| 0.27 mM NmrA | 30μM AreA663–727 | 1.00 (0.1) | 0.94 | 14.6 (0.2) | 76.50 | 32 | |
| 2 mM NAD+ | 112 μM FNM1B+C-term | 0.6 (0.2) | NA | NA | −3.6 (1.5) | 6.03 | 0.62 |
| 2 mM NAD+ | 112 μM FNM1B+C-term | 0.6 (0.1) | NA | −4.4 (1.11) | 3.65 | 0.59 |
The binding of AreA637–727 and AreA663–727 to NmrA was measured in 50 mM potassium phosphate pH 7.2, 5 mM DTT buffer. Shown are the values for n, the stoichiometry of binding; KD(app), the apparent equilibrium dissociation constant; ΔHobs, the observed enthalpy; and ΔS0, the standard entropy change for single-site binding, for sets of three individual assays for each protein construct. The c values for the NmrA:AreA Zf interaction fall within the range of 1–1000 that allows the isotherms to be accurately deconvoluted with reasonable confidence to derive K values.22 The c values for the FNM1B+C-term: NAD+ interaction are below 1 therefore the isotherms cannot be accurately deconvoluted to calculate the KD for the interaction. The average KD(app) values for the NmrA:AreA Zf interaction are shown ± S.D. NA = not applicable.
NmrA was digested with PNMB in the presence of 0.5 mM NAD+, generating FNM1B and a C-terminal fragment of approximately 6.6 kDa. The FNM1B fragment was purified as described in Materials and Methods and when analyzed by SDS-PAGE using a 15% separating gel, was found to copurify with the 6.6 kDa Mr fragment. Mass spectrometry determined the accurate Mr of this protein to be 6.625 kDa, a value identical (within experimental error) to that of the C-terminal fragment produced by the digestion of NmrA by PNMB. Densitometry measurements using PROGENESIS software (Nonlinear Dynamics, UK) showed that the FNM1B and C-terminal fragments of NmrA purified with a 1:1 stoichiometry. These observations indicate that when PNMB cleaves NmrA within its C-terminal surface exposed loop, the FNM1B and C-terminal fragments produced remain associated and that this association is not disrupted by MONO Q FPLC. In the following description the designation FNM1B+C-term refers to NmrA that has been digested with PNMB and the proteolytic fragments purified by MONO Q FPLC. In contrast to nondigested NmrA [Fig. 7(A)], in ITC experiments where FNM1B+C-term was titrated onto AreA663–727 no significant heat exchanges over baseline controls were observed [Fig. 7(B)], implying that FNM1B+C-term and AreA663–727 did not interact. This observation can be rationalized by considering the crystal structure of NmrA in a complex with the AreA663–727. The AreA Zf interacts mainly through α-helices 1, 6, and 11 of NmrA in a combination of polar and hydrophobic contacts.9 Of the four hydrogen bonds between the AreA Zf and NmrA, three involve amino acids located within the C-terminal 27 amino acids of NmrA. Cleavage within the unordered surface loop will break the covalent link to the C-terminal peptide and is therefore likely to cause changes to its orientation relative to the FNM1B fragment. Such changes could disrupt the potential polar contacts of FNM1B+C-term with AreA663–727 and account for their observed inability to interact.
To test the hypothesis that NmrA and FNM1B+C-term have some structural differences, we determined their far-UV CD spectra (Fig. 8). Inspection of Figure 8 shows that there are small differences in the far-UV CD spectra in the region 202–222 nm, consistent with a subtle change in structure of FNM1B+C-term relative to NmrA. In ITC experiments FNM1B+C-term bound NAD+ [Table II and Fig. 7(C)], however, the calculated stoichiometry of the interaction (approximately 0.6) was lower than the value of 1 known from structural studies12 and the KD could not be reliably calculated as the ‘c’ value fell below 1.21 The reason for the aberrant stoichiometry and low c value is possibly due to the subtle structural changes revealed by the far-UV CD spectrum of FNM1B+C-term. These changes may cause a reduction in the half life of the ability of FNM1B+C-term to bind NAD+. The necessary time taken (72 h) to generate, purify and use FNM1B+C-term in ITC experiments makes it likely that the protein solution used was a mixture of active and inactive molecules. However, the ITC data do confirm directly that purified FNM1B+C-term retains the ability to bind NAD+.
Figure 8.
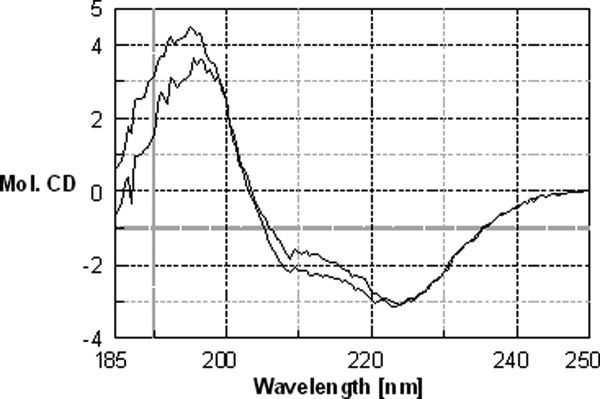
Far-UV circular dichroism analysis of NmrA and FNM1B+C-term. The secondary structure of NmrA and FNM1B+C-term at a concentration of 0.5 mg mL−1 in 50 mM KPO4 pH 7.2, 1 mM DTT was probed by far-UV CD spectroscopy. CD spectra for each protein were recorded from 10 accumulative scans of protein in buffer and buffer alone at 200°C using a Jasco J-810 spectropolarimeter. The buffer baseline was subtracted from the experimental value, the data adjusted for protein concentration and the results shown as molecular CD units. The trace with the greatest signal between 202 and 222 nm is from nondigested NmrA.
Discussion
Proteolytic processing of fungal transcription factors has been reported previously. For example, the A. nidulans zinc finger-containing transcription factor PacC undergoes two step proteolytic processing in response to alkaline ambient pH.23 However, this differs from the case of NmrA where proteolysis negates the activity of a transcription repressor, in that PacC is processed to produce the biologically active form of a transcription activator.23 Proteolytic processing of a transcription repressor has been reported in Human, and the possibility of regulating transcription factors by target-specific proteolysis has been suggested as a general mechanism.24 For example, activation of the nuclear factor κ-B (NF-κ-B) is modulated by its interaction with the repressor protein IκB-α, and the activity of IκB-α is itself controlled by ubiquitination and subsequent proteasome-mediated degradation.24-26
Depletion of NmrA under conditions of nitrogen starvation has been reported previously.8 In these experiments western blot analysis was used to monitor the levels of NmrA over a 4 h period of nitrogen starvation and demonstrated almost complete loss of NmrA. These observations are entirely consistent with the diminution in the levels of wild-type NmrA seen in the fluorescence microscopy experiments reported here. However the diminution in the NmrA levels over a time course of nitrogen starvation conditions seen in the western blot experiments was not associated with the formation of proteolytic products corresponding to FNM1A, FNM1B or FNM2. This apparent anomaly can be explained by considering the genotype of the A. nidulans strain used in the western blot experiment. The strain used contained a recombinant nmrA gene situated at the normal locus and under the transcriptional control of the native nmrA promoter and the 3/ untranslated region of the trpC gene. The recombinant nmrA gene encoded an NmrA protein that had a Flag tag fused to its C-terminus and the NmrA fusion protein was identified in western blots by the use of anti-Flag tag antibodies. Due to the ordered proteolysis of NmrA, the first site to be digested will be in the C-terminal disordered surface loop and this will have the effect of severing the covalent link between the N-terminal NmrA fragments and the Flag epitope. Consequently proteolytic intermediates FNM1A, FNM1B, and FNM2 of NmrA would be rendered invisible in a western blot analysis that used an anti-Flag tag antibody.
The finding that the sequence of the hypothetical protein Q5BAR4 is not highly conserved and only sporadically identified in BLAST searches of closely related species was unexpected. It may be that in this respect A. nidulans is atypical and that the ordered proteolysis of NmrA is an unusual aspect of the control of NMR confined to this species. However protein BLAST searches reveal that homologues, albeit with substantial sequence divergence, appear to be present in Gibberella zeae and A. terreus. It is possible that in other species trypsin-like enzymes that are not revealed in BLAST searches with Q5BAR4 fulfill the same function. In this respect, we note that Bovine trypsin has 36.5% identity with Q5BAR4 but was not identified in the top 100 proteins hits in such a BLAST search. Nonetheless Bovine trypsin digests NmrA in a manner indistinguishable in SDS-PAGE analysis from PNMB (data not shown). In vivo confirmation that PNMB corresponds to Q5BAR4 requires the generation and analysis of an A. nidulans strain deleted for the gene AN2366.2. Further in vitro proof could be provided by analyzing the proteolytic properties of purified of recombinant Q5BAR4 encoded by AN2366.2.
We have speculated previously that NmrA may have an additional role as a redox sensor.10 If the function of a redox sensing ability is to form a link between changes to cellular metabolism and the control of transcription, one might expect that NmrA would be more likely to have its biological effects modulated by binding reduced dinucleotides.27 The rationale for this argument is that it follows from the high (mM range) NAD(H) concentration and the high NAD+/NADH ratio (500–700)28,29 that conversion of NAD+ to NADH will have the greatest relative effect on the NADH level.27 In this respect we note that the Human protein HSCARG, which belongs to the NmrA-like structural family, binds NADPH with a 360-fold lower KD than that for NADP+.15 Given these observations, a redox sensing role for NmrA is less likely.
Interestingly, the resistance of the N-terminal 32 kDa proteolytic fragment of NmrA (FNM1A/B) to further digestion is enhanced in vitro by binding oxidized dinucleotides. However, the biological significance of this observation is not clear as the initial proteolytic cleavage event does not appear to be affected by dinucleotide binding and is sufficient to abrogate the ability of NmrA to bind to the AreA Zf. It could be argued therefore that the dinucleotide binding properties of NmrA have no in vivo significance and are simply an evolutionary ‘echo’ of its membership of the superfamily containing the short-chain dehydrogenase-reductases (SDRs).12 However the dinucleotide binding motif GlyXXGlyXXGly characteristic of bi-domain SDRs is replaced by the sequence AsnXXGlyXXAla in NmrA. It appears therefore that although there have been substantial evolutionary changes to the amino acid sequence, there has been selective pressure for NmrA to retain the ability to bind oxidized dinucleotides. Most of the dinucleotide pool is believed to be protein bound29 in vivo, and this latter observation has led to the caveat that ‘estimates of the actual substrate availability, for example, for NAD(P)+-mediated signaling processes are to be viewed with caution, at least, when based upon pyridine nucleotide concentrations in cellular extracts’.29 As NmrA appears to be produced in vivo under most physiological conditions, there is no obvious reason why it should not form part of the cellular pool of protein that binds the majority of the dinucleotide pool in vivo. Therefore, it is not unreasonable to infer that a significant proportion of the in vivo NmrA pool is likely to have bound NAD+ under most physiological conditions. In a highly speculative scenario it is possible that the FNM1B+C-term protein of NmrA may have an in vivo biological role distinct from the repressing role of intact NmrA. If this were the case then in the presence of PNMA, B and C dinucleotide binding may increase the half life of any biological function associated with FNM1B+C-term.
In conclusion, we propose that the dynamic interplay between the production of NmrA and its subsequent ordered proteolysis facilitates a rapid and finely tuned response to changes in the concentration and nature of the nitrogen source supporting growth.
Materials and Methods
Materials
Chemicals and solvents were purchased from local suppliers and were of AnalaR or greater purity. All other molecular biology and biochemistry reagents (which were used in accordance with the manufacturers recommendations) and chromatography columns were purchased from Sigma, Invitrogen or GE Healthcare. Secondary antibodies were from Abcam UK and mouse antibody to chick α tubulin was from Fitzgerald Industries International, USA.
Fungal strains
Two fungal strains were used in this research. The wild type strain R21 (yA2, pabaA1), and the nmrA deletion strain MH8935 (yA1, nmrA::Bleo, amdS:: lacZ, pyroA4).
Molecular biology and biochemistry
All other routine molecular biology protocols followed individual manufacturer's recommendations or were as previously described.10,11
Production and affinity purification of rabbit anti-NmrA antibodies
Rabbit antibodies were raised against purified NmrA according to standard protocols.30 NmrA (350 mg) was covalently bound to NHS-activated sepharose (25 ml) as previously described31 and used as an affinity column to purify NmrA-specific antibodies from rabbit serum. Filtered antibody serum was loaded onto the NmrA:NHS-sepharose column and, following a wash with 300 mL of 0.2 M sodium acetate to remove nonspecifically bound proteins, antibodies were eluted from the column in two steps. First the column was eluted with 200 mL of 3.6 M MgCl2, 0.2 M sodium acetate pH 6.5 collecting 10 mL fractions and second, following a wash with 250 mL of 10 mM potassium phosphate buffer pH 6.8 the column was further eluted with 500 mL of 100 mM glycine, pH 2.5, and collecting 10 mL fractions. Antibody-containing fractions were pooled and dialyzed twice against 5 L of PBS pH 7.5 and stored in 100 μL aliquots at −80oC. ELISA measurements showed that a 1 in 6,400 dilution of the antibody gave a signal that was at least 100-fold higher than background levels.
Fluorescence confocal microscopy and statistical analysis of fluorescence data
A. nidulans wild-type strain R21 and nmrA deletion strain MH8935 were used throughout the microscopy work. Growth of mycelium on cover slips for fluorescence confocal microscopy followed the method of Osmani et al.32 except that Novozyme Vinoflow FCE, prepared as described by Szewczyk et al.,33 was used to digest the cell walls to allow entry of antibodies. In these experiments the carbon source was 0.5% w/v quinic acid, 0.05% w/w glucose. Quinic acid was used because it is an abundant biomass utilized by A. nidulans when growing in its natural environment – the supplementation with low levels of glucose was to aid the initial germination of the conidiospores. Nonspecific sites were blocked for 30 min by incubation buffer (PBS containing 5% w/v BSA). NmrA was visualized by the sequential use of rabbit anti-NmrA antibodies and a secondary goat anti-rabbit IgG antibody conjugated to Cy5. A monoclonal mouse antibody to chick tubulin that cross reacts with A. nidulans tubulin was used in conjunction with a goat polyclonal antibody to mouse IgG conjugated to FITC to visualize the boundaries of the hyphae. An overnight incubation with primary antibodies (1:1000) and RNase (1:200 of a 10 mg mL−1 solution) was carried out at 4°C in incubation buffer. After washing three times with PBS, secondary antibodies (1:1000) and DAPI (1:1000 of a 10 mg mL−1 solution) were applied in incubation buffer for 2 h at room temperature. Cover slips were mounted in 50% glycerol. To measure the relative concentration of NmrA present in the mycelium under different nitrogen growth regimes, three representative areas of mycelial growth were selected on each of three separate cover slips for each growth condition prepared on different days. The fluorescence intensities within 20 randomly placed, fixed sized areas, spread over the three representative areas of mycelial growth on each cover slip were recorded for each location giving a final sample size of 60 measurements for each growth condition. All statistical analysis was carried out using SPSS. Data are presented as mean ± the standard error of the mean of n determinations. The fluorescence measurements were initially analyzed by the ANOVA test which indicated statistically significant differences at the P < 0.05 level between some of the data groups, and subsequently individual T-tests were performed to identify the source of these differences. When measuring the cytoplasmic values, the contribution from the NmrA channel was omitted from the screen representation and all measurements were taken within the boundaries defined by the fluorescence from the tubulin. Similarly the intensity resulting from NmrA within the nucleus was measured within the area defined by the DAPI fluorescence. All measurements were taken with a Leica TCS SP2 confocal microscope and processed using Leica LCS Lite software (Leica Microsystems, Germany).
Protein purification
NmrA and C-terminal fragments of AreA including the Zf were purified as previously described.9 The proteases PNMA and PNMB were separated and partially purified according to the following protocol: 25 g of frozen mycelium grown at 37oC in minimal medium containing 10 mM ammonium tartrate and subsequently transferred to nitrogen free medium for 4 h, was ground to a fine powder in liquid nitrogen and soluble proteins extracted by stirring on ice in 0.1 M Tris pH 7.5 for 1 h. Following centrifugation at 10,000 x g for 30 min the clarified supernatant was filtered through a 0.45 μM filter and chromatographed on a 5 mL Hi-Trap FF DEAE column in conjunction with an AKTA explorer chromatography station (GE Healthcare). A 20 column volume 0.0 to 1.0 M NaCl gradient in 0.1 M Tris pH 7.5 was used to elute bound proteins in 2 mL fractions after a 30 column volume wash. The column flow through and wash was collected as 10 mL fractions. Protease-containing fractions were identified by SDS-PAGE analysis of 10 μL reaction volumes, in which 2 μL of individual fractions were incubated with 2 μg of purified NmrA for 30 mins at 25°C. Appropriate pools containing the proteases PNMA and PNMB were pressure concentrated 50-fold and further chromatographed on a Superdex 200 HR 10/30 column (GE Healthcare) equilibrated and eluted in 50 mM potassium phosphate pH 7.2, 1 mM DTT, 150 mM NaCl collecting 0.5 mL fractions. Proteolytically active fractions were subsequently pooled. The NmrA proteolytic fragment FNM1B was generated by digesting full-length NmrA with purified PNMB in the presence of 0.5 mM NAD+ in buffer consisting of 50 mM potassium phosphate pH 7.2, 1 mM DTT at 25oC. The digestion was monitored by SDS-PAGE until greater than 95% of the full-length NmrA had been digested, at which point the reaction was terminated by the addition of benzamidine to 10 mM. FNM1B was subsequently purified from PNMB and the benzamidine by chromatography on a MONO Q 10.100 GL FPLC column. Digested NmrA (typically 45 mg) was applied to the MONO Q column in 50 mM potassium phosphate pH 7.2, 1 mM DTT and after a three column volume wash was eluted at 3 mL min−1 with a 0 to 1.0 M NaCl gradient collecting 2 mL fractions. After pooling suitable fractions from the MONO Q salt gradient, NaCl was removed by dialysis against 50 mM potassium phosphate pH 7.2, 1 mM DTT and typically yielded approximately 25 mg of the FNM1B proteolytic fragment.
Protease assays
In the standard assay for protease activity, 2 μg of NmrA was incubated at 25oC for 30 min in 50 mM potassium phosphate pH 7.2, 1 mM DTT, with 1μg of cell-freeA. nidulans extract in a final volume of 10 μL. Following digestion, the products were analyzed by SDS-PAGE using a 10% separating gel. The same buffer conditions were used for longer digestions times in the range 3–20 h to detect less-sensitive protease sites. To attempt a semiquantitative measure of protease activity using NmrA as a substrate, 1 unit of activity is defined as the amount of protease necessary to convert 1 μg of NmrA to FNM1 in 2 h at 25oC in a reaction mixture containing 2 μg NmrA in 50 mM potassium phosphate pH 7.2, 1 mM DTT in a final reaction volume of 10 μL. Amounts of product formation were estimated by eye after separation by SDS-PAGE and staining for protein with coomassie blue. In selected assays of some crude cell-free extracts benzamidine was added to a final concentration of 1 mM to inhibit the activity of the protease PNMB.
Edman protein sequencing
Proteins resolved by SDS-PAGE were electroblotted onto PVDF membranes with 10 mM CAPS buffer (cyclohexlyamino-1-propanesulphonic acid), pH 11.0, containing 10% methanol. Membranes were then stained with Coomassie blue and the bands excised for N-terminal Edman protein sequence analysis using an LF3000 sequencer (Beckman, Fullerton, CA, USA).
Protein extraction
Protein was extracted from gel slices as follows. Destained gel bands were diced into small pieces (ca. 1 mm2) and placed in a 0.2 mL tube. A solution consisting of 50% hexafluoro-2-propanol HFIP, 20% formic acid (aq) was added to just cover the gel slices (∼50 μL). Extraction was performed for 2 h at room temperature before the supernatant was removed to a clean 0.2 mL tube and reduced to 10 μL in vacuo before being subjected to LC-MS or MALDI analysis. The recoveries of full length and protease generated fragments of NmrA from this procedure were uniformly low and sufficient quantities were generated by eluting multiple-gel slices.
MALDI-TOF MS
MALDI-TOF analysis was performed using a Voyager DE-STR MALDI-TOF mass spectrometer (Applied Biosystems, Framingham, MA, USA). The instrument was operated in linear mode for whole protein analysis and in reflectron mode for peptide digests. Monoisotopic peptide mass fingerprints were generated and average protein masses (Mr) assigned using Data Explorer software (Applied Biosystems). For whole protein mass measurements four sequential 0.5 μL aliquots of intact protein extract were dried onto a target plate pre-spotted with 1 μL of sinapinic acid matrix solution (10 mg ml−1 in 50% acetonitrile, 0.1% trifluoroacetic acid). Tryptic peptides for matrix-assisted laser desorption/ionization (MALDI-TOF) mass fingerprinting were eluted from a Zip-Tip (Milipore) directly onto a target plate using a matrix solution of alpha cyano-4-hydroxycinnamic acid (10 mg ml−1) in 50% acetonitrile, 0.1% trifluoroacetic acid (2 μL). The tryptic digest data was analyzed by two independent means, Mascot identity database searching and BLAST homology searching of de novo generated sequences in conjunction with the program DeNovoX (Thermo. The de novo sequencing of peptides was attempted from their MS/MS spectra without prior reference to any database. The output of this process was manually inspected and the ‘best’ 25–30 peptide MS/MS sequences were extracted to a list for BLAST homology searching in both directions using the Swiss-Prot database.
Whole protein LC-MS
The 10 μL protein extract was diluted to 50 μL using 0.1% formic acid (aq) and the acidified protein solution loaded onto a home-made trap column (approximate dimensions: 2 mm long X 1 mm diameter) packed with C18 sorbent (3M Empore, 60A pore, 12 μ particle size). Loading was performed using the syringe pump of the mass spectrometer at 10 μL min-1. The loaded trap column was then flushed with 100 μL of 0.1% formic acid to remove nonbound material. For electrospray protein analysis the trap was switched in-line with the flow of an HPLC (Agilent 1100) pumping at 100 μL/min and flow-split to produce a trap column flow of 10 μL min-1. Buffer A contained 0.1% formic acid (aq) and buffer B was acetonitrile with 0.1% formic acid. A linear elution gradient from 4% to 64% B in 22 min was used to elute the bound protein from the C-18 trap. The flow from the column was coupled to an ‘Ion Max’ ion source (ThermoElectron, Bremen, Germany) and sprayed directly into an LTQ-FT mass spectrometer (ThermoElectron, Bremen, Germany). LC-MS data was collected using the FTMS analyser in full scan and normal mass range mode at a resolution of 200,000 (at m/z = 400). The mass spectrometer collected data from scans over the mass range m/z = 300 – 2000. Mass spectrum plots and spectral deconvolution were generated using the QualBrowser program (ThermoElectron, Bremen, Germany). Protein spectra were deconvoluted to determine the average molecular mass of the protein (Mr) assuming ‘averagine’ (0.2678% sulfur) composition.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) is a nondestructive and noninvasive (label-free) technique that can be used to give a direct measure of the KD and additionally provides information on the enthalpy (ΔH) and stoichiometry (n) of ligand binding.34-36 In these ITC experiments one protein solution is placed in a reaction vessel and a second protein is titrated in from a syringe whilst constantly stirring the mixture. Any heat absorbed (endothermic reaction) or released (exothermic reaction) when ligand binds to the protein is measured directly. Standard Gibbs free energy (ΔG0) and entropy (ΔS0) of ligand binding are subsequently calculated from the following equations: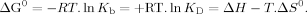
ITC experiments were performed at 25°C using a high precision VP-ITC system (Microcal). Proteins were dialyzed into 50 mM potassium phosphate pH 7.2, 1 mM DTT, and the concentrations of the reagents used in the injector and cell are shown in Table II and the legend to Figure 7. The heat evolved following each 10 μL injection was obtained from the integral of the calorimetric signal. The heat due to the binding reaction was obtained as the difference between the heat of reaction and the corresponding heat of dilution. Analysis of data was performed using Microcal Origin software.
Circular dichroism spectroscopy
The far-UV CD spectrum of NMRA and FNM1B+C-term was recorded from an average of 10 accumulative scans at 20°C from 250 to 185 nm using a Jasco J-810 spectropolarimeter and cuvettes with a 0.01 cm pathlength. Samples of NmrA and FNM1B in 50 mM potassium phosphate pH 7.2, 1 mM DTT were used at concentration of approximately 0.5 mg ml−1. The buffer baseline was subtracted from the experimental value, the data adjusted for protein concentration and the results converted to molecular CD units.
Acknowledgments
We thank Meryl Davis and Michael Hynes for the A. nidulans strain MH8935 and Tom Chadwick for help and advice with statistical analysis.
Glossary
Abbreviations:
- CD
circular dichroism
- ITC
iso-thermal titration calorimetry
- LC/MS
liquid chromatography-mass spectrometry
- MALDI-TOF MS
Matrix-assisted laser desorption ionization time-of-flight mass spectrometry
- SDR
short-chain dehydrogenase reductase
- Zf
zinc finger.
References
- 1.Wilson RA, Arst HN., Jr Mutational analysis of AREA, a transcriptional activator mediating nitrogen metabolite repression in Aspergillus nidulans and a member of the “streetwise” GATA family of transcription factors. Microbiol Mol Biol Rev. 1998;62:586–596. doi: 10.1128/mmbr.62.3.586-596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morozov IY, Galbis-Martinez M, Jones MG, Caddick MX. Characterization of nitrogen metabolite signalling in Aspergillus via the regulated degradation of areA mRNA. Mol Microbiol. 2001;42:269–277. doi: 10.1046/j.1365-2958.2001.02636.x. [DOI] [PubMed] [Google Scholar]
- 3.Morozov IY, Martinez MG, Jones MG, Caddick MX. A defined sequence within the 3′ UTR of the areA transcript is sufficient to mediate nitrogen metabolite signalling via accelerated deadenylation. Mol Microbiol. 2000;37:1248–1257. doi: 10.1046/j.1365-2958.2000.02085.x. [DOI] [PubMed] [Google Scholar]
- 4.Muro-Pastor MI, Gonzalez R, Strauss J, Narendja F, Scazzocchio C. The GATA factor AreA is essential for chromatin remodelling in a eukaryotic bidirectional promoter. EMBO J. 1999;18:1584–1597. doi: 10.1093/emboj/18.6.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Platt A, Langdon T, Arst HN, Jr, Kirk D, Tollervey D, Sanchez JM, Caddick MX. Nitrogen metabolite signalling involves the C-terminus and the GATA domain of the Aspergillus transcription factor AREA and the 3′ untranslated region of its mRNA. EMBO J. 1996;15:2791–2801. [PMC free article] [PubMed] [Google Scholar]
- 6.Platt A, Ravagnani A, Arst H, Jr, Kirk D, Langdon T, Caddick MX. Mutational analysis of the C-terminal region of AREA, the transcription factor mediating nitrogen metabolite repression in Aspergillus nidulans. Mol Gen Genet. 1996;250:106–114. doi: 10.1007/BF02191830. [DOI] [PubMed] [Google Scholar]
- 7.Andrianopoulos A, Kourambas S, Sharp JA, Davis MA, Hynes MJ. Characterization of the Aspergillus nidulans nmrA gene involved in nitrogen metabolite repression. J Bacteriol. 1998;180:1973–1977. doi: 10.1128/jb.180.7.1973-1977.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong KH, Hynes MJ, Todd RB, Davis MA. Transcriptional control of nmrA by the bZIP transcription factor MeaB reveals a new level of nitrogen regulation in Aspergillus nidulans. Mol Microbiol. 2007;66:534–551. doi: 10.1111/j.1365-2958.2007.05940.x. [DOI] [PubMed] [Google Scholar]
- 9.Kotaka M, Johnson C, Lamb HK, Hawkins AR, Ren J, Stammers DK. Structural analysis of the recognition of the negative regulator NmrA and DNA by the zinc finger from the GATA-type transcription factor AreA. J Mol Biol. 2008;381:373–382. doi: 10.1016/j.jmb.2008.05.077. [DOI] [PubMed] [Google Scholar]
- 10.Lamb HK, Leslie K, Dodds AL, Nutley M, Cooper A, Johnson C, Thompson P, Stammers DK, Hawkins AR. The negative transcriptional regulator NmrA discriminates between oxidized and reduced dinucleotides. J Biol Chem. 2003;278:32107–32114. doi: 10.1074/jbc.M304104200. [DOI] [PubMed] [Google Scholar]
- 11.Lamb HK, Ren J, Park A, Johnson C, Leslie K, Cocklin S, Thompson P, Mee C, Cooper A, Stammers DK, Hawkins AR. Modulation of the ligand binding properties of the transcription repressor NmrA by GATA-containing DNA and site-directed mutagenesis. Protein Sci. 2004;13:3127–3138. doi: 10.1110/ps.04958904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stammers DK, Ren J, Leslie K, Nichols CE, Lamb HK, Cocklin S, Dodds A, Hawkins AR. The structure of the negative transcriptional regulator NmrA reveals a structural superfamily which includes the short-chain dehydrogenase/reductases. EMBO J. 2001;20:6619–6626. doi: 10.1093/emboj/20.23.6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamb HK, Stammers DK, Hawkins AR. Dinucleotide-sensing proteins: linking signaling networks and regulating transcription. Sci Signal. 2008;1:e38. doi: 10.1126/scisignal.133pe38. [DOI] [PubMed] [Google Scholar]
- 14.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng X, Dai X, Zhao Y, Chen Q, Lu F, Yao D, Yu Q, Liu X, Zhang C, Gu X, Luo M. Restructuring of the dinucleotide-binding fold in an NADP(H) sensor protein. Proc Natl Acad Sci USA. 2007;104:8809–8814. doi: 10.1073/pnas.0700480104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Zhang J, Li H, Li Y, Ren J, Luo M, Zheng X. An NADPH sensor protein (HSCARG) down-regulates nitric oxide synthesis by association with argininosuccinate synthetase and is essential for epithelial cell viability. J Biol Chem. 2008;283:11004–11013. doi: 10.1074/jbc.M708697200. [DOI] [PubMed] [Google Scholar]
- 17.Kumar PR, Yu Y, Sternglanz R, Johnston SA, Joshua-Tor L. NADP regulates the yeast GAL induction system. Science. 2008;319:1090–1092. doi: 10.1126/science.1151903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhat PJ, Murthy TV. Transcriptional control of the GAL/MEL regulon of yeast Saccharomyces cerevisiae: mechanism of galactose-mediated signal transduction. Mol Microbiol. 2001;40:1059–1066. doi: 10.1046/j.1365-2958.2001.02421.x. [DOI] [PubMed] [Google Scholar]
- 19.Starich MR, Wikstrom M, Arst HN, Jr, Clore GM, Gronenborn AM. The solution structure of a fungal AREA protein-DNA complex: an alternative binding mode for the basic carboxyl tail of GATA factors. J Mol Biol. 1998;277:605–620. doi: 10.1006/jmbi.1998.1625. [DOI] [PubMed] [Google Scholar]
- 20.Starich MR, Wikstrom M, Schumacher S, Arst HN, Jr, Gronenborn AM, Clore GM. The solution structure of the Leu22-->Val mutant AREA DNA binding domain complexed with a TGATAG core element defines a role for hydrophobic packing in the determination of specificity. J Mol Biol. 1998;277:621–634. doi: 10.1006/jmbi.1997.1626. [DOI] [PubMed] [Google Scholar]
- 21.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 22.Hawkins AR, Lamb HK, Smith M, Keyte JW, Roberts CF. Molecular organisation of the quinic acid utilization (QUT) gene cluster in Aspergillus nidulans. Mol Gen Genet. 1988;214:224–231. doi: 10.1007/BF00337715. [DOI] [PubMed] [Google Scholar]
- 23.Hervas-Aguilar A, Rodriguez JM, Tilburn J, Arst HN, Jr, Penalva MA. Evidence for the direct involvement of the proteasome in the proteolytic processing of the Aspergillus nidulans zinc finger transcription factor PacC. J Biol Chem. 2007;282:34735–34747. doi: 10.1074/jbc.M706723200. [DOI] [PubMed] [Google Scholar]
- 24.Lin YC, Brown K, Siebenlist U. Activation of NF-kappa B requires proteolysis of the inhibitor I kappa B-alpha: signal-induced phosphorylation of I kappa B-alpha alone does not release active NF-kappa B. Proc Natl Acad Sci USA. 1995;92:552–556. doi: 10.1073/pnas.92.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 26.Hu J, Haseebuddin M, Young M, Colburn NH. Suppression of p65 phosphorylation coincides with inhibition of IkappaBalpha polyubiquitination and degradation. Mol Carcinog. 2005;44:274–284. doi: 10.1002/mc.20142. [DOI] [PubMed] [Google Scholar]
- 27.Fjeld CC, Birdsong WT, Goodman RH. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc Natl Acad Sci USA. 2003;100:9202–9207. doi: 10.1073/pnas.1633591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J. 1967;103:514–527. doi: 10.1042/bj1030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollak N, Dolle C, Ziegler M. The power to reduce: pyridine nucleotides--small molecules with a multitude of functions. Biochem J. 2007;402:205–218. doi: 10.1042/BJ20061638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harlow E, Lane D. Antibodies: a laboratory manual. Woodbury, NY: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- 31.Lamb HK, Thompson P, Elliott C, Charles IG, Richards J, Lockyer M, Watkins N, Nichols C, Stammers DK, Bagshaw CR, Cooper A, Hawkins AR. Functional analysis of the GTPases EngA and YhbZ encoded by Salmonella typhimurium. Protein Sci. 2007;16:2391–2402. doi: 10.1110/ps.072900907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osmani SA, Engle DB, Doonan JH, Morris NR. Spindle formation and chromatin condensation in cells blocked at interphase by mutation of a negative cell cycle control gene. Cell. 1988;52:241–251. doi: 10.1016/0092-8674(88)90513-2. [DOI] [PubMed] [Google Scholar]
- 33.Szewczyk E, Nayak T, Oakley CE, Edgerton H, Xiong Y, Taheri-Talesh N, Osmani SA, Oakley BR. Fusion PCR and gene targeting in Aspergillus nidulans. Nat Protoc. 2006;1:3111–3120. doi: 10.1038/nprot.2006.405. [DOI] [PubMed] [Google Scholar]
- 34.Cooper A, Johnson CM. Isothermal titration microcalorimetry. Methods Mol Biol. 1994;22:137–150. doi: 10.1385/0-89603-232-9:137. [DOI] [PubMed] [Google Scholar]
- 35.Cooper A. Microcalorimetry of protein-protein interactions. Methods Mol Biol. 1998;88:11–22. doi: 10.1385/0-89603-487-9:11. [DOI] [PubMed] [Google Scholar]
- 36.Cooper A. In: Protea comprehensive treatise. Allen G, editor. Stamford, CT: JAI Press Inc; 1999. pp. 217–270. Thermodynamics of protein folding and stability. [Google Scholar]