

Abstract
We have studied the formation and the stability of ionomer complexes from grafted copolymers (GICs) in solution and the influence of GIC coatings on the adsorption of the proteins β-lactoglobulin (β-lac), bovine serum albumin (BSA), and lysozyme (Lsz) on silica and polysulfone. The GICs consist of the grafted copolymer PAA28-co-PAPEO22 {poly(acrylic acid)-co-poly[acrylate methoxy poly(ethylene oxide)]} with negatively charged AA and neutral APEO groups, and the positively charged homopolymers: P2MVPI43 [poly(N-methyl 2-vinyl pyridinium iodide)] and PAH∙HCl160 [poly(allylamine hydrochloride)]. In solution, these aggregates are characterized by means of dynamic and static light scattering. They appear to be assemblies with hydrodynamic radii of 8 nm (GIC-PAPEO22/P2MVPI43) and 22 nm (GIC-PAPEO22/PAH∙HCl160), respectively. The GICs partly disintegrate in solution at salt concentrations above 10 mM NaCl. Adsorption of GICs and proteins has been studied with fixed angle optical reflectometry at salt concentrations ranging from 1 to 50 mM NaCl. Adsorption of GICs results in high density PEO side chains on the surface. Higher densities were obtained for GICs consisting of PAH∙HCl160 (1.6 ÷ 1.9 chains/nm2) than of P2MVPI43 (0.6 ÷ 1.5 chains/nm2). Both GIC coatings strongly suppress adsorption of all proteins on silica (>90%); however, reduction of protein adsorption on polysulfone depends on the composition of the coating and the type of protein. We observed a moderate reduction of β-lac and Lsz adsorption (>60%). Adsorption of BSA on the GIC-PAPEO22/P2MVPI43 coating is moderately reduced, but on the GIC-PAPEO22/PAH∙HCl160 coating it is enhanced.
Keywords: Grafted copolymer, Ionomer complexes, Protein adsorption, Silica, Polysulfone
Introduction
In order to effectively suppress protein adsorption on surfaces (primary adsorption) [1] by a polymer brush coating, the brush should be sufficiently dense to prevent penetration by protein molecules. Relatively high brush densities can be obtained by time-consuming and laborious methods, i.e., chemical grafting of polymer chains to the surfaces (“grafting to” and “grafting from” methods), or by adsorption of grafted block copolymers (also called comb copolymers) [2–4] instead of the linear polymers. Using such polymers, the density of the neutral, brush forming chains is partly pre-determined by the density of their chemical grafting to the backbone. Grafted copolymers have been shown to be effective in suppressing protein adsorption on various surfaces [2, 5]. Surface modification with comb copolymers can be obtained in two ways: the grafted copolymers can be adsorbed at solid–liquid interfaces [2] or mixed into a casting melt, e.g., during the production of antifouling membranes [6, 7]; however, simple adsorption of such chains on a solid–liquid interface may result in low surface coverage due to the steric repulsion from earlier adsorbed chains, and hence in a low overall brush density. Comb copolymers used as additives to the casting melt consist of hydrophilic and hydrophobic blocks. During the membrane preparation (precipitation in a water-based coagulation bath), hydrophilic blocks segregate and position themselves in the membrane–liquid interface [6, 7]. The degree of reduction of protein adsorption depends on the structure of the grafted copolymers, i.e., the grafting density [1], the length of the grafted chains [8], the size of the protein molecules, ionic strength, and surface charge [9].
In previous papers [5, 10–12] we reported on the formation of brush layers by adsorption of reversible, self-assembled micellar structures known as “Complex Coacervate Core Micelles” (C3Ms) [13], also called “Block Ionomer Complexes” [14] or “Polyion Complex Micelles” [15]. C3Ms are formed upon mixing oppositely charged polyelectrolytes, of which at least one contains a neutral soluble block. Due to electrostatic attraction the oppositely charged blocks form the coacervate core which is stabilized by the neutral corona blocks [13, 16, 17]. Upon addition of homopolymers to oppositely charged diblock copolymers, charged loose structures with low aggregation numbers, so-called soluble complex particles, are initially formed. At a critical composition neutral C3Ms are formed and the number of soluble complex particles decreases to zero at the preferred micellar composition (PMC). The charge composition (mixing ratio) of C3Ms may be defined by the ratio (f−) of the negatively chargeable groups and the sum of the negatively and positively chargeable groups. A mechanism of the adsorption of C3Ms on solid–liquid interfaces has been proposed by van der Burgh et al. [12] and Voets et al. [18]. According to this model, upon adsorption at the interface, C3Ms unfold in such a way that their corona forms a brush on top of a coacervate layer attached directly to the solid surface (Fig. 1a). The effect of a C3M coating on protein adsorption strongly depends on its composition, the surface properties, and the salt concentration [5]. The densities of the brushes, thus formed by the C3M systems consisting of linear polyelectrolytes, were found to be rather low and often insufficient to fully suppress adsorption of proteins [5]. In the present paper, we focus on further improvement of the density of the C3M brush. Here, we combine the high density of the neutral chains in grafted copolymers with the formation of C3Ms (Fig. 1c). We anticipate that the obtained brush density is higher as compared to the adsorption of the grafted copolymer alone (Fig. 1b). This approach may allow for a better reduction of protein adsorption, independent on the properties of the native surfaces.
Fig. 1.

Schematic representation of polymer brushes formed upon adsorption of a C3Ms consisting of a linear block copolymer and a linear polyelectrolyte, b a grafted copolymer, c GICs consisting of a grafted copolymer and a linear polyelectrolyte
As indicated previously [19], formation of micelles with grafted copolymers has severe consequences for the resulting structure of the aggregate, i.e., there may not be a distinguished core–corona structure, characteristic of C3Ms. More likely, attraction between individual oppositely charged chains will dominate resulting in low aggregation numbers. To distinguish these aggregates from regular C3Ms, we introduced the term “grafted ionomer complexes” (GICs).
The aim of this work is to investigate the formation and stability of GICs in solution, their adsorption on silica and polysulfone, and the effect of the adsorbed GIC layer on protein adsorption.
Homopolymers used in this study are poly(allylamine hydrochloride) and poly(N-methyl 2-vinyl pyridinium iodide). These polyelectrolytes differ in length (160 and 43 monomers, respectively) and properties. Poly(allylamine hydrochloride) was extensively studied for its ability to form stable polyelectrolyte multilayers [20–22], also at low salt concentrations, which are considered for many applications, e.g., to enhance the tensile strength of paper [23], encapsulation [24], drug delivery [25], or corrosion protection [26]. The derivatives of poly(vinyl pyridine) are applied in, e.g., catalysis [27–29], removal of heavy metals [30–33] and organic pollutants [34–36], water purification and disinfection [34, 37–39], or production of materials with antibacterial properties [40–43].
A silica surface was chosen as a well-defined model hydrophilic surface. Polysulfone was selected to mimic a polymeric material frequently used in the production of membranes for water purification. The repeating unit of polysulfone is shown in Fig. 2. Polysulfone is a hydrophobic, transparent, rigid, and high-strength thermoplastic material. It is resistant to salt, acids, and bases (pH 2–13) as well as to surfactants and hydrocarbon oils [44].
Fig. 2.

Polysulfone repeating unit
Materials and methods
Chemicals
Poly(2-vinyl pyridine) (P2VP43, M n = 4.15 kg/mol, PDI = 1.09) was purchased from Polymer Source Inc. The synthesis of poly(acrylic acid)-co-poly(acrylate methoxy poly(ethylene oxide)) (PAA28-co-PAPEO22, M n = 8.86 kg/mol, PDI = 1.4, M grafts = 450 g/mol, corresponding to eight to nine EO monomers) has been described elsewhere [45]. The schematic representation of this molecule is shown in Fig. 3. Iodomethane (99%) and poly(allylamine hydrochloride) (PAH·HCl160, M n = 15 kg/mol, ≥95%) were purchased from Sigma. A high molecular weight polysulfone (UDEL P3500 Resin) was a kind gift from AMOCO. Lysozyme (Lsz) from chicken egg white (L6876-5G), β-lactoglobulin (β-lac) from bovine milk, approximately 90% (L0130-5G), and bovine serum albumin (BSA), minimum 98% (A7030-10G) were purchased from Sigma. Selected properties of these proteins are summarized elsewhere [5].
Fig. 3.

A schematic representation of PAA28-co-PAPEO22 molecule. Subscripts denote numbers of repeating blocks: acrylic acid (AA): x × m = 28 and acrylate methoxy poly(ethylene oxide) (APEO): x × n = 22
Sodium chloride (NaCl), sodium hydroxide (NaOH, 1 M), and hydrochloric acid (HCl, 1 M) were purchased from Sigma. All chemicals were used as received. Silicon wafers (Boron doped, orientation 100, and resistivity 7–15 Ω cm−1) were purchased from WaferNet, Inc., USA.
Quaternization of P2VP
Poly(2-vinyl pyridine) (P2VP43) was quaternized according to the following procedure: 1 g of the polymer was dissolved in 35 ml of N,N-dimethylmethanamide. Three milliliters of iodomethane were added and the mixture was allowed to react for 48 h at 60 °C, under nitrogen gas flow and stirring. A second portion of 3 ml iodomethane was added 24 h after the beginning of the reaction. Subsequently, the quaternized polymer was precipitated with ether, filtered, washed several times with fresh portions of ether, and dried overnight in a vacuum oven at 48 °C. The degree of quaternization was determined from the maximum in a dynamic light scattering titration of a freshly quaternized batch of P2MVPI43 with a well-defined, oppositely charged copolymer, and verified by comparison with a titration performed with a P2MVPI43 batch of known degree of quaternization [46]. The degree of quaternization of the new batch was determined to be approximately 89%.
Characterization of GICs in solution
Stock solutions of PAA28-co-PAPEO22, P2MVPI43, and PAH·HCl160 in MilliQ water were prepared. Prior to the measurements, solutions of the required polymer and salt concentrations were prepared from stock solutions. The pH was adjusted to 7 ± 0.1 with 1 and 0.1 M NaOH and HCl, when necessary.
Zeta potential measurements
Zeta potentials of GICs were measured with a Nanosizer (Nano ZS, Malvern Instruments) in order to determine the zero charge composition, i.e., the composition at which the formed particles are electrically neutral. Samples at different mixing ratios (f−) of PAA28-co-PAPEO22 and P2MVPI43, and PAA28-co-PAPEO22 and PAH·HCl160 were prepared in 1 mM NaCl solution. The total polymer concentrations were approximately 1 g/l. Samples were measured approximately 12 h after mixing to ensure equilibrium. For each mixing ratio (f−) we report averages over at least four measurements. Calculations were done using Smoluchowski’s equation.
Dynamic light scattering
Dynamic light scattering (DLS) measurements were performed with an ALV light scattering instrument equipped with an ALV_500 digital correlator and a 300 mW argon ion laser (532 nm) at 20 ± 0.5 °C. Titrations were performed with a Schott Titronic T200 burette with a TA05 exchange unit. The temperature was controlled with a thermostatic bath (Haake Phoenix II–C30P). Decalin was used as a refractive index matching medium. The titrations were performed at 90° detection angle in order to determine the PMC as well as the stability of the micelles against increasing salt concentration. To determine the PMC, the PAA28-co-PAPEO22 solution was titrated with a P2MVPI43 or a PAH·HCl160 solution. The concentration of PAA28-co-PAPEO22 varied from 0.1 to 1.3 g/l, the concentration of P2MVPI43 varied from 1 to 5 g/l, and the concentration of PAH·HCl160 varied from 0.6 to 2.5 g/l. All polymer solutions were prepared in 1 mM NaCl. To evaluate the stability of GICs against salt, 2 M NaCl solution, pH 7, was titrated into GIC solutions.
Preparation of protein solutions
Protein solutions were prepared prior to each experiment at the required NaCl concentration using MilliQ water. The pH was adjusted to 7 ± 0.1 with 1 and 0.1 M NaOH or HCl.
Preparation of the surfaces
Preparation of silica surfaces is described in detail elsewhere [5].
Polysulfone-coated silica surfaces were prepared as follows: A Si wafer was cut into strips of approximately 1 × 5 cm2. Subsequently, the strips were cleaned with a freshly prepared Piranha solution (1 part of 35% H2O2 and 2 parts of 95% H2SO4) for 2–3 min and rinsed with MilliQ water. Next, the strips were rinsed with acetone and dried with nitrogen gas. The polysulfone layer was created by spin coating (RDE-/SPINCOATER Motor controller, Eco Chemie B.V.) the polymer solution (5 g/l in chloroform) onto the cleaned surfaces. Coated strips were kept 1 h at 220 °C to increase the adhesion of the spin-coated layer to the Si strips. After cooling, the coated strips were stored in separate, dry, clean, and closed vials. The quality of the resulting layer was controlled with scanning electron microscopy (JEOL SEM-6480LV). Polysulfone surfaces used in the experiments were not older than 4–5 days.
The thickness of the silica and polysulfone layers was determined with ellipsometry (SE 400, SENTECH Instruments GmbH, Germany). The complex refractive index of the silicon layer was set to n = 3.85, k = 0.02, and the refractive indexes of silica and polysulfone layer were 1.46 and 1.63, respectively. The thickness was measured at several positions of the strip, and an average of at least five measurements has been taken for calculation of the sensitivity factor used in reflectometry.
Reflectometry
The adsorbed amounts were determined with a fixed angle optical reflectometer [47]. Changes in the signal (ΔS) measured can be related to the adsorbed amount, Γ (milligram per square meter), according to:
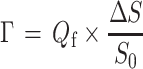 |
1 |
where Q f is the sensitivity factor (milligram per square meter), S 0 is the initial (baseline) signal, and ΔS = S−S 0 is a change in the signal upon adsorption on the surface. For each measurement Q f was calculated with “Prof. Huygens” 1.2b (Dullware Software), using the refractive index increments dn/dc of the adsorbates as determined in separate measurements and the thickness of the optical spacer (the substrate layer).
An example of the output of a typical reflectometry experiment is shown in Fig. 4. At the beginning of the experiment, a surface placed in the reflectometer cell is rinsed with solvent (impinging jet flow, flow rate ≈ 2 ml/min, cell volume ≈ 3 ml) to establish the baseline signal, S 0. Once the baseline is established the experiment starts. The sample solution is introduced into the cell (A). Adsorption of the solute is monitored until a plateau is reached (B) followed by rinsing with solvent and introduction of the next sample (C), and rinsing (D) until again a constant signal is obtained (E). All reported values were calculated based on amounts that remained adsorbed on the surface after rinsing with solvent, unless mentioned otherwise. Each measurement, including protein adsorption was repeated at least three times and averaged values are reported.
Fig. 4.

Example of a typical reflectometry experiment. Adsorption of β-lac on polysulfone coated with GIC-PAPEO22/PAH160. A Introduction of the GIC solution, B rinsing with solvent, C introduction of a β-lac solution, D rinsing with solvent, E final plateau. The experiments were carried out in 1 mM NaCl, pH 7 ± 0.1. Concentrations of GICs and β-lac were 100 mg/l. Once a stable initial signal is reached, the baseline is recorded for 2–5 min from the beginning of the experiment
Determination of the refractive index increment (dn/dc)
Refractive index increments (dn/dc) of the polymers were determined with a differential refractive index detector (Shodex RI-71). Prior to the measurements, the instrument was calibrated with NaCl. GICs and polymer solutions were prepared and measured in 10 mM NaCl. Calibration and dn/dc values were determined based on at least five different concentrations of each sample. The values of dn/dc measured for PAA28-co-PAPEO22 and GICs corrected for salt according to the method described elsewhere [5], and the values measured for polyelectrolytes and proteins [5] used in further calculations are listed in Table 1.
Table 1.
Values of refractive index increments used for calculations
| Compound | Solvent | dn/dc [milliliter per gram] |
|---|---|---|
| Proteins: | ||
| Lsz[5] | Buffer | 0.250 |
| β-lac[5] | Buffer | 0.196 |
| BSA[5] | Buffer | 0.204 |
| Polymers: | ||
| PAA28-co-PAPEO22 | Salt | 0.156 |
| PAH·HCl160 | Salt | 0.170 |
| P2MVPI43 | Salt | 0.204 |
| GICs: | ||
| GIC-PAPEO22/P2MVPI43 | Salt | 0.167 |
| GIC-PAPEO22/PAH160 | Salt | 0.129 |
Buffer 50 mM phosphate buffer, Salt 10 mM NaCl
Results and discussion
Formation and characterization of GICs in solution
Formation of micellar particles by mixing a grafted copolymer with an oppositely charged polyelectrolyte has been studied by DLS titrations as a function of the mixing ratio f−. The PMC, i.e., the mixing ratio at which the polymeric micelles (GICs) are formed [17] can be determined from the maximum scattering intensity. Experimental results of the scattering intensity for mixtures of PAA28-co-PAPEO22 and P2MVPI43 or PAH·HCl160 are shown as a function of the mixing ratio f− in Fig. 5a and b, respectively. The mixing ratio f− is defined as the fraction of the negatively chargeable groups in GICs assuming that 89% monomers in P2VP43 and all monomers in PAH·HCl160 are charged:
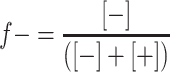 |
2 |
[−] and [+] are the amount of negatively and positively chargeable groups, respectively.
Fig. 5.

Formation of GICs in bulk as determined with DLS titrations and zeta potential measurements. Left Y axis zeta potential, right Y axis scattering intensity [kHz] divided by the total concentration, c [g/l], of the polymer. (White circles) DLS titration curves of PAA28-co-PAPEO22 with P2MVPI43 (a) and PAH·HCl160 (b) in 1 mM NaCl, pH 7, (black circles) zeta potentials of GIC-PAPEO22/P2MVPI43 (a) and GIC-PAPEO22/PAH160 (b) measured at various mixing ratios (Emphasis>)) in 1 mM NaCl, pH 7. Lines were added to guide the eye. Instruments settings for DLS titrations, namely the aperture of the detector (pinhole), were different for titration with P2MVPI43 and PAH·HCl160. For titration with P2MVPI43, the aperture was 200 μm and for titration with PAH·HCl160 the aperture was 400 μm. The pinhole size was increased to improve the signal measured for weakly scattering PAA28-co-PAPEO22 + PAH·HCl160 complexes
For linear polymers, C3Ms are formed upon mixing the oppositely charged polyelectrolytes at stoichiometric charge ratio. During this process, the relatively long neutral chains form a corona that prevents the complex coacervate core to grow to macroscopic dimensions [17]. At f− = 0.5, one expects electrically neutral C3M particles. For grafted copolymers, the mechanism of micelle formation is expected to be different. The PAA28-co-PAPEO22 copolymer consists of a chain of alternating negatively charged acrylic acid groups and short PEO grafts with a length of eight to nine PEO groups. Thus, it is likely that the structure of the polymeric micelle differs from the classical spherical core–shell structures for C3Ms composed of linear polymers. We anticipate that due to the limited flexibility, the grafted chains form aggregates whose structure limits their bending. One of the possible structures is a linear aggregate.
The maxima of the measured scattering intensities are not very well defined, in particular for GIC-PAPEO22/PAH160, suggesting a slow aggregation process and a relatively small amount of the formed complexes. The hydrodynamic radii (R h) of GIC-PAPEO22/P2MVPI43 and GIC-PAPEO22/PAH160, as determined with DLS titrations, are approximately 6 and 22 nm, respectively. The radius of GICs with the short P2MVP homopolymer is exceptionally small. It excludes a mechanism with the formation of a complex coacervate core and a neutral corona which would result in a size of the aggregates independent of the length of the homopolymer. It is more likely that the difference in size is directly related to the length of the homopolymers. A possible explanation is that one homopolymer molecule assembles with the grafted copolymer in stoichiometric amounts. The difference in persistence length is then the origin of a size ratio of about three instead of four. In order to unravel the formation and structure of these assemblies, additional measurements were carried out. These are presented and discussed in a forthcoming paper.
The relative scattering intensity of GIC-PAPEO22/PAH160 is approximately 75% lower than that of GIC-PAPEO22/P2MVPI43 suggesting that, despite the same total polymer concentrations, PAA28-co-PAPEO22 forms a lower number of micelles with PAH∙HCl160 than with P2MVPI43. In order to observe the formation of the GIC-PAPEO22/PAH160, the aperture of the detector had to be increased twice to strengthen the weak scattering signal. As a result, the (unnormalized) intensity plotted for GIC-PAPEO22/PAH160 in Fig. 5 appears to be much higher than the intensity plotted for GIC-PAPEO22/P2MVPI43.
Determination of the zeta potentials for different mixing ratios (f−) reveals that, in solution, GIC-PAPEO22/PAH160 are electrically neutral at f− = 0.46. The isoelectric point corresponds to the maximum in the scattering intensity (PMC); however, for assemblies with the short homopolymer, GIC-PAPEO22/P2MVPI43, the maximum scattering intensity corresponds to a composition at which particles carry an excess of negative charge. In this case the broad maximum corresponds to about f− = 0.45, but the isoelectric point is found at a specific ratio, i.e., f− = 0.37. A possible explanation for the observed deviation of the isoelectric point from the maximum of scattering intensity for GIC-PAPEO22/P2MVPI43 system may be the small size of the aggregates as a consequence of the short length of the homopolymer that does not allow accommodating all charged blocks on the single homopolyelectrolyte and to the stiffness of the polymer chains with bulky side groups, which does not allow for full contact between oppositely charged groups. As a result, the maximum scattering intensity corresponding to the highest number of assemblies is obtained if the grafted copolymer in the aggregate is in excess.
As for the measurements of the coated surfaces electroneutrality of the coating is preferred, in this study we have chosen to use grafted ionomer complexes formed at a f− corresponding to the zero charge composition determined with zeta potential measurements, i.e., f− = 0.37 and f− = 0.45 for GIC-PAPEO22/P2MVPI43 and GIC-PAPEO22/PAH160, respectively.
Stability against salt
The stability of the GICs in solution depends on the strength of the electrostatic attraction between the oppositely charged polyelectrolytes and, hence, is influenced by the salt concentration [13]. Experimental results of titrations of GIC-PAPEO22/P2MVPI43 and GIC-PAPEO22/PAH160 with 2 M NaCl at pH 7 are shown in Fig. 6a and b, respectively.
Fig. 6.

Effect of salt on the stability of GIC-PAPEO22/P2MVPI43 (a) and GIC-PAPEO22/PAH 160 (b) as determined with DLS titrations. Left Y axis scattering intensity (I [kHz]) divided by the total polymer concentration (c [g/l]), right Y axis hydrodynamic radii. (White circles) Rh [nm], (black circle) I/c [kHz/(g/l)]. Experiments were carried out at pH 7. For both titrations, the aperture was 200 μm, and therefore for titration with PAH·HCl160 the initial scattering intensity at the iep appears to be much lower than in Fig. 5
As the salt concentration increases from 0.01 to 0.2 M NaCl, the scattering intensity of GIC-PAPEO22/P2MVPI43 in solution decreases steeply and stabilizes at approximately 30% of the value at 1 mM NaCl. The hydrodynamic radius (R h) remains stable up to approximately 0.08 M NaCl and increases at higher salt concentrations indicating swelling and partial disintegration of GICs. Under the same conditions the scattering intensity of GIC-PAPEO22/PAH160 in solution decreases slowly and R h remains relatively stable up to 0.6 M NaCl. It increases at higher salt concentrations. Thus, GICs are sensitive to salt concentration and start to disintegrate already above 0.01 M NaCl.
Adsorption of GICs on silica and polysulfone
Adsorption of GICs on silica and polysulfone was studied with fixed angle optical reflectometry. Experimental results are presented in Fig. 7a and b.
Fig. 7.

Reversibility of the adsorption of GIC coatings on silica (a) and polysulfone (b). Arrows indicate the addition of the solvent. Concentrations of GICs in solution were 100 mg/l. Experiments were carried out at 10 mM NaCl, pH 7 ± 0.1
The initial adsorption of GICs with P2MVPI43 and PAH∙HCl160 shows many similarities; however, at a later stage the differences between the two systems are more pronounced. For both systems, the initial adsorption is fast and then levels off to reach a relatively stable value within 10 min, with the exception of adsorption of GIC-PAPEO22/PAH160 on polysulfone which reaches a stable value after approximately 1 h. The adsorbed amounts are higher on polysulfone than on silica. Upon rinsing with solvent, which is indicated by arrows in Fig. 7a and b, any reversibly adsorbed fraction is removed. The adsorbed amount of GIC-PAPEO22/PAH160 is higher than GIC-PAPEO22/P2MVPI43 and its adsorption on silica and polysulfone is practically irreversible. The reversibly adsorbed fraction of GIC-PAPEO22/P2MVPI43 is approximately 23% and 25% on both silica and polysulfone, respectively. We have observed similar trends for micelles consisting of a grafted block copolymer [19] of a length comparable to that of the grafted copolymer discussed here. Both for grafted copolymers and grafted block copolymers a higher fraction of loosely attached aggregates was observed for aggregates containing oppositely charged blocks, of which the lengths are of the same order of magnitude. On the contrary, for both cases aggregates composed of charged blocks with significantly different lengths adsorbed practically irreversibly. We have attributed these differences in adsorbed amounts after rinsing with solvent to the stability of the aggregates and to the wettability of the native surface [11]. Adsorption of regular C3Ms includes an unfolding mechanism in order to adapt the spherical geometry of the micelles to the flat geometry of the surface. This mechanism has been previously described by van der Burgh et al. [12] and Voets et al. [48]; however, assemblies containing a grafted copolymer do not unfold. Their backbone lies flat on the surface and charge repulsion is eliminated by the oppositely charged homopolymer. The short and dense PEO grafts are naturally perpendicular to the surface and form a thin neutral layer between solution and adsorbed backbone assemblies. Formation of this structure is strongly enhanced due to the stiffness of the grafted copolymer chain. The stiff grafted chain is more likely to form elongated structures to limit bending. The grafted copolymer also contains a short hydrophobic sticker at one end of the polymer. The presence of that hydrophobic chain (C12H25), indicated in Fig. 3, may promote adsorption. It is likely that assemblies of these hydrophobic stickers lie flat on the surface and do not disrupt the geometry of the coating.
As shown in Fig. 8, after rinsing with solvent, adsorption of GIC-PAPEO22/P2MVPI43 on silica hardly depends on salt concentration, if at all; however, on polysulfone the adsorbed amount decreases as the salt concentration increases from 1 to 10 mM NaCl. At 50 mM NaCl, the adsorbed amounts on silica and polysulfone are comparable indicating similar structure of the coating. The lower adsorption on silica, as compared to polysulfone, may be due to electrostatic repulsion, as both GICs and silica are negatively charged under the experimental conditions. Adsorption of GIC-PAPEO22/PAH160 on silica increases with increasing salt concentration whereas on polysulfone it remains stable beyond 10 mM NaCl. Adsorbed amounts of these aggregates on both surfaces are very similar.
Fig. 8.

Adsorption of GIC-PAPEO22/P2MVPI43 (a) and GIC-PAPEO22/PAH160 (b) coatings on silica and polysulfone surfaces, and the average grafting densities of PEO chains on both surfaces at various concentrations of NaCl. Concentrations of GICs in solutions were 100 mg/l, pH 7. Lines were added to guide the eye
Effect of GIC coatings on protein adsorption
Reduction of protein adsorption by GICs adsorbed on silica and polysulfone surfaces was determined with fixed angle optical reflectometry. For comparison, we also measured the adsorption of proteins on the corresponding bare surfaces. Adsorbed amounts of proteins on silica and polysulfone as a function of the NaCl concentration are presented in Fig. 9a and b, respectively.
Fig. 9.

Adsorption of proteins on bare silica (a) and polysulfone (b) surfaces. Concentrations of proteins in solutions were 100 mg/l, pH 7 ± 0.1. Lines were added to guide the eye
On silica, adsorbed amounts of BSA and Lsz are almost equal and invariant with salt concentration beyond 10 mM NaCl. Adsorbed amounts of these proteins increase with decreasing salt concentration below 10 mM NaCl. The pattern for β-lac is very different. β-lac adsorbs in much larger amounts and adsorption increases with increasing salt concentration reaching a similar value as those for BSA and Lsz at 50 mM NaCl. On polysulfone, between 1 and 10 mM NaCl adsorption of β-lac and BSA slightly increases and that of Lsz strongly decreases. Beyond 10 mM NaCl (up to 50 mM NaCl) the adsorbed amount of BSA is invariant with salt concentration, and those of β-lac and Lsz strongly increase with increasing salt concentration.
Protein adsorption on a solid–liquid interface is a result of the balance between electrostatic and non-electrostatic forces. It is determined by the properties of both the protein (size, charge, wetting behavior) and the surface (hydrophobicity, charge, heterogeneity). The process is strongly influenced by the ionic strength of the solution and valence of the present ions [11, 49, 50]. Upon increasing the salt concentration, electrostatic interactions between the protein and the surface become weaker due to screening of charges; however, hydrophobic interactions remain essentially unchanged. This may explain why we observe a decrease of the adsorbed amount of protein on hydrophilic silica with increasing salt concentration and constant or increasing adsorbed amounts of proteins on hydrophobic polysulfone.
In Figs. 10 and 11 we present experimentally determined reduction factors of protein adsorption on surfaces coated with GIC-PAPEO22/P2MVPI43 and GIC-PAPEO22/PAH160, respectively. We define the reduction of protein adsorption on coated surface as: Reduction (%) = (1−Γ/Γ 0) × 100%, where Γ 0 is the amount of protein adsorption on the native surface.
Fig. 10.

Reduction of protein adsorption by pre-adsorbed GIC-PAPEO22/P2MVPI43 layer on silica (a) and polysulfone (b). Concentrations of proteins and GICs in solutions were 100 mg/l, pH 7 ± 0.1. Lines were added to guide the eye
Fig. 11.

Reduction of protein adsorption by pre-adsorbed GIC-PAPEO22/PAH160 layer on silica (a) and polysulfone (b). Concentrations of proteins and GICs in solutions were 100 mg/l. pH 7 ± 0.1. Lines were added to guide the eye
From the results in Figs. 10 and 11 it follows that the protein resistance of a coating of GICs with P2MVPI43 and PAH·HCl160 as the homopolymer differ significantly. On a GIC-PAPEO22/P2MVPI43 layer the adsorption of all proteins is strongly suppressed. An excellent reduction (>90%) was obtained on a silica surface for salt concentrations of 1 and 50 mM NaCl. On polysulfone the effect of salt concentration is more pronounced. As the salt concentration is increased from 1 to 50 mM NaCl, the adsorption reduction factors for β-lac and Lsz increase from about 68% and 82%, respectively to >95%. On both surfaces the reduction of BSA adsorption is much lower than of the other proteins and passes a minimum at 10 mM NaCl. This suggests the presence of some interactions between BSA and the coating that weaken as the salt concentration increases. The adsorbed amount of GIC-PAPEO22/P2MVPI43 corresponds to an area of 1.67 to 0.67 nm2 per PEO graft, as the salt concentration increases from 1 to 50 mM NaCl on both surfaces, respectively. This density should not allow for the coating to be penetrated by BSA [4 × 4 × 14 (nm3)]. In solution, both β-lac and BSA form soluble complexes with P2MVPI43 [5]. Aggregates formed with β-lac are much bigger and much more stable than the ones formed with BSA and may result in erosion of the coating [5]; however, the interactions between polyelectrolytes within the GIC-PAPEO22/P2MVPI43 layer are very strong, and its structure does not allow for the formation of such complexes. Thus, the nature of BSA attraction to this coating remains unclear.
GIC-PAPEO22/PAH160 pre-adsorbed on silica resulted in high reduction of β-lac and Lsz adsorption at 10 mM NaCl. Adsorption of β-lac was strongly suppressed independently of the salt concentration, whereas reduction of Lsz adsorption improved as the salt concentration increased from 1 to 50 mM NaCl, going through a minimum at 10 mM NaCl. On polysulfone, reduction of β-lac and Lsz adsorption was moderate and practically invariant with salt concentration. On both surfaces, reduction of BSA adsorption differs significantly from the other two proteins. On silica, the highest reduction was observed at 10 mM NaCl, and it decreased as the salt concentration increased to 50 mM NaCl. Similarly on polysulfone, the highest reduction of BSA adsorption was measured at 10 mM NaCl (approximately 20%); however, at 1 and 50 mM NaCl BSA adsorption on a coated surface was higher than on the bare polysulfone (negative reduction in Fig. 11b). These results indicate that the layer formed upon adsorption of GIC-PAPEO22/PAH160 is electrically neutral but seems to have a specific interaction with BSA that results in attraction of this protein to the coating. The adsorbed amount of GIC-PAPEO22/PAH160 corresponds to an average area of 0.63 to 0.53 nm2 per PEO graft, for 1 and 50 mM NaCl on both surfaces, respectively. This density should be sufficient to prevent penetration by proteins providing the formation of a perfect brush.
The grafted copolymer used in this study (PAA28-co-PAPEO22) resembles the grafted block copolymer (PAA21-b-PAPEO14) discussed in our previous paper [19]. The main difference between the two is the distribution of the PEO grafts (eight to nine EO monomers in both copolymers) along the backbone. In the grafted copolymer, the grafts are randomly distributed along the charged chain (Fig. 3), and in the block copolymer the charged monomers and grafted uncharged monomers form separate blocks. Both copolymers have additional hydrophobic chains of the same length, C12H25. The difference in distribution of charged and grafted monomers in these two copolymers have an enormous impact on the structure of the resulting aggregates formed with an oppositely charged polyelectrolyte and, more importantly, on the structure of the adsorbed layer. In the aggregates formed with a grafted block copolymer and P2MVPI43, the hydrophobic tail is exposed, and the secondary aggregation occurs due to hydrophobic interactions between the tails. The R h of the resulting particles in the bulk is approximately 100 nm [19], and when adsorbed they effectively suppress protein adsorption on coated surfaces regardless of the properties of the native surface. In aggregates formed with grafted copolymers and P2MVPI43 (GIC-PAPEO22/P2MVPI43), the hydrophobic tails are “hidden” between the PEO grafts, and the secondary aggregation is only very limited (R h ∼ 22 nm). Moreover, the limited flexibility of the PAA28-co-PAPEO22 chain suggests that GICs are elongated structures rather than globular. As a result, the adsorbed GIC layer is thinner and the properties of the native surfaces influence suppression of protein adsorption. The structure of the resulting GICs is also influenced by the length and the flexibility of the oppositely charged blocks.
A relatively low reduction of BSA adsorption on GIC-PAPEO22/PAH160 results from the factors discussed above: thin adsorbed GIC layer, relatively short brush forming PEO grafts, and a PAH chain accessible for interactions with the proteins in the bulk. Ball et al. [51] reports on the formation of the soluble complexes between BSA and PAH. The largest aggregates formed at a specific ratio of BSA to PAH were shown to be electrically neutral. At other mixing ratios, the net charge of the complexes corresponds to the charge carried by the molecule present in excess. At a given mixing ratio, the size of the aggregates was shown to depend on the salt concentration and to increase if the salt concentration decreases. It was shown that these complexes are thermodynamically stable, and it was concluded that their formation is entropically driven. Apparently, similar complexation does not occur between β-lac or Lsz and PAH·HCl160 under the experimental conditions discussed here. Moreover, we have not observed removal of the GIC layer from the surface upon contact with BSA that is observed during the formation of such aggregates [19]. Thus, the nature of the BSA attraction to the discussed coating remains unclear.
Conclusions
We have studied the formation and the stability of grafted ionomer complexes (GICs) in solution and the influence of GIC coatings on the adsorption of the proteins β-lac, BSA, and Lsz on silica and polysulfone. In solution, these GICs were characterized by means of dynamic light scattering. The size of the GICs depend on the length of the homopolymer (8 nm for GIC-PAPEO22/P2MVPI43 and 22 nm for GIC-PAPEO22/PAH160 indicating that no core–corona structure is formed as for C3Ms). GICs partly disintegrate at salt concentrations above 10 mM NaCl. Adsorption of GICs on silica and polysulfone results in high density PAPEO chains. Higher density (1.6 ÷ 1.9 PEO grafts/nm2) was obtained for GICs consisting of PAH∙HCl160 than of P2MVPI43 (0.6 ÷ 1.5 PEO grafts/nm2). Both GIC coatings strongly suppress the adsorption of all proteins on silica (>90%). On polysulfone we observe reduction of β-lactoglobulin and lysozyme adsorption (>60%), only limited reduction of adsorption by GIC-PAPEO22/P2MVPI43, and attraction of bovine serum albumin to the GIC-PAPEO22/PAH160 coating probably due to the strong interaction between BSA and PAH and insufficient screening of the short PEO grafts.
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Halperin A. Polymer brushes that resist adsorption of model proteins: design parameters. Langmuir. 1999;15:2525–2533. [Google Scholar]
- 2.Pasche S, De Paul SM, Voros J, Spencer ND, Textor M. Poly(L-lysine)-graft-poly(ethylene glycol) assembled monolayers on niobium oxide surfaces: a quantitative study of the influence of polymer interfacial architecture on resistance to protein adsorption by ToF-SIMS and in situ OWLS. Langmuir. 2003;19:9216–9225. [Google Scholar]
- 3.VandeVondele S, Voros J, Hubbell JA. RGD-grafted poly-L-lysine-graft-(polyethylene glycol) copolymers block non-specific protein adsorption while promoting cell adhesion. Biotechnology and Bioengineering. 2003;82:784–790. doi: 10.1002/bit.10625. [DOI] [PubMed] [Google Scholar]
- 4.Zhang ZP, Ma HW, Hausner DB, Chilkoti A, Beebe TP. Pretreatment of amphiphilic comb polymer surfaces dramatically affects protein adsorption. Biomacromolecules. 2005;6:3388–3396. doi: 10.1021/bm050446d. [DOI] [PubMed] [Google Scholar]
- 5.Brzozowska AM, Hofs B, de Keizer A, Fokkink R, Cohen Stuart MA, Norde W. Reduction of protein adsorption on silica and polystyrene surfaces due to coating with complex coacervate core micelles. Colloids Surf A Physicochem Eng Asp. 2009;347:146–155. [Google Scholar]
- 6.Asatekin A, Kang S, Elimelech M, Mayes AM. Anti-fouling ultrafiltration membranes containing polyacrylonitrile-graft-poly (ethylene oxide) comb copolymer additives. Journal of Membrane Science. 2007;298:136–146. [Google Scholar]
- 7.Ma XL, Su YL, Sun Q, Wang YQ, Jiang ZY. Preparation of protein-adsorption-resistant polyethersulfone ultrafiltration membranes through surface segregation of amphiphilic comb copolymer. Journal of Membrane Science. 2007;292:116–124. [Google Scholar]
- 8.Pasche S, Textor M, Meagher L, Spencer ND, Griesser HJ. Relationship between interfacial forces measured by colloid-probe atomic force microscopy and protein resistance of poly(ethylene glycol)-grafted poly(L-lysine) adlayers on niobia surfaces. Langmuir. 2005;21:6508–6520. doi: 10.1021/la050386x. [DOI] [PubMed] [Google Scholar]
- 9.Pasche S, Voros J, Griesser HJ, Spencer ND, Textor M. Effects of ionic strength and surface charge on protein adsorption at PEGylated surfaces. The Journal of Physical Chemistry. B. 2005;109:17545–17552. doi: 10.1021/jp050431+. [DOI] [PubMed] [Google Scholar]
- 10.Brzozowska AM, Zhang Q, de Keizer A, Norde W, Cohen Stuart MA. Protein adsorption on silica and polysulfone surfaces coated with complex coacervate core micelles with poly(vinyl alcohol) as a neutral brush forming block. Colloids Surf A Physicochem Eng Asp. 2010;368:96–104. [Google Scholar]
- 11.Hofs B, Brzozowska A, de Keizer A, Norde W, Cohen Stuart MA. Reduction of protein adsorption to a solid surface by a coating composed of polymeric micelles with a glass-like core. J Colloid Interface Sci. 2008;325:309–315. doi: 10.1016/j.jcis.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 12.van der Burgh S, Fokkink R, de Keizer A, Cohen Stuart MA. Complex coacervation core micelles as anti-fouling agents on silica and polystyrene surfaces. Colloids Surf A Physicochem Eng Asp. 2004;242:167–174. [Google Scholar]
- 13.Cohen Stuart MA, Besseling NAM, Fokkink RG. Formation of micelles with complex coacervate cores. Langmuir. 1998;14:6846–6849. [Google Scholar]
- 14.Kabanov AV, Bronich TK, Kabanov VA, Yu K, Eisenberg A. Soluble stoichiometric complexes from poly(N-ethyl-4-vinylpyridinium) cations and poly(ethylene oxide)-block-polymethacrylate anions. Macromolecules. 1996;29:6797–6802. [Google Scholar]
- 15.Kataoka K, Togawa H, Harada A, Yasugi K, Matsumoto T, Katayose S. Macromolecules. 1996;29:8556–8557. [Google Scholar]
- 16.Borisov OV, Zhulina EB. Effect of salt on self-assembly in charged block copolymer micelles. Macromolecules. 2002;35:4472–4480. [Google Scholar]
- 17.van der Burgh S, de Keizer A, Cohen Stuart MA. Complex coacervation core micelles. Colloidal stability and aggregation mechanism. Langmuir. 2004;20:1073–1084. doi: 10.1021/la035012n. [DOI] [PubMed] [Google Scholar]
- 18.Voets IK, de Vos WA, Hofs B, de Keizer A, Stuart MAC, Steitz R, Lott D. Internal structure of a thin film of mixed polymeric micelles on a solid/liquid interface. The Journal of Physical Chemistry. B. 2008;112:6937–6945. doi: 10.1021/jp709758p. [DOI] [PubMed] [Google Scholar]
- 19.Brzozowska AM, de Keizer A, Norde W, Detrembleur C, Cohen Stuart MA. Grafted block complex coacervate core micelles and their effect on protein adsorption on silica and polystyrene. Colloid and Polymer Science. 2010;288:1081–1095. doi: 10.1007/s00396-010-2228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kovacevic D, van der Burgh S, de Keizer A, Cohen Stuart MA. Specific ionic effects on weak polyelectrolyte multilayer formation. The Journal of Physical Chemistry. B. 2003;107:7998–8002. [Google Scholar]
- 21.Ladam G, Schaad P, Voegel JC, Schaaf P, Decher G, Cuisinier F. In situ determination of the structural properties of initially deposited polyelectrolyte multilayers. Langmuir. 2000;16:1249–1255. [Google Scholar]
- 22.Tuong SD, Lee H, Kim H. Chemical fixation of polyelectrolyte multilayers on polymer substrates. Macromolecular Research. 2008;16:373–378. [Google Scholar]
- 23.Ankerfors C, Lingstrom R, Wagberg L, Odberg L. A comparison of polyelectrolyte complexes and multilayers: their adsorption behaviour and use for enhancing tensile strength of paper. Nordic Pulp & Paper Research Journal. 2009;24:77–86. [Google Scholar]
- 24.Radtchenko IL, Sukhorukov GB, Leporatti S, Khomutov GB, Donath E, Mohwald H. Assembly of alternated multivalent ion/polyelectrolyte layers on colloidal particles. Stability of the multilayers and encapsulation of macromolecules into polyelectrolyte capsules. Journal of Colloid and Interface Science. 2000;230:272–280. doi: 10.1006/jcis.2000.7068. [DOI] [PubMed] [Google Scholar]
- 25.Qiu XP, Donath E, Mohwald H. Permeability of ibuprofen in various polyelectrolyte multilayers. Macromolecular Materials and Engineering. 2001;286:591–597. [Google Scholar]
- 26.Khaled M, Abu-Sharkh B, Amr E, Yilbas BS, Manda A, Abulkibash A. Corrosion properties of 316L stainless steel coated with polyelectrolyte multilayers of varying anionic acidity. Corrosion Engineering, Science and Technology. 2007;42:356–362. [Google Scholar]
- 27.Jagtap SR, Raje VP, Samant SD, Bhanage BM. Silica supported polyvinyl pyridine as a highly active heterogeneous base catalyst for the synthesis of cyclic carbonates from carbon dioxide and epoxides. Journal of Molecular Catalysis A: Chemical. 2007;266:69–74. [Google Scholar]
- 28.Ren LZ, Meng LJ. Suzuki coupling reactions catalyzed by poly(N-ethyl-4-vinylpyridinium) bromide stabilized palladium nanoparticles in aqueous solution. Express Polymer Letters. 2008;2:251–255. [Google Scholar]
- 29.Yarapathi RV, Kurva S, Tammishetti S. Synthesis of 3, 4-dihydropyrimidin-2(1H) ones using reusable poly(4-vinylpyridine-co-divinylbenzene)-Cu(II)complex. Catalysis Communications. 2004;5:511–513. [Google Scholar]
- 30.Arslan M, Yigitoglu M, Soysal A. Removal of chromium(VI) from aqueous solutions using poly(4-vinyl pyridine) beads. Journal of Applied Polymer Science. 2006;101:2865–2870. [Google Scholar]
- 31.Ortiz-Palacios J, Cardoso J, Manero O. Production of macroporous resins for heavy-metal removal. I. Nonfunctionalized polymers. Journal of Applied Polymer Science. 2008;107:2203–2210. [Google Scholar]
- 32.Totolin V, Manolache S, Rowell RM, Denes FS. Removal of Cu + 2 ions from solution using cellulose-poly(acrylic acid) and poly(4-vinyl) pyridine based materials that were deposited using plasma technique. Journal of Natural Fibers. 2007;4:57–65. [Google Scholar]
- 33.Yavuz E, Senkal BF, Bicak N. Poly(acrylamide) grafts on spherical polyvinyl pyridine resin for removal of mercury from aqueous solutions. Reactive and Functional Polymers. 2005;65:121–125. [Google Scholar]
- 34.Kawabata N, Hayashi T, Matsumoto T. Removal of bacteria from water by adhesion to cross-linked poly(vinylpyridinium halide) Applied and Environmental Microbiology. 1983;46:203–210. doi: 10.1128/aem.46.1.203-210.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawabata N, Ohira K. Removal and recovery of organic pollutants from aquatic environment. 1. Vinylpyridine-divinylbenzene copolymer as a polymeric adsorbent for removal and recovery of phenol from aqueous solution. Environmental Science & Technology. 1979;13:1396–1402. [Google Scholar]
- 36.Kawabata N, Tsuchida Y, Nakamori Y, Kitamura M. Adsorption of phenol on clustered micro-sphere porous beads made of cross-linked poly-4-vinylpyridine. Reactive and Functional Polymers. 2006;66:1641–1648. [Google Scholar]
- 37.Kawabata N. Capture of micro-organisms and viruses by pyridinium-type polymers and application to biotechnology and water puryfication. Progress in Polymer Science. 1992;17:1–34. [Google Scholar]
- 38.Kawabata N, Hayashi T, Nishikawa M. Effect of the structure on insoluble pyridinium-type polymer on its ability to capture bacteria in water. Bulletin. Chemical Society of Japan. 1986;59:2861–2863. [Google Scholar]
- 39.Kawabata N, Nishiguchi M. Antibacterial activity of soluble pyridinium-type polymers. Applied and Environmental Microbiology. 1988;54:2532–2535. doi: 10.1128/aem.54.10.2532-2535.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kantouch A, El-Sayed AA. Polyvinyl pyridine metal complex as permanent antimicrobial finishing for viscose fabric. International Journal of Biological Macromolecules. 2008;43:451–455. doi: 10.1016/j.ijbiomac.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 41.Krishnan S, Ward RJ, Hexemer A, Sohn KE, Lee KL, Angert ER, Fischer DA, Kramer EJ, Ober CK. Surfaces of fluorinated pyridinium block copolymers with enhanced antibacterial activity. Langmuir. 2006;22:11255–11266. doi: 10.1021/la061384v. [DOI] [PubMed] [Google Scholar]
- 42.Tiller JC, Lee SB, Lewis K, Klibanov AM. Polymer surfaces derivatized with poly(vinyl-N-hexylpyridinium) kill airborne and waterborne bacteria. Biotechnology and Bioengineering. 2002;79:465–471. doi: 10.1002/bit.10299. [DOI] [PubMed] [Google Scholar]
- 43.Tiller JC, Liao C-J, Lewis K, Klibanov AM. Design surfaces that kill bacteria in contact. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5981–5985. doi: 10.1073/pnas.111143098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parker, D, Bussink, J, van de Grampel, HT, Wheatley, GW, Dorf, EU, Ostlinning, E, and Reinking, K. (2002) Ullmann’s encyclopedia of industrial chemistry, polymers, high temperature, vol. 28. Wiley-VCH, Weinheim, pp 30080
- 45.Aquil A, Vasseur S, Duguet E, Passirani C, Benoît JP, Roch ARM, Jérôme R, Jérôme C. PEO coated magnetic nanoparticles for biomedical application. Eur Polym J. 2008;44:3191–3199. [Google Scholar]
- 46.Voets IK, de Vries R, Fokkink R, Sprakel J, May RP, de Keizer A, Cohen Stuart MA. Towards a structural characterization of charge-driven polymer micelles. Eur Phys J E. 2009;30:351–359. doi: 10.1140/epje/i2009-10533-4. [DOI] [PubMed] [Google Scholar]
- 47.Dijt JC, Cohen Stuart MA, Hofman JE, Fleer GJ. Colloids and surfaces. 1990;51:141–158. [Google Scholar]
- 48.Voets IK, de Vos WA, Hofs B, de Keizer A, Cohen Stuart MA, Steitz R, Lott D. Internal structure of a thin film of mixed polymeric micelles on a solid/liquid interface. J Phys Chem B. 2008;112:6937–6945. doi: 10.1021/jp709758p. [DOI] [PubMed] [Google Scholar]
- 49.Murashov VV, Leszczynski J. Adsorption of the phosphate groups on silica hydroxyls: an ab initio study. J Phys Chem A. 1999;103:1228–1238. [Google Scholar]
- 50.Tao WA, Gilpin RK. Liquid chromatographic studies of the effect of phosphate on the binding properties of silica-immobilized bovine serum albumin. J Chromatogr Sci. 2001;39:205–212. doi: 10.1093/chromsci/39.5.205. [DOI] [PubMed] [Google Scholar]
- 51.Ball V, Winterhalter M, Schwinte P, Lavalle P, Voegel JC, Schaaf P. Complexation mechanism of bovine serum albumin and poly(allylamine hydrochloride) J Phys Chem B. 2002;106:2357–2364. [Google Scholar]