

Abstract
Amphiphilic block copolymers consisting of hydrophilic poly(ethylene glycol) and hydrophobic polyester bearing pendent cyclic ketals were synthesized by ring-opening copolymerization of ε-caprolactone (CL) and 1,4,8-trioxaspiro-[4,6]-9-undecanone (TSU) using α-hydroxyl, ω-methoxy, polyethylene glycol as the initiator and stannous octoate as the catalyst. Compositional analyses indicate that TSU was randomly distributed in the hydrophobic blocks. When the TSU content in the copolymers increased, the polymer crystallinity decreased progressively and the glass transition temperature increased accordingly. Hydrophobic, anticancer drug, camptothecin (CPT), was successfully encapsulated in the block copolymer nanoparticles. The CPT encapsulation efficiency and release kinetics were strongly dependent on the copolymer composition and crystallinity. CPT release from nanoparticles constructed from copolymers containing 0, 39 and 100 mol% TSU in the hydrophobic block followed the same trend, with an initial burst of ~40% within one day followed by a moderate and slow release lasting up to 7 days. At a TSU content of 14 mol%, CPT was released in a continuous and controlled fashion with a reduced initial burst and a 73% cumulative release by day 7. In vitro cytoxicity assay showed that the blank nanoparticles were not toxic to the cultured bone metastatic prostate cancer cells (C4-2B). Compared to the free drug, the encapsulated CPT was more effective in inducing apoptotic responses in C4-2B cells. Modulating the physical characteristics of the amphiphilic copolymers via copolymerization offers a facile method for controlling the bioavailability of anticancer drugs ultimately increasing effectiveness and minimizing toxicity.
Keywords: Amphiphilic block copolymer, cyclic ketal, crystallinity, nanoparticles, controlled release, camptothecin
1. Introduction
Polymeric nanoparticles are attractive delivery vehicles for cytotoxic drugs for cancer treatment because they can be engineered to exhibit maximized anti-cancer activities, reduced side effects and a controlled release over a prolonged period of time[1–3]. Packaging anti-cancer drugs into polymeric nanoparticles offers an alternative strategy to enhance drug stability, increase drug solubility and simultaneously improve pharmacokinetics and pharmacodynamics[4, 5]. Due to their inherent biocompatibility, block copolymers based on poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG-b-PCL) have been investigated widely as carrier systems for hydrophobic drugs via their preferential association with the hydrophobic PCL segments. The presence of the hydrophilic PEG block not only stabilizes the particles, but also avoids undesirable systemic clearance of the nanoparticles[6–9].
Many physical, chemical and biological barriers associated with PEG-b-PCL based drug delivery systems have to date limited their successful implementation in clinical protocols[10–12]. In particular, the crystalline nature of PCL prevents homogeneous distribution of the drugs within the particle interior. As a result, undesirable drug aggregation within the particles or on the particle surface has been unavoidable[13, 14]. Furthermore, the lack of variation in the chemical structure in the PCL backbone has prohibited the drug encapsulation and its release profile from being modulated readily. Altogether, these limitations contribute to low drug encapsulation efficiency and uncontrolled burst release kinetics that can produce detrimental side effects to healthy tissues. We hypothesize that alteration of the cyrstallinity of the polymeric nanocarriers offers a simple and adaptive method to fine-tune the pharmacokinetics of delivery of cancer therapeutics. Previous investigations have shown that the introduction of pendent cyclic ketal groups to linear PCL can effectively disrupt its crystalline structure and reduce its overall crystallinity[15]. Thus, by incorporating the bulky cyclic ketals to the hydrophobic segments of the amphiphilic block copolymers, stable nanoparticulate systems with intermediate crystallinity, enhanced drug-polymer interaction and reduced burst release can be formulated.
We are interested in studying the effects of the polymer composition and crystallinity on the encapsulation and release of camptothecin (CPT), a model hydrophobic drug for these studies. CPT is a classical chemotherapeutic that effectively triggers an apoptotic response in cancer cells by inhibiting DNA topoisomerase I activity [16, 17]. However, CPT is difficult to administer clinically because of its poor water-solubility and instability at neutral pH [16, 18]. In this study, we report a straightforward approach for efficient integration of CPT into polymeric nanoparticles in order to improve the drug stability and bioavailability. To this end, amphiphilic block copolymers carrying pendent cyclic ketals were synthesized by ring opening polymerization of ε-caprolactone (ε-CL) and 1,4,8-trioxaspiro-[4,6]-9-undecanone (TSU) using methoxy poly(ethylene glycol) (mPEG) as the initiator and stannous octoate (Sn(Oct)2) as the catalyst. A series of copolyesters with varying amounts of TSU were obtained by adjusting the monomer feed ratio. The polymer composition and molecular weight were characterized by nuclear magnetic resonance (NMR) and size exclusion chromatography (SEC), while their thermal properties and physical state were evaluated using differential scanning calorimetry (DSC) and wide angle X-ray diffraction (WAXD). CTP-loaded nanoparticles prepared via a nanoprecipitation process were analyzed in terms of their morphology and particle size as well as CPT localization in the particles. The effects of block copolymer composition and chain flexibility on CPT loading and release were investigated systematically. Cytotoxicity of blank nanoparticles and CPT-loaded nanoparticles was tested in vitro against LNCaP cells that are susceptible to apoptosis, as well as PC3 cells that are relatively resistant to chemotherapy.
2. Experimental Section
2.1. Materials
All chemicals were obtained from Sigma-Aldrich (Milwaukee, WI) and were used as received unless otherwise indicated. Alpha-hydroxyl, ω-methoxy, poly(ethylene glycol) (mPEG, Mn = 5,000 g/mol) was dried under vacuum at 100 °C in the presence of anhydrous phosphorous pentoxide (P2O5) overnight prior to use. Epsilon-caprolactone (ε-CL) was dried over calcium hydride (CaH2) for 48 h at room temperature and was distilled under reduced pressure just before use. PC-3 cells, T-medium and F-12K-medium were purchased from ATCC (Manassas, VA). All other cell culture reagents, SYTO-13 and propidium iodide nucleic acid stains were purchased from Invitrogen (Carlsbad, CA).
2.2. Polymer Synthesis
Caprolactone derivative containing a cyclic ketal, 1,4,8-trioxaspiro[4,6]-9-undecanone (TSU), was synthesized and characterized following previously reported procedures[15, 19]. Amphiphilic block copolymers were prepared by ring opening polymerization (ROP) of CL and TSU using mPEG as the initiator and Sn(Oct)2 as the catalyst. In all polymerizations, the amount of mPEG and the total amount of monomer were maintained constant while the monomer feed ratio was varied systematically (Table 1). Briefly, mPEG, ε-CL and TSU were placed in an oven-dried, round-bottom flask that was connected to a schlenk line. Sn(Oct)2 (0.1 mol% relative to the free –OH groups in mPEG) was dissolved in anhydrous toluene and was added drop-wise to the monomer/initiator mixture. The reactants were azeotropically dried under reduced pressure at 70 °C for 2 h. The mixture was heated subsequently to 110 °C and was maintained at this temperature under vacuum with constant stirring for 24 h. The polymerization was terminated by cooling the reaction mixture to room temperature. The polymerization product was solubilized in dichloromethane and precipitated into a large excess of hexane repeatedly. The precipitated copolymer was isolated by centrifugation at 4000 RPM for 10 min and was dried at room temperature under vacuum for 48 h. The products were stored in a desiccator containing P2O5 under vacuum. The resulting copolymers are abbreviated as ECTx, with x being the amount (in grams) of TSU added to the reaction mixture (Table 1).
Table 1.
Characteristics of ECT copolymers synthesized by ring opening copolymerization of ε-CL and TSU using mPEG (Mn=2,000 g/mol) as the initiator, and Sn(Oct)2 (0.1mol% of the hydroxyl group in mPEG) as the catalyst. The polymerization was carried out in bulk at 110 °C for 24 h.
| ID | Feed ratio (g) | Yield (%) | Polymer compositiona | Mnb (kg/mol) | Mwb (kg/mol) | PDIb | ||
|---|---|---|---|---|---|---|---|---|
| PEG | CL | TSU | ||||||
| ECT0 | 1.0 | 10.0 | 0.0 | 87.3 | EG113CL427 | 56.7 | 78.3 | 1.38 |
| ECT2 | 1.0 | 8.0 | 2.0 | 85.2 | EG113CL328TSU55 | 44.9 | 54.8 | 1.22 |
| ECT5 | 1.0 | 5.0 | 5.0 | 78.2 | EG113CL208TSU133 | 44.7 | 59.1 | 1.32 |
| ECT10 | 1.0 | 0 | 10.0 | 57.4 | EG113TSU261 | 44.0 | 58.9 | 1.34 |
Composition of CL and TSU calculated by 1H NMR.
Mn, Mw, and PDI (polydispersity index) determined by GPC.
2.3. Nanoparticle Preparation and Drug Encapsulation
Drug-free nanoparticles were prepared using the acetone/water solvent mixture. Briefly, polymer dissolved in acetone (10 mg/mL) was injected slowly into a magnetically stirred (500 RPM) aqueous phase of 5 mL DI water. Acetone was allowed to evaporate overnight under constant stirring at room temperature. CPT-loaded nanoparticles were prepared using the THF/DMSO/H2O solvent mixture[13]. Polymer and CPT were dissolved in THF/DMSO (v/v 4/1) at concentrations of 10 mg/mL and 0.1 mg/mL, respectively. The organic phase (1 mL), prepared by mixing the above solutions at a volume ratio of 1/2, was added subsequently drop-wise to DI H2O (5 mL) under gentle stirring at room temperature. After extensive dialysis against multiple exchanges of DI H2O for 24 h, the resulting dispersion was centrifuged at 4000 RPM for 25 min to eliminate drug and polymer aggregates. The supernatant was collected for further characterizations. Freshly prepared nanoparticles were immediately used for in vitro release and cytotoxicity experiments.
2.4. Chemical and Physical Characterizations
2.4.1. Chemical Composition and Molecular Weight
1H and 13C NMR spectra were recorded on a Bruker DRX-400 spectrometer at room temperature. Polymers were dissolved in CDCl3 and tetramethylsilane (TMS) was used as the internal reference with a chemical shift of 0 ppm. 13C NMR spectra were obtained at room temperature with 4096 scans, an acquisition time 1.8 s, pulse width 2.2 μs (22.5°), and a delay of 3 s between pulses. Polymer molecular weight and polydispersity index (PDI) were determined by size exclusion chromatography (GPC) on a Waters GPC equipped with a Waters 515 HPLC pump, a Waters 2414 refractive index detector, a Waters 2996 photodiode array detector and two Styragel columns (HR1 and HR4; 7.8 × 300 mm for each) packed with 5 μm particles capable of measuring molecular weight in the range of 100–5,000 g/mol for HR1 and 5,000–500,000 for HR4, respectively. THF was used as a mobile phase with a flow rate of 1.0 mL/min. The measurements were carried out at 40 °C with an injection volume of 20 μL. Sample solutions were filtered through a 0.2 μm filter before the injection. Data was collected and analyzed with Waters Empower software. Narrow polystyrene standards in the range of 1,700–706,000 g/mol (Polysciences Inc., Warrington, PA) were used for the calibration.
2.4.2. Thermal Properties
Differential scanning calorimetry (DSC) was performed on a Mettler Toledo DSC (823e) calibrated with Indium and Gallium. The melting temperature (Tm) and the melting enthalpy (ΔHm) were recorded from the second scan between −10 and 200°C in a nitrogen atmosphere at a heating rate of 10°C/min. The glass transition temperature (Tg) was determined from the midpoint of the transition zone during the second heating cycle from −75 to −0 °C.
2.4.3. Crystallinity
Wide angle X-ray diffraction (WAXD) was carried out using a Rigaku X-ray diffractometer with a Ni-filtered Cu Kα radiation (λ = 0.1546 nm) at room temperature at a scan rate of 4 °/min. The selected voltage and current are 40 kV and 20 mA, respectively. Three drops of polymer solution (1 wt % in chloroform) were added to a clean cover glass and were allowed to air-dry at room temperature for 2 days followed by vacuum drying for 2 days. Drug-free and CPT-loaded nanoparticles were freeze-dried before WAXD analysis. CPT powder was used directly for WAXD measurement.
2.4.4. Particle Size
The average diameter of the blank and CPT-loaded nanoparticles (dispersed in DI H2O) was determined by a Malvern Zetasizer nanoZS apparatus (Malvern Instruments Ltd.). Z-average particle size and the polydispersity index (PDI) were measured at 25°C using dynamic light scattering combined with Malvern’s DTS software (v.5.02).
2.4.5. Morphological Examination
Blank ECT nanoparticles were visualized using a cryogenic scanning electron microscope (CryoSEM) following previously reported procedures[20, 21]. Briefly, the aqueous particle suspension was subjected to rapid freezing (> 10,000 deg/s) under high pressure (>2000 bar). The frozen sample was immediately transferred to the preparation chamber (Gatan ALTO 2500) in liquid nitrogen slush under vacuum, where water was allowed to sublime at −90 °C for 5 min. The dry sample was subsequently coated with gold-palladium at −125 °C, and visualized in a Hitachi S-4700 FESEM (Tokyo, Japan) at 1 kV with an emission current of 30 μA and a working distance of 3–6 mm.
2.5. CPT Loading and Release
2.5.1. CPT Loading
To determine the drug content in the nanoparticles, an aliquot of drug-loaded nanoparticle suspension was withdrawn and freeze-dried. The dry powder was weighted accurately before being thoroughly dissolved in 1 mL of THF/DMSO (v/v, 4/1). The CPT concentration in the organic solvent was determined by a FluoroMax-3 spectrofluorometer at an excitation wavelength of 374 nm and an emission wavelength of 432 nm [18, 22, 23]. The encapsulation efficiency (as a % of total) was calculated by dividing the amount of CPT loaded into the particles with the amount of CPT initially added during the particle preparation. The drug loading efficiency was defined as the amount of CPT (mg) loaded per gram of the polymeric nanoparticles. All measurements were performed in triplicate, and the results were expressed as the mean ± standard deviation.
2.5.2. In vitro Release
In order to avoid the dissolution saturation of CPT in the release medium, CPT-loaded nanoparticles were diluted with PBS to a final concentration such that the maximum CPT concentration in the release media is approximately 2 μg/mL. The particle suspension (3 mL) then was loaded into a hydrated dialysis cassette with a molecular weight cut-off of 10 kDa. The cassette was subsequently immersed in the release media (500 mL PBS) that was gently agitated at room temperature. At a predetermined time, 50 mL of the release media was withdrawn and was replaced with 50 mL of fresh PBS to maintain a constant volume of 500 mL. The CPT concentration in the release media was determined by a FluoroMax-3 spectrofluorometer as described above. All analyses were carried out in triplicate and the cumulative release (as a % of total) was calculated by dividing the amount of CPT released up to a chosen measurement time with the total amount of CPT initially loaded.
2.6. In Vitro Cell Toxicity Assay
C4-2B cells [24] were maintained in T-75 tissue culture flasks at 37°C in 5.0% (v/v) CO2 in T-medium supplemented with 5% (v/v) fetal bovine serum (FBS) and 100 U/ml penicillin G sodium and 100 μg/ml streptomycin sulfate in 0.085% (w/v) saline (PS) as described previously 25]. PC-3 cells were cultured under the same conditions in F-12K medium containing 10% (v/v) FBS. Medium was changed every other day. Cells were passaged routinely using 0.25% (w/v) trypsin with EDTA 4· Na. Trypsinized cells (C4-2B: 1 × 105 cells per well; PC-3: 5 × 104 cells per well) were suspended in 1 mL of culture medium and plated into a 24 well plate for the apoptosis assay or a 4 well Lab-Tek II Chambered Coverglass (Nalgene Nunc, Napersville, IL) for live/dead stainings. Cultures were maintained for 24 h prior to the addition of free CPT or CPT-loaded ECT2 nanoparticles. CPT-loaded nanoparticles were prepared in filtered (0.4 μm) DI H2O and added to the medium to produce a final CPT concentration (based on the initial loading) varying from 0 to 0.4 μg/ml for C4-2B cells and 0 to 1.6 μg/mL for PC-3 cells. Separately, free CPT, dissolved in DMSO and diluted with filtered DI H2O, was added to the cell culture medium to afford a final CPT concentration in the same range. After 24 h incubation, cells and medium were collected in 1.5 mL microcentrifuge tubes. C4-2B cells were collected by gentle pipetting while PC-3 cells were released by 0.25% (w/v) trypsin (100 μl) for 5 minutes. In both cases, cells were collected by centrifugation for 10 min at 5000 RPM. Apoptosis was detected using a Cell Death Detection ELISAPLUS kit (Roche Applied Science, Mannheim, Germany) following previously reported protocols [5]. Colorimetric detection was carried out according to manufacturer’s instructions using MRX microplate reader (Dynex Technologies, Chantilly, VA) at 405 nm excitation/490 nm emission. The level of apoptosis was expressed as the ratio between the colorimetric readout from CPT-treated cells and that from cells cultured under the same conditions in normal medium. Vehicle controls (water: 5% (v/v), DMSO: 0.1% (v/v), and blank nanoparticles: (95 μg/mL) were also subjected to the same apoptosis assay. Cells cultured in the presence of blank nanoparticles and the filtered water were further subjected to live/dead assay. After 24 h, the medium was removed and 1 mL of 1:1000 (v/v) each SYTO-13 and propidium iodide (PI) in PBS was applied to the cells. The cultures were incubated for 15 minutes then visualized using confocal microscopy on a Zeiss LSM 510 VIS (Carl Zeiss, Maple Grove, MN).
Statistical Analysis
Statistical data analysis was performed using the student t-test with p<0.05 as the minimal level of accepted significance.
3. Results and Discussion
Instead of resorting to sophisticated chemical conjugation approaches [26, 27], we elect to investigate the potential of controlling CPT encapsulation and release via the alteration of the physical characteristics (i.e. crystallinity) of the drug carriers. This was accomplished by incorporating a bulky cyclic ketal group to an amphiphilic block copolymer based on mPEG-b-PCL. Although ring opening polymerization of TSU has been reported previously[19], TSU-containing, amphiphilic block copolymers have not been synthesized and their utility as nanoparticles for cancer drug delivery has not been demonstrated.
3.1. Polymer Synthesis
A series of amphiphilic block copolymers were synthesized successfully by ring opening copolymerization of CL and TSU using the terminal hydroxyl of mPEG (Mn 5,000 g/mol) as the initiator and Sn(Oct)2 as the catalyst (Scheme 1) via a coordination-insertion mechanism (Scheme 1) [28]. Copolymers were prepared with a TSU/CL feed ratio (w/w) of 0:10, 2:8, 5:5 and 10:0, while keeping the amount of the initiator and the catalyst constant (Table 1). The resulting polymers are denoted as ECTx, where x is the weight of TSU used in feed. 1H NMR spectra for the copolymers show distinct peaks from the constituent building blocks, and the relative peak intensity increases as the amount of TSU in the monomer feed increases accordingly (Figure 1). 1H NMR spectrum for ECT0 shows representative methylene signals from the CL repeating units (-CH2CH2CH2CH2CH2COO- at 1.65 ppm and -CH2OCO- at δ 4.06 ppm). Characteristic peaks from TSU repeating units, including the ethylene ketal signals (-OCH2CH2O- at 3.95 ppm) and the methylene protons adjacent to the cyclic ketal (k+j in Figure 1) at 1.95–2.05 ppm clearly are visible on the spectrum of ECT10. The representative methylene protons (-CH2CH2O-, δ = 3.64 ppm) from mPEG repeating units are present in all copolymers. The copolymer composition and molecular weight were calculated by comparing the relative intensity of the methylene protons from mPEG at 3.64 ppm and the typical proton signals for each monomer (δ 3.95 ppm, 4H, for TSU and δ 4.06 ppm, 2H, for CL) on the basis of PEG’s molecular weight. Our results (Table 1) show that the chemical compositions of the copolymers can be readily controlled by varying the monomer feed ratio. Amphiphilic copolymers with TSU content in the hydrophobic block varying from 0, 14, 39 to 100 mol% for ECT0, ECT2, ECT5, and ECT10 were synthesized successfully.
Scheme 1.

Synthesis of TSU containing block copolymers via ring opening copolymerization of ε-CL and TSU using mPEG as the initiator and Sn(Oct)2 as the catalyst.
Figure 1.

1H NMR spectra of ECT copolymers with varying TSU content. Polymer composition and molecular weight were calculated by comparing integrals of characteristic peaks of the PEG blocks at 3.64 ppm and the typical protons for the monomers (3.95 ppm for TSU and 4.06 ppm for ε-CL). The molecular weight of PEG served as the reference.
The molecular weight and PDI of the ECT copolymers also were estimated by SEC using THF as the mobile phase and polystyrene as the molecular weight standards. The results are in good agreement with the 1H NMR analysis. The SEC chromatograph (Figure 3) of a representative copolymer ECT2 shows a monomodal distribution of various molecular weight species, demonstrating the absence of homopolymers. The TSU-containing copolymers investigated in this study exhibit similar molecular weight of ~44 kg/mol and polydispersity while the molecular weight of ECT0 is slightly higher (~56 kg/mol). Finally, 13C NMR spectra were acquired in order to determine the specific sequences of CL and TSU in the hydrophobic block[29]. 13C NMR spectra of ECT copolymers exhibited several well-separated peaks in the carbonyl region (from δ 173.8 to 173.0 ppm, Figure 2) that were sensitive to the chemical environment surrounding the carbonyl carbon. For ECT0, only one peak was observed for the carbonyl carbon at δ of 173.58 ppm. This peak can be assigned to the CL unit adjacent to another CL unit in the polymer. Similarly, only one peak located at δ of 173.30 ppm was detected for ECT10 due to the universal presence of TSU-TSU sequences in this copolymer. When both CL and TSU units are present, a splitting of the carbonyl signals was obvious. Signals at 175.58, 173.44 and 173.30 ppm correspond to CL-CL, CL-TSU, and TSU-TSU sequences; and their relative intensities correlate to the chemical composition of the copolymer. These results indicate that the CL and TSU units are randomly distributed in the hydrophobic blocks of the ECT copolymers.
Figure 3.

SEC chromatograph of a typical copolymer (ECT2). THF was used as the mobile phase. Mn = 44,900 g/mol; PDI = 1.22.
Figure 2.

13C NMR spectra (the carbonyl region) of ECT copolymers with different compositions showing a random distribution of CL and TSU units in the hydrophobic block.
Collectively, our results demonstrate that the terminal OH group of mPEG is capable of ring opening copolymerization of CL and TSU simultaneously, producing in a random distribution of TSU and CL units along the polymer backbone. The amount of TSU/CL incorporated easily was modulated by the monomer feed ratio and the molecular weight of the block copolymers was controlled by the relative concentration of monomers and the initiator. The presence of a bulky cyclic ketal group in TSU did not reduce significantly the reactivity of TSU towards ring opening copolymerization with CL, in agreement with results from previous studies[19]. Therefore, random copolymerization of CL and TSU initiated by mPEG offers a straightforward method to create novel amphiphilic copolyesters whose material properties are significantly different from those of the parent homopolymers (see below).
3.2. Thermal Transitions and Crystallinity
Because drug encapsulation and release are strongly dependent on the physical properties of the polymeric nanocarriers, the ECT copolymers were characterized by DSC and WAXD prior to the particle preparation and CPT loading. DSC thermograph (Figure 4) of ECT0 revealed a broad transition centered around −60 °C that is characteristic of the glass transition temperature (Tg) of the PCL block[28]. The Tg for the PEG block was not detected due to its relatively short length in comparison to the hydrophobic block (Table 1). With the addition of TSU in the copolymer, the glass transition temperature progressively approached that for a copolymer with 100% TSU content in the hydrophobic blocks. Compared to PCL in ECT0, PTSU exhibited a much higher Tg (−25 °C) in ECT10. The presence of a single Tg confirms the miscibility of the amorphous phase and reinforces the notion that the CL and TSU repeats are randomly distributed in the copolymers. The elevation of Tg with the increase in TSU incorporation can be attributed to the enhanced dipole-dipole interactions and the bulkiness of the cyclic ketal side group, both of which effectively restrict the rotational motion of the polymers [30, 31].
Figure 4.

DSC thermographs (second scan) of ECT copolymers showing the evolution of the glass transition temperature (top) and the melting temperature/melting enthalpy (bottom) as a function of copolymer composition. Heating rate: 10 °C/min.
A moderate degree of crystallinity contributes to nanoparticle stability and confers greater drug retention by decreasing drug diffusion from the particle interior during the encapsulation process [5]. It can be seen from Figure 4 that while ECT0 is semi-crystalline with a distinct melting transition around 40–52°, ECT10 is completely amorphous with no detectable melting endotherm. Due to its higher molecular weight in comparison to mPEG, the PCL block was anticipated to crystallize first, effectively restricting the crystallization of the PEG blocks[32]. With the exception of ECT10, all other ECT copolymers exhibit two melting peaks (Tm1 and Tm2) and their locations and intensity shifted as the copolymer composition changed (Table 2). An increase in TSU content in the copolymers led to a gradual decrease Tm2. The presence of two melting peaks can be attributed to the difference in the chain fold number in the lamellae and/or to the formation of imperfect crystal structures of varying sizes [33]. When TSU content in the hydrophobic block increases, the overall melting enthalpy decreases accordingly (Table 2). The random insertion of TSU in the hydrophobic block essentially hindered the folding and close packing of the PCL chains, producing smaller and less defined crystal structures with reduced overall polymer crystallinity.
Table 2.
Characteristics of ECT copolymers and nanoparticles.
| ID | Tga (°C) | Tma |
ΔHma (J/g) | Particle size (nm)b |
LEc (mg/g) | EEc (%) | ||
|---|---|---|---|---|---|---|---|---|
| Tm1 (°C) | Tm2 (°C) | NP | CPT-NP | |||||
| ECT0 | −62.5 | 40.2 | 51.7 | −89.24 | 106 | 190 | 16.4±1.3 | 84.5±3.7 |
| ECT2 | −59.7 | 45.7 | 50.1 | −62.61 | 104 | 126 | 17.2±0.5 | 82.2±6.4 |
| ECT5 | −48.8 | 38.8 | 48.2 | −14.7 | 118 | 187 | 9.6±1.5 | 48.7±7.7 |
| ECT10 | −26.1 | - | - | −2.23 | 120 | 190 | 12.3±0.8 | 61.7±4.2 |
Glass transition temperature (Tg) and melting temperature (Tm) were determined from the second heating cycle. The melting enthalpy (ΔH) was calculated by integrating the corresponding melting peaks.
Mean particle diameter of blank NP and CPT-loaded NP determined by dynamic light scattering. Three measurements on the same NP compositions gave perfectly overlapping histograms.
LE: loading efficiency; EE: encapsulation efficiency. Values reported are mean ± standard deviation (n=3).
The WAXD pattern for ECT0 (Figure 5) shows the characteristic peaks at 2θ of 21.3, 22.0, and 23.7° for the crystalline PCL blocks and lacks the typical PEG crystal patterns [32], further confirming the inability of PEG to crystallize in ECT copolymers. When TSU was present in the copolymer at a moderate amount, the crystalline peaks of PCL could be detected, although at a lower intensity. When the TSU content exceeds a certain threshold (ECT5), the ability of PCL segments to crystallize is severely hindered. With 100% TSU (ECT10) in the hydrophobic block, the polymeric powder projects a diffuse, amorphous halo in WAXD. These results further confirm the disruption of the regularity of PCL molecular structure by the random insertion of TSU units in the hydrophobic block, hindering the ability of PCL to crystallize [32]. The DSC and WAXD results were substantiated further by visual observation. While ECT0 was obtained as a white powder, ECT5 appeared as a waxy solid, and ECT10 was a tacky material even though the copolymers have comparable molecular weights.
Figure 5.

WAXD patterns of ECT copolymers with different compositions. With an increase in TSU content, the characteristic diffraction peaks for PCL decrease in intensity. Only a diffuse and amorphous halo was detected for ECT5 and ECT10.
3.3. Nanoparticle Formation and Drug Encapsulation
We are interested in utilizing ECT copolymers as the nanocarriers for the delivery and controlled release of CPT or other hydrophobic drugs. The amphiphilic nature of these copolyesters allows for the formation of stable colloidal nanoparticles with stealth PEG shells. Drug-free nanoparticles were assembled using an acetone/water solvent system [34]. Particle size analysis by DLS indicates that the blank particles exhibit an average diameter of 104 nm to 120 nm, depending on the polymer composition (Figure 6). The CryoSEM (Figure 7) revealed the presence of spherical nanoparticles with narrow size distribution and smooth surfaces. Our particles can be produced reproducibly in large quantities and are relatively stable because long term storage in an aqueous media did not significantly change the overall particle size (data not shown).
Figure 6.

DLS histograms of blank nanoparticles (top) and CPT-loaded nanoparticles (bottom).
Figure 7.

CryoSEM image of blank ECT2 nanoparticles assembled from ECT2. The nanoparticles are spherical in shape and exhibit a relative narrow size distribution and smooth surfaces.
The initial attempt to encapsulate CPT into ECT nanoparticles using an acetone/water mixed solvent system failed due to CPT’s poor solubility and its strong tendency to self-aggregate. CPT was loaded successfully into the polymeric nanoparticles using a DMSO/THF/H2O solvent system. Optimal particle formation and drug encapsulation were achieved when the CPT/polymer feed ratio was 2 wt% and the copolymer concentration in the organic phase was 10 mg/mL. The improved solubility of ECT polymers in DMSO/THF produced a more swollen hydrophobic environment, resulted in a larger average particle diameter as compared to the blank particles prepared in acetone/water. Overall, CPT-loaded nanoparticles exhibited narrower particle size distribution than did the corresponding blank particles (Figure 6). Compared to other polymer compositions (ECT0, ECT5 and ECT10), ECT2 gave rise to nanoparticles of smaller diameter (Figure 6, Table 2). The CPT-loaded nanoparticles were stable upon dilution and prolonged incubation under the experimental conditions employed in this study.
CPT-loaded nanoparticles, along with free CPT and the blank particles, were subjected to WAXD analysis (Figure 8) in order to assess the physical state of the drug incorporated in the nanoparticles [13, 35]. While the blank ECT2 and CPT-loaded ECT2 nanoparticles showed the diffraction peaks of PCL segments, the characteristic diffraction pattern for free CPT was missing in CPT-loaded ECT2 nanoparticles. The absence of CPT signals from CPT-loaded nanoparticles suggests the molecular dispersion of CPT in the nanoparticles. Alternatively, the amount of CPT encapsulated (see below) in the nanoparticles may be beyond the detection limit of the diffractometer.
Figure 8.
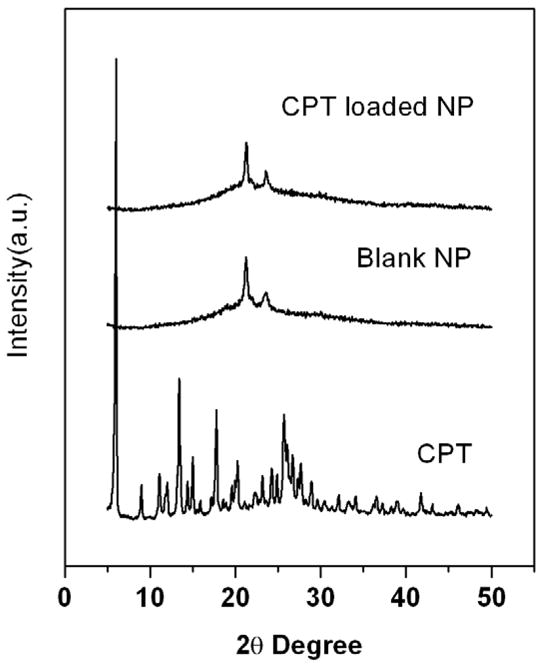
WAXD diagrams of free CPT (bottom), blank nanoparticles (middle) and CPT-loaded nanoparticles assembled from ECT2 (top).
The presence of a conjugated pyridine moiety in CPT allows its concentration to be accurately determined by spectrofluorometer [18, 22, 23]. No CPT precipitation was expected under the experimental conditions employed. While the highest CPT loading efficiency was observed for polymers with the lowest TSU incorporation, namely ECT0 and ECT2 (16.4 ± 1.3 mg/g and 17.2 ± 0.5 mg/g, respectively), polymers containing 113 TSU repeating units (ECT5) exhibited the lowest CPT loading. An intermediate CPT loading was observed for polymers with 100% TSU in the hydrophobic block (ECT10). The encapsulation efficiency followed the same trend. Collectively, our results suggest that CPT was entrapped effectively within the hydrophobic interior of the nanoparticles assembled from ECT2. The effective compartmentalization of CPT by ECT2 nanoparticles, as well as their smaller average particle dimension compared to other compositions, was the direct result of a perfect balance of crystallinity and chain dynamics in ECT2.
Factors that affect drug encapsulation in amphiphilic block copolymer nanoparticles include (1) the crystallinity and viscosity of the hydrophobic segments; (2) the volume of the hydrophobic blocks; (3) drug solubility and drug association in water; and (4) the affinity of the loaded drug with the hydrophobic blocks [13, 36]. The hydrophobic blocks of all ECT copolymers investigated here exhibit comparable length. We do not anticipate significant difference in the affinities of CPT with CL and TSU units either. In our case, the physical characteristics of the copolymers play an important role in modulating CPT encapsulation. Considering ECT5 and ECT10, the significant reduction in polymer crystallinity creates more opportunity for CPT molecules to disperse in the dynamic polymer matrix. On the other hand, the reduction in particle crystallinity inevitably makes it difficult to condense the polymer chains into the confined spherical geometry, giving rise to larger overall particles. Similarly, the dynamic nature of the hydrophobic interior permits the free diffusion of CPT into the surrounding media during the encapsulation process, rendering the overall CPT retention in these particles relatively low. It was anticipated that the homogeneous dispersion of CPT in ECT0 nanoparticles was significantly hindered due to the high crystallinity of ECT0. The relatively high loading efficiency seen with ECT0 is not surprising because CPT can form large and relatively stable crystalline aggregates within the nanoparticles at or near the particle surface. With only 14 mol% TSU incorporation, ECT2 can foster homogeneous drug distribution within the nanoparticles and maintain sufficient particle stability, a property attributable to the presence of microcrystalline domains that reinforce the particles. Nanoparticles prepared from ECT2 showed the highest loading and encapsulation efficiency among all TSU-containing polymers. Although ECT0 and ECT2 have comparable loading and encapsulation efficiencies, their release kinetics are significantly different (see below). We note that the low drug loading capacity may render the current formulation impractical for in vivo applications. Careful selection of the solvent pairs that maximize the drug solubility or the introduction of aromatic moieties to the polymer side chains that foster drug polymer interactions may lead to enhanced drug loading.
3.4. In vitro release
In vitro CPT release from ECT nanoparticles was evaluated by incubating CPT-loaded nanoparticles in a physiological buffer under sink conditions for up to 7 days. The results are summarized in Figure 9. It is worth mentioning that the amount of CPT loaded into the polymers, if completely released, was well below the saturation limit of CPT in aqueous media [24]. Interestingly, the release profiles for ECT0, 5 and 10 overlap with each other despite their different chemical compositions. Approximately 56–60% CPT was released during the first two days for ECT0, 5 and 10. In all cases, a burst release was followed by a slower and more moderate release after day 2. Over 90% CPT initially loaded was released at day 7. Such release kinetics is very similar to literature reported results [37, 38]. For ECT0, the burst release can be attributed to the drug that is aggregated at the periphery of the crystalline region that can be readily released upon exposure to an aqueous media. The reduced crystallinity and increased fluidity for ECT5 and ECT10 nanoparticles led to poor drug retention in the particles and preferential drug deposition at the regions near and on the hydrophilic corona. Under these circumstances, the drug molecules do not have to overcome any diffusion barrier once the drug-loaded particles are exposed to the aqueous medium.
Figure 9.

In vitro release profiles of CPT from ECT nanoparticles in PBS (pH 7.4) at ambient temperature.
A sustained release with a reduced initial burst was observed for nanoparticles derived from ECT2, in sharp contrast to a previous report in which severe burst release was observed [39]. After one day of incubation, only 25% of the drug initially loaded was released. The improved drug retention and uniform drug distribution within the nanoparticles effectively diminished the initial burst release. From day 2 to day 4, CPT was released from ECT2 at a linear rate of approximately 5% per day. Thereafter, a higher rate of release was observed (9% per day) until day 7, when the experiment was terminated. Importantly, only 73% CPT was released from ECT2, at day 7, in contrast to over 90% from other formulations.
Factors that control drug encapsulation also play important roles in modulating the drug release kinetics. In order for the encapsulated drug to be released into the media, the drug molecules have to overcome the diffusion barriers established by the polymeric matrix. Because no polymer degradation was observed during the course of the release study, the different physical state of both the associating segments and the drug was the overriding factor. For ECT0, CPT was excluded from the crystalline, hydrophobic interior, potentially forming microscopic aggregate close to the short PEG shell. Therefore, CPT molecules did not have to overcome a significant diffusion barrier in order to be released into the media. On the other hand, when the crystallinity is too low, the particle is much less compact and more diffuse. Consequently, the encapsulated CPT is exposed readily to the aqueous environment once the particle interior is swollen and can be readily released. When the nanoparticles exhibit an intermediate crystallinity, CPT is homogeneously dispersed in the amorphous regions of the particle interior that is stabilized by the presence of residual crystalline structure of the PCL block. In fact, the microcrystals serve as a physical crosslink to stabilize the nanoparticle.
For ECT2, the first stage of release (day 1) corresponds to the initial burst due to the small amount of CPT physically adsorbed near the PEG shell. The second stage of relatively slow release can be attributed to the period when CPT had to overcome diffusion barriers and meander through the amorphous regions of the semi-crystalline interior. Once over 45% of CPT was released, the interior compartment of the nanoparticles became more accessible to the release medium, facilitating further particle swelling. The rate of CPT release from ECT2 during the last four days is statistically higher (p<0.05) than that from ECT0, 5 and 10. As mentioned before, particles formed from ECT2 were the smallest (120 nm). Smaller particles have more surface area per volume, thus theoretically more rapid release. The observation that ECT2 exerts the most control over the release of CPT reinforces the notion that a perfect balance of particle crystallinity and chain flexibility is needed in order to allow for a controlled release of the encapsulated drug.
In Vitro Cell Viability
To assess the applicability of CPT-loaded ECT nanoparticles for the treatment of osteoblastic bony lesions of prostate cancer, C4-2B cells derived from the LNCaP series of prostate cancer cells were used to determine the toxicity of blank and CPT-loaded nanoparticles [24]. Because ECT2 exhibited the best control over CPT release under the experimental conditions employed in the current study, their toxicity was assessed. The blank ECT2 nanoparticles at the highest concentration (95 μg/mL) did not elicit any toxic effects to the cells after 24 h of incubation, as evidenced by the healthy cell morphology and the predominantly green staining (live cells) in Figure 10A and 10B. Apoptosis assay results agree well with the live/dead assay, confirming that all vehicle controls, including filtered DI H2O (5%, v/v), DMSO (0.1%, v/v) and the blank nanoparticles (95 μg/mL), were essentially non-cytotoxic to C4-2B cells (Figure 10C). Treatment of C4-2B cells with an increasing concentration of free CPT or CPT-loaded nanoparticles resulted in a dosage-dependent cell killing (Figure 11A). Compared to the free drug, the encapsulated CPT induced a higher level of apoptosis than the free drug at all concentrations tested. With an increase in drug concentration, a gradual increase in cell death was observed for free CPT. In contrast, the apoptosis curve for the encapsulated CPT undergoes a sharp upward transition between 0.1 and 0.2 μg/mL, and becomes leveled off when CPT concentration is greater than 0.3 μg/mL. At the highest CPT concentration (0.4 μg/mL), the encapsulated drug exhibited a normalized apoptosis (relative to the controls) of 16.4 ± 2.1, 4 times more effective in causing cell death than the free drug. More importantly, only ~24% of CPT initially loaded was released from the nanoparticles during the first 24 h of incubation, suggesting that the nanoparticle formulations are even more potent on a per drug release basis.
Figure 10.

Cytotoxicity of blank ECT2 nanoparticles and CPT-loaded nanoparticles to the cultured C4-2B cells. Representative live/dead staining of C4-2B cells cultured in the presence of filtered DI H2O (5%, v/v, A) and blank ECT2 nanoparticles (95μg/mL, B) for 24 h. Live (green) and dead (red) cell nuclei were stained with SYTO 13 and propidium iodide, respectively. C: Apoptosis assay of vehicle controls, including filtered DI H2O (5%, v/v), DMSO (0.1%, v/v) and blank ECT2 nanoparticles (95 μg/mL).
Figure 11.

Dosage-dependent apoptosis response of C4-2B cells (A) and PC-3 cells (B) to free CPT and CPT-loaded, ECT2 nanoparticles.
In human plasma, at pH 7.4 and 37 °C, the half life of the active CPT with the intact lactone ring is only ~11 min. At equilibrium, the lactone content was reduced to 0.2% [18]. Free CPT, once converted into the carboxylate form, no longer is effective in preventing DNA replication, thus therapeutically inactive. Abundant literature show that when incorporated into the interior of the block copolymer nanoparticles, the undesirable hydrolysis of the lactone ring can be avoided [13, 37, 40]. Therefore, the effectiveness of the encapsulated CPT in killing C4-2B cells can be potentially attributed to the improved drug stability [41]. Moreover, when CPT-loaded nanoparticles were added to the cell culture, drug molecules were released gradually in a controlled fashion. Such controlled release also may enhance the potency of the drug. Finally, the possibility of CPT-loaded nanoparticle be internalized by the cells cannot be ruled out. Polymeric nanoparticles with an average diameter in the range of 100–120 nm have been shown to be readily taken up by various types of cells, including prostate cancer LNCaP cells [6, 42–44], within 2 h of incubation. Under the experimental conditions employed, sustained release of CPT within the intracellular milieu is possible after the nanoparticles are internalized. The acidic intracellular conditions have been shown to maintain CPT in its active lactone form [41].
Another human prostate cancer cell line, PC-3, was also subjected to the same CPT treatment and the results for apoptosis assay are summarized in Figure 11B. With an increase in free CPT concentration, the level of apoptosis increased smoothly, reaching a plateau at a drug concentration of 0.4 μg/mL. Compared to C4-2B cells, PC-3 cells exhibited a considerably lower level of apoptosis at the concentration range investigated. Contrary to the case with C4-2B cells, the nanoparticle-encapsulated CPT induced comparable apoptotic responses to the free drug. It is well known that PC-3 cells are resistant to CPT treatment. [45]. Cancer cells develop resistance to chemotherapy by adapting to cellular stress and toxic insults via the increased drug efflux, decreased drug influx, DNA repair activation, detoxification and blockage of apoptosis [46]. It is not surprising that our nanoparticle formulations, consisting of single active drug component without the specific targeting modality and co-factors or modulators, are not efficient in combating drug resistance [47]. Follow up studies beyond the scope of this work will examine strategies to enhance drug loading and improve the efficacy to treat drug resistant cancer cells.
4. Conclusion
Ring opening copolymerization of CL and TSU using mPEG as the initiator offers a versatile technique for the synthesis of amphiphilic block copolymers containing randomly distributed CL and TSU units in the hydrophobic blocks. The presence of pendent cyclic ketal groups effectively disrupted the close packing of PCL chains, resulting in a decrease in polymer crystallinity. These copolyesters self-assembled into spherical nanoparticles with a hydrophobic interior that can sequester the anti-cancer drug CPT and a hydrophilic PEG corona that stabilizes the colloidal suspension. Our results show that CPT loading and release are directly related to the physical state of the copolymers and can be modulated readily through copolymerization. When the copolymer contains 14.3 mol% TSU in the hydrophobic block, there exists a perfect balance of particle stability and chain flexibility, allowing for homogeneous dispersion of CPT within the hydrophobic interior that is physically stabilized by the microcrystalline domains. Consequently, higher drug loading, slower and more controlled CPT release were observed. In vitro cytotoxicity assays using C4-2B cells as the target showed that ECT-based nanoparticles are biocompatible and the encapsulated CPT exhibits higher anticancer activity against C4-2B cells than the free drug. The current investigation underlines the importance of the physical characteristics of the block copolymer nanocarriers in fine-tuning the drug encapsulation and the release kinetics.
Acknowledgments
We thank G. Hassnain Jaffari and Dr. S. Ismat Shah for their assistance with the WAXD measurements. We are grateful to Dr. Steve Bai for his discussions on the NMR results and Deborah Powell for her assistance with CryoSEM. This work was supported in part by NSF (DMR: 0643226 to XJ) and NIH (NCRR: P20 RR017716 to XJ; NCI: P01 CA098912 to MCF-C).
References
- 1.Panyam J, Labhasetwar V. Adv Drug Deliver Rev. 2003;55:329. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- 2.Allen TM, Cullis PR. Science. 2004;303:1818. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 3.Rabinow BE. Nat Rev Drug Discov. 2004;3:785. doi: 10.1038/nrd1494. [DOI] [PubMed] [Google Scholar]
- 4.Davis ME, Chen Z, Shin DM. Nat Rev Drug Discov. 2008;7:771. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 5.Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. J Control Release. 2005;109:169. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 6.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Proc Nat Acad Sci USA. 2008;105:17356. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho HK, Lone S, Kim DD, Choi JH, Choi SW, Cho JH, Kim JH, Cheong IW. Polymer. 2009;50:2357. [Google Scholar]
- 8.Lee JS, Hwang SJ, Lee DS, Kim SC, Kim DJ. Macromol Res. 2009;17:72. [Google Scholar]
- 9.Noh T, Kook YH, Park C, Youn H, Kim H, Oh ET, Choi EK, Park HJ, Kim C. J Polymer Sci A Polym Chem. 2008;46:7321. [Google Scholar]
- 10.Yang XQ, Deng WJ, Fu LW, Blanco E, Gao JM, Quan DP, Shuai XT. J Biomed Mat Res A. 2008;86A:48. doi: 10.1002/jbm.a.31537. [DOI] [PubMed] [Google Scholar]
- 11.Kim SY, Cho SH, Lee YM, Chu LY. Macromol Res. 2007;15:646. [Google Scholar]
- 12.Hu Y, Xie JW, Tong YW, Wang CH. J Control Release. 2007;118:7. doi: 10.1016/j.jconrel.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 13.Zhang LY, Hu Y, Jiang XQ, Yang CZ, Lu W, Yang YH. J Control Release. 2004;96:135. doi: 10.1016/j.jconrel.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 14.Zhang LY, Yang M, Wang Q, Li Y, Guo R, Jiang XQ, Yang CZ, Liu BR. J Control Release. 2007;119:153. doi: 10.1016/j.jconrel.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Tian D, Dubois P, Jerome R. Macromolecules. 1997;30:1947. [Google Scholar]
- 16.Hertzberg RP, Caranfa MJ, Hecht SM. Biochemistry. 1989;28:4629. doi: 10.1021/bi00437a018. [DOI] [PubMed] [Google Scholar]
- 17.Cragg GM, Newman DJ. J Nat Prod. 2004;67:232. doi: 10.1021/np030420c. [DOI] [PubMed] [Google Scholar]
- 18.Mi ZH, Burke TG. Biochemistry. 1994;33:10325. doi: 10.1021/bi00200a013. [DOI] [PubMed] [Google Scholar]
- 19.Tian D, Dubois P, Grandfils C, Jerome R. Macromolecules. 1997;30:406. [Google Scholar]
- 20.Jha AK, Hule RA, Jiao T, Teller SS, Clifton RJ, Duncan RL, Pochan DJ, Jia XQ. Macromolecules. 2009;42:537. doi: 10.1021/ma8019442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farran AJE, Teller SS, Jha AK, Hule R, Jiao T, Clifton RJ, Pochan DJ, Duncan RL, Jia XQ. Tissue Eng. PMID: 20064012, (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, Chen L, Li L, Hu X, Cai Y. Int J Pharm. 2004;287:13. doi: 10.1016/j.ijpharm.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Chung MK, Han SS, Kim JC. Regul Toxicol Pharm. 2006;45:273. doi: 10.1016/j.yrtph.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 24.Gurski LA, Jha AK, Zhang C, Jia XQ, Farach-Carson MC. Biomaterials. 2009;30:6076. doi: 10.1016/j.biomaterials.2009.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savore C, Zhang C, Muir C, Liu RT, Wyrwa J, Shu J, Zhau HE, Chung LWK, Carson DD, Farach-Carson MC. Clin Exp Metast. 2005;22:377. doi: 10.1007/s10585-005-2339-3. [DOI] [PubMed] [Google Scholar]
- 26.Tong R, Cheng JJ. Polym Rev. 2007;47:345. [Google Scholar]
- 27.Tong R, Cheng JJ. Angew Chem Int Edit. 2008;47:4830. doi: 10.1002/anie.200800491. [DOI] [PubMed] [Google Scholar]
- 28.Albertsson AC, Varma IK. Biomacromolecules. 2003;4:1466. doi: 10.1021/bm034247a. [DOI] [PubMed] [Google Scholar]
- 29.Tian D, Dubois P, Jerome R. Macromolecules. 1997;30:2575. [Google Scholar]
- 30.Sperling LH. Introduction to Physical Polymer Science. 3. Hoboken, NJ: Wiley-Interscience; 2001. [Google Scholar]
- 31.Strobl G. The Physics of Polymers. 3. Berlin, Germany: Springer; 2007. [Google Scholar]
- 32.He CL, Sun J, Deng C, Zhao T, Deng MX, Chen XS, Jing XB. Biomacromolecules. 2004;5:2042. doi: 10.1021/bm049720e. [DOI] [PubMed] [Google Scholar]
- 33.An JH, Kim HS, Chung DJ, Lee DS, Kim S. J Mater Sci. 2001;36:715. [Google Scholar]
- 34.Zhang Y, Wang CC, Yang WL, Shi B, Fu SK. Colloid Polym Sci. 2005;283:1246. [Google Scholar]
- 35.Soo PL, Luo LB, Maysinger D, Eisenberg A. Langmuir. 2002;18:9996. [Google Scholar]
- 36.Forrest ML, Won CY, Malick AW, Kwon GS. J Control Release. 2006;110:370. doi: 10.1016/j.jconrel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 37.Min KH, Park K, Kim YS, Bae SM, Lee S, Jo HG, Park RW, Kim IS, Jeong SY, Kim K, Kwon IC. J Control Release. 2008;127:208. doi: 10.1016/j.jconrel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 38.Chang YC, Chu IM. Eur Polym J. 2008;44:3922. [Google Scholar]
- 39.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. J Control Release. 2001;70:1. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe M, Kawano K, Yokoyama M, Opanasopit P, Okano T, Maitani Y. Int J Pharm. 2006;308:183. doi: 10.1016/j.ijpharm.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 41.Liu J, Jiang ZZ, Zhang SM, Saltzman WM. Biomaterials. 2009;30:5707. doi: 10.1016/j.biomaterials.2009.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desai MP, Labhasetwar V, Walter E, Levy RJ, Amidon GL. Pharm Res. 1997;14:1568. doi: 10.1023/a:1012126301290. [DOI] [PubMed] [Google Scholar]
- 43.Cartiera MS, Johnson KM, Rajendran V, Caplan MJ, Saltzman WM. Biomaterials. 2009;30:2790. doi: 10.1016/j.biomaterials.2009.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qaddoumi MG, Ueda H, Yang J, Davda J, Labhasetwar V, Lee VHL. Pharm Res. 2004;21:641. doi: 10.1023/b:pham.0000022411.47059.76. [DOI] [PubMed] [Google Scholar]
- 45.Akao Y, Banno Y, Nakagawa Y, Hasegawa N, Kim TJ, Murate T, Igarashi Y, Nozawa Y. Biochem Bioph Res Co. 2006;342:1284. doi: 10.1016/j.bbrc.2006.02.070. [DOI] [PubMed] [Google Scholar]
- 46.Jabbari E. Pharm Res. 2009;26:612. doi: 10.1007/s11095-008-9802-1. [DOI] [PubMed] [Google Scholar]
- 47.Khdair A, Chen D, Patil Y, Ma LN, Dou QP, Shekhar MPV, Panyam J. J Control Release. 2010;141:137. doi: 10.1016/j.jconrel.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]