

Abstract
Isoforms of protein kinase C (PKC) have been shown to modulate some cellular responses such as pathological secretion and generation of inflammatory mediators during acute pancreatitis (AP). We propose that PKC also participates in premature zymogen activation within the pancreatic acinar cell, a key event in the initiation of AP. This hypothesis was examined in in vivo and cellular models of caerulein-induced AP using PKC activators and inhibitors. Phorbol ester, 12-O-tetradecanoylphorbol-13-acetate (TPA, 200 nM), a known activator of PKC, enhanced zymogen activation at both 0.1 nM and 100 nM caerulein, concentrations which mimic physiological and supraphysiological effects of the hormone cholecystokinin, respectively, in preparations of pancreatic acinar cells. Isoform-specific PKC inhibitors for PKC-δ and PKC-ε reduced supraphysiological caerulein-induced zymogen activation. Using a cell-free reconstitution system, we showed that inhibition of PKC-δ and -ε, reduced zymogen activation in both zymogen granule-enriched and microsomal fractions. In dispersed acinar cells, 100 nM caerulein stimulation caused PKC-δ and -ε isoform translocation to microsomal membranes using cell fractionation and immunoblot analysis. PKC translocation was confirmed with in vivo studies and immunofluorescence microscopy in pancreatic tissues from rats treated with or without 100 nM caerulein. PKC-ε redistributed from an apical to a supranuclear region following caerulein administration. The signal for PKC-ε overlapped with granule membrane protein, GRAMP-92, an endosomal/lysosomal marker, in a supranuclear region where zymogen activation takes place. These results indicate that PKC-δ and -ε isoforms translocate to specific acinar cell compartments and modulate zymogen activation.
Keywords: translocation, PKC activator phorbol ester, PKC inhibitor
The pathophysiology of acute pancreatitis (AP) is determined by many sequential events in the exocrine pancreas. These include activation of zymogens in the acinar cell, decreased apical secretion of digestive zymogens, release of inflammatory cytokines, inflammation, edema, and cell death. Isoforms of the serine/threonine kinase, protein kinase C (PKC), have been shown to modulate some of these AP responses (8, 28, 29). Of the ten known PKC isoforms, four have been identified in pancreatic acinar cells (acini): conventional PKC-α [1,2 diacylglycerol (DAG)- and Ca2+-sensitive], novel PKC-δ and PKC-ε, (DAG-sensitive and Ca2+-insensitive), and atypical PKC-ζ (DAG- and Ca2+-insensitive) (2, 23).
The role PKC isoforms play has largely been determined using cellular and in vivo experimental models of AP. These models often use supramaximal concentrations of the hormone cholecystokinin (CCK) or its ortholog caerulein to induce AP (28, 29, 33). The cellular model of acute pancreatitis, using dispersed groups of pancreatic acini, has been particularly useful for studying the early events in AP. The effects of ethanol, a known factor in induction of AP, have been studied in these models (29). Pretreatment with pharmacological inhibitors of PKC isoforms in these model systems has revealed that PKC-δ alone mediates physiological secretion, whereas diverse pathological responses, ranging from lack of exocytosis to generation of inflammatory mediators, are regulated by more than one PKC isoform. Conventional PKC-α, for example, has been implicated in mediating pathological secretory processes in experimental pancreatitis, whereas the novel PKC-δ and PKC-ε isoforms have been shown to regulate expression of inflammatory mediators (8, 28, 29, 33). The function of PKC-ζ has yet to be determined in the pancreatic acinar cell.
In the present study, we investigated a potential role for PKC isoforms in regulating premature zymogen activation, using both the in vitro caerulein model of AP and a cell-free reconstitution system in conjunction with PKC activators and inhibitors. Two concentrations of caerulein were used to stimulate acinar cells: 0.1 nM and 100 nM to mimic physiological and supraphysiological effects of CCK, respectively (25, 27). Inhibition of PKC-δ and -ε decreased caerulein-stimulated zymogen activation. Furthermore, immunoblot studies showed that both isoforms translocated from the cytosol to microsomal membranes upon supraphysiological caerulein stimulation. With in vivo studies, using immunofluorescence, we observed a redistribution of PKC-ε from the apical region of the cell to a supranuclear region. PKC-ε distribution poststimulation overlapped with cellular compartments positive for the lysosomal/endosomal marker, granule membrane protein (GRAMP)-92. Our laboratory has previously shown that a subset of GRAMP-92-containing compartments overlap with compartments positive for the trypsinogen activation peptide (TAP) (19), a marker for zymogen activation. Our study suggested that relevant PKC isoforms were present in areas of the cell where zymogen activation took place. These studies demonstrated that PKC-δ and -ε modulated caerulein-stimulated protease activation in the pancreatic acinar cell.
MATERIALS AND METHODS
Preparation of isolated pancreatic acini
Acini were isolated as previously described (7). Briefly, fasted male Sprague-Dawley rats (50 –150 g) (Charles River Laboratories, Wilmington, MA) were euthanized by CO2 with a protocol approved by the Yale University and the Veterans Affairs Animal Care and Use Committees. Acinar media was prepared as follows (in mM): 10 HEPES (pH 7.4), 95 NaCl, 4.7 KCl, 0.6 MgCl2, 1 NaH2PO4, 10 glucose, 2 glutamine, plus 0.1% BSA, 1× MEM-amino acids (GIBCO, San Jose, CA), and 1.3 CaCl2. The pancreas was collected in 15 ml of calcium-free acinar media. The pancreas was then minced in a minimal volume of calcium-free medium for 5 min and washed three times with calcium free medium. The minced tissue was then placed into a 50-ml flask with 12 ml of acinar medium containing 50 U/ml of type 4 collagenase (Worthington, Freehold, NJ) for 60 min at 37°C with shaking (120 revolution/min). The digest was filtered through a 300 – 400-μm mesh (Sefar American, Depew, NY) and washed with acinar medium. Isolated acini (groups of 20 –100 acinar cells) were distributed among the 24 wells (0.5 ml suspension/well) of a 24-well Falcon tissue culture plate (Becton Dickinson, Franklin Lakes, NJ). All reagents were purchased from Sigma (St. Louis, MO) unless otherwise noted.
Acinar experimental protocol
Acini were recovered for 120 min at 37°C under constant O2 with shaking (90 revolution/min). Medium was changed at 60 min. At 120 min, acini were treated without changing medium. Acini were treated with caerulein (0.1 nM and 100 nM) for 60 min (unless otherwise noted). Samples were collected, placed in 1.5-ml centrifuge tubes (USA Scientific, Waltham, MA) and centrifuged for 1 min at 30 g. A sample (50 μl) of the resulting cell-free supernatant was removed to a 0.5-ml microcentrifuge tube to assay for secreted amylase. The remaining 450 μl of cells plus media were retained for zymogen activation assays and determination of total amylase. All samples were stored at −80°C.
PKC activator
Phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA; 200 nM) or the biologically-inactive form of TPA, TPA-4α (200 nM), was added 15 min before caerulein stimulation. In some treatment groups, the broad spectrum PKC inhibitor GF-109203X (10 μM) was added 120 min before addition of TPA. During the recovery period, GF-109203X was readded after the media change.
PKC inhibitors
Acinar cells were treated with 10 μM of each of the following: GF-109203X, isoform-specific PKC-δ translocation inhibitor (δV1-1: S-F-N-S-Y-E-L-G-S-L), isoform-specific PKC-ε translocation inhibitor (εV1-2: E-A-V-S-L-K-P-T), and a control scrambled peptide (L-S-E-T-K-P-A-V) for 120 min before caerulein stimulation. Each of these peptides was synthesized as an amino terminal extension to a Drosophila antennapedia peptide (R-QI-K-I-W-F-Q-N-R-R-M-K-W-K-K) to make it cell permeable (28, 29).
Enzymatic activity assays
Enzyme activities were carried out as previously described (7). Briefly, samples were thawed, homogenized, and centrifuged. To each well of a 24-well plate (Greiner Bio-one Cellstar TC-Plate) we added the following: 100 μl of postnuclear supernatant and 350 μl of trypsin assay buffer [50 mM Tris (pH 8.1), 150 mM NaCl, 1 mM CaCl2, 0.01% BSA]. The assay was initiated by the addition of 50 μl of 400 μM enzyme substrate (fluorometric trypsin substrate; Peptides International, Louisville, KY and fluorometric chymotrypsin substrate; Calbiochem, San Diego, CA) diluted in trypsin assay buffer (40 μM final). The plate was read by using a fluorometric microtiter plate reader (model HTS 7000; Perkin-Elmer Analytical Instruments, Shelton, CT; 380-nm excitation; 440-nm emission; 20 reads/10 min).
Amylase assay
Amylase activity was determined with a commercial kit (Phaebadas kit; Pharmacia Diagnostic, Rochester, NY) as described (7). Amylase secretion was calculated as the percent total release [medium/(medium + cells)].
Reconstitution experiments
Pancreatic fractionation was carried out as described (34). Briefly, the pancreas was divided in two and each half homogenized in 10 volumes (~5 ml) of 300 mM sucrose, 1 mM DTT. Homogenates were centrifuged at 500 g for 5 min to generate a postnuclear supernatant (PNS). All preparations and storage were at 4°C unless otherwise stated. The PNS fraction was centrifuged to generate various membrane fractions as previously described (34). Briefly, PNS was serially centrifuged at 3,000 g (10 min), 15,000 g (10 min), and 180,000 g (60 min) over a 2 M sucrose cushion. The interface of each centrifugation was collected, i.e., zymogen granule, mitochondrial, and microsomal fractions, respectively. Only the zymogen granule and microsomal fractions were retained for further study. All particulate fractions were washed and diluted as described (34).
Cytosol was prepared from PNS as described (34). Briefly, PNS was centrifuged at 180,000 g for 60 min on a 2 M sucrose cushion. The supernatant was removed and dialyzed in cytosol buffer: 300 mM sucrose, 25 mM Tris, pH 7.2, and 100 mM KCl for 60 min using a dialysis cassette (Slide-A-Lyzer Dialysis Cassette, 3500 MWCO, 0.5–3 ml capacity; Pierce Biotech, Rockford, IL). The dialyzed fraction was centrifuged in tubes precoated with cytosol buffer at 288,000 g for 15 min to pellet any small vesicles. The resultant supernatant (cytosol) was collected.
Enzyme activity in cell fractions was assayed as previously described (34). To each well of a 24-well plate the following was added: 350 μl of assay buffer (50 mM Tris, pH 7.6, and 150 mM KCl), 50 μl of zymogen granule-enriched or microsomal fractions, and 50 μl of 400 μM enzyme substrate (Peptides International). After 15-min incubation at room temperature, 50 μl of buffer (control) or cytosol were added to each well and incubated for a further 15 min. ATP (5 mM) was then added, and fluorescence emissions were recorded [excitation wavelength 380 nm, emission 440 nm; 20 recordings over 10 min with a HTS 7000 fluorimeter (Perkin-Elmer)]. Enzyme activity was normalized to amylase content and expressed as fold activation vs. the zymogen granule/microsomal fraction plus cytosol plus ATP condition.
In previous studies, PKC was found to be predominantly in the cytosol and thought to be in an active or semiactive form before stimulation (14, 16, 28). Thus cytosol was preincubated for 15 min with isoform-specific PKC inhibitors before addition to the well as described earlier in this section for the assay of zymogen activation in cellular fractions. The PKC inhibitors used in the cell-free system were not conjugated to Drosophila antennapedia peptide since cell permeability was not required.
Preparation of cells for immunoblot
Cells were prepared as described in Preparation of isolated pancreatic acini, with the following modifications: after 30-min supraphysiological caerulein treatment, cells and medium (500 μl) were collected. Samples were allowed to sediment by gravity, and the cell free medium was removed. An equal volume (500 μl) of homogenization buffer [25 mM HEPES, pH 7.4, 300 mM sucrose, 1 mM benzamidine, complete protease inhibitor cocktail (EDTA-free) (1 tablet per 25 ml stock solution; Roche, Mannheim, Germany)] was added, and cells were homogenized in a conical 1.5-ml Eppendorf tube with a pestle and centrifuged at 1,000 g for 1 min. The PNS was removed and further centrifuged at 3,000 g for 5 min; the pellet was collected as the zymogen granule fraction. This fraction was washed with homogenization buffer (500 μl) and spun two more times. The supernatant from the 3,000 g step was centrifuged at 233,000 g, and the supernatant was collected as the cytosolic fraction, the pellet as the microsomal fraction. To solubilize proteins, the zymogen granule and microsomal pellets were resuspended in Laemmli buffer (125 μl of 1×). A sample (50 μl) of 6× Laemmli buffer was added to 300 μl of cytosol. Samples were heated to 95°C for 5 min and then stored at −80°C.
Immunoblot analysis
Western blot analysis was performed to detect PKC isoforms. Briefly, samples were separated on 10% SDS-PAGE gels (Bio-Rad, Hercules, CA) and transferred to Immobilon-P membranes (Millipore, Billerica, MA). Membranes were blocked for 60 min at room temperature with Blotto [Tris-buffered saline (TBS), 5% nonfat dry milk, 0.05% Tween-20]. Membranes were then probed with primary antibody (rabbit anti-PKC-δ or -ε, 1:200; Santa Cruz Biotechnology, Santa Cruz, CA) in Blotto for 120 min at room temperature, washed, and incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (Sigma) for 60 min at room temperature. Membranes were washed in TBS, and autoradiography was performed by using a SuperSignal West pico chemiluminescence kit (Pierce).
Immunofluorescence microscopy
Sprague-Dawley rats were anesthetized, and a supraphysiological dose of caerulein (40 μg/kg) was administered intraperitoneally. After 60 min, rat tissues were fixed by transcardiac perfusion of 2% paraformaldehyde and 10 mM benzamidine to optimize tissue morphology. Rat pancreata were fixed for an additional 30 min at room temperature, sucrose infiltrated overnight at 4°C, and frozen in Tissue Tek optimal cutting temperature compound (Sakura, Torrance, CA). Cut sections (5 microns thick) were permeabilized in 0.05% saponin and quenched in 500 mM NH4Cl and 3% normal goat serum. Primary antibodies (rabbit anti-PKC-δ or -ε, 1:100; mouse anti-GRAMP-92, 1:20; Santa Cruz Biotechnology) were incubated with tissue sections for 120 min at room temperature in a humidified chamber. Primary antibodies were detected with AlexaFluor488-labeled goat anti-rabbit (1:1,000) or AlexaFluor555-labeled goat anti-mouse (1:1,000) secondary antibodies, and sections were mounted in Prolong Gold with DAPI (Molecular Probes). Labeled tissues were examined by epifluorescence using an Axioplan microscope (Carl Zeiss, Thornwood, NY), and digital images were collected with a Spot camera and analyzed with software version 4.0.9 (Diagnostic Instruments, Sterling Heights, MI). Digital images were assembled and labeled in Adobe Photoshop (Adobe Systems, San Jose, CA). Further images were obtained by use of a Zeiss LSM510 laser scanning confocal microscope and analyzed using LSM510 image browser software.
Statistical analysis
Data represent means ± SE of at least three individual experiments unless otherwise noted, with each experiment being performed in at least duplicate. A Student’s t-test analysis was used to determine statistical significance, and P values of <0.05 were assigned significance.
RESULTS
Phorbol ester, TPA, enhances caerulein-induced zymogen activation in pancreatic acini
Our initial study investigated whether PKC stimulation could affect basal or caerulein-stimulated zymogen activation. Pancreatic rat acini were pre-treated with the phorbol ester TPA (200 nM), a known activator of PKC isoforms (33). Both trypsin and chymotrypsin activities, normalized to amylase content and expressed as fold vs. maximal, were used as measures of zymogen activation using specific fluorogenic substrates. TPA pretreatment alone had no effect, but TPA pretreatment followed by caerulein stimulation led to increased zymogen activation both at 0.1 nM and 100 nM caerulein (Fig. 1, A and B). The enhancement of caerulein-stimulated zymogen activation was not seen with the biologically inactive form of TPA, TPA 4-α (200 nM), or TPA in the presence of GF-109203X (10 μM), a broad-spectrum PKC inhibitor.
Fig. 1.

PKC activator phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA) enhances caerulein-induced zymogen activation. Trypsin (A) and chymotrypsin (B) activities and amylase secretion (C) were measured. Data for trypsin and chymotrypsin were normalized to total amylase content and expressed as fold vs. maximal; amylase secretion was expressed as % total. Dispersed pancreatic acinar cells were treated 15 min before caerulein stimulation with phorbol ester (200 nM), its biologically inactive form TPA-4α or TPA following 120 min pretreatment with broad-spectrum PKC inhibitor GF-109203X (GF) (10 μM); all treatments were added in DMSO. DMSO was added to non-TPA or GF-109203X-treated cells to control for nonspecific DMSO effects. Following pretreatment, cells were either unstimulated or stimulated with caerulein (CER) (0.1 nM or 100 nM) for 60 min. *P < 0.05 vs. corresponding caerulein alone condition (n = 3).
Although low concentrations of caerulein (0.1 nM) stimulation alone caused little, if any, zymogen activation, when combined with TPA, zymogen activation results were comparable to that seen with supraphysiological caerulein alone (Fig. 1, A and B). Supraphysiological caerulein-induced zymogen activation itself almost doubled with TPA pretreatment. The PKC inhibitor, GF-109203X, reduced activation but not to basal levels. The incomplete response could indicate that a component of zymogen activation may be PKC insensitive or that higher concentrations of GF-109203X might be required to completely inhibit PKC in this condition.
TPA appeared to have little effect on caerulein-stimulated amylase secretion (Fig. 1C). A slight increase in amylase secretion in low-dose (0.1 nM) caerulein-stimulated cells was seen, but this was not statistically significant.
PKC inhibitors reveal a role for PKC in caerulein-induced zymogen activation
In previous studies, PKC isoforms were shown to be activated by CCK and caerulein in pancreatic acinar cells (16, 28, 29). Furthermore, as shown in this study, TPA enhanced 0.1 nM caerulein-induced zymogen activation to levels comparable to that seen with 100 nM caerulein alone, indicating that PKC sensitized the cell to caerulein treatment. To examine whether endogenously activated PKC mediated supraphysiological caerulein-induced zymogen activation, dispersed acinar cells were pretreated with increasing concentrations of GF-109203X followed by supraphysiological caerulein stimulation (Fig. 2). Caerulein-induced trypsin and chymotrypsin activity was reduced in the presence of 1 μM and 10 μM GF-109203X, with significance at 10 μM, a concentration used routinely in the literature (16, 28, 29), indicating that PKC was involved in caerulein-stimulated zymogen activation.
Fig. 2.

Broad-spectrum protein kinase C (PKC) inhibitor GF-109203X reduces supraphysiological caerulein-induced trypsinogen and chymotrypsinogen activation. Trypsin (open bars) and chymotrypsin (shaded bars) activities were measured and data normalized to amylase content and expressed as fold vs. caerulein. Dispersed pancreatic acinar cells were stimulated with supra-physiological caerulein for 60 min following 120-min pretreatment with broad-spectrum PKC inhibitor GF-109203X (1 and 10 μM); DMSO was added to noninhibitor-treated controls. *P < 0.05 vs. caerulein (n = 3).
To address which PKC isoforms mediated zymogen activation, acini were pretreated with isoform-specific PKC inhibitors and then stimulated with supraphysiological concentrations of caerulein. Inhibition of the PKC-α isoform with the conventional PKC inhibitor Gö 6976 did not affect zymogen activation (data not shown). This observation was consistent with the report that PKC-α was not activated by supraphysiological caerulein or CCK (28). Preliminary studies showed that inhibition of PKC-ζ with a specific pseudosubstrate did not affect zymogen activation either (data not shown). Thus PKC-α and -ζ isoforms were not studied further in the context of zymogen activation. Inhibition of PKC-δ or -ε, however, significantly reduced zymogen activation (Fig. 3, A and B) but did not affect stimulated amylase secretion (not shown). When cells were incubated with a combination of inhibitors (either δ/ε or δ/ε/GF-109203X), zymogen activation was significantly reduced, but the effect was similar to that seen with δ, ε, or GF-109203X inhibition alone, suggesting that the effects of inhibiting PKC isoforms was not additive (Fig. 3C). Finally, preincubation of cells with a scrambled peptide similar to the isoform-specific PKC translocation inhibitors did not affect zymogen activation, indicating that the reduction in zymogen activation due to PKC inhibition was specific.
Fig. 3.
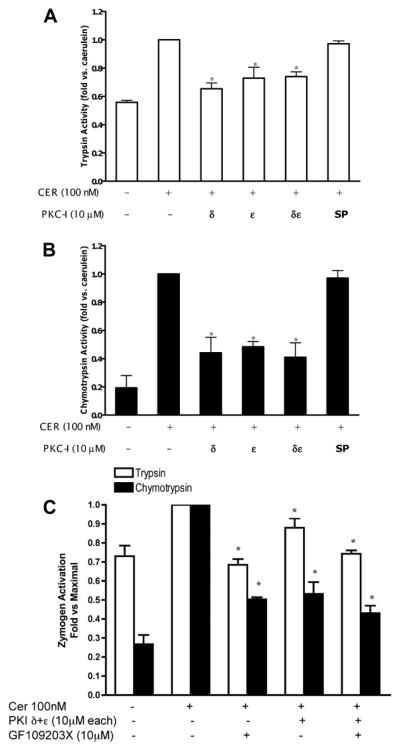
PKC-δ and -ε isoform-specific inhibitors reduce supraphysiological caerulein-induced trypsinogen and chymotrypsinogen activation. Trypsin (A) and chymotrypsin (B) activities were measured and data normalized to amylase content and expressed as fold vs. caerulein. Dispersed pancreatic acinar cells were stimulated with supraphysiological caerulein for 60 min following 120-min pretreatment with isoform-specific PKC inhibitors, PKC translocation inhibitors δV1-1 or εV1-2 (10 μM) or scrambled peptide (SP) (10 μM). C: combined effects of PKC translocation inhibitors δV1-1 and εV1-2 with GF-109203X (10 μM) on trypsin and chymotrypsin activities. DMSO was added to noninhibitor-treated controls. *P < 0.05 vs. caerulein (n = 5).
PKC-δ and -ε isoforms mediate zymogen activation in both zymogen granule-enriched and microsomal fractions in a cell-free system
PKC has been shown to phosphorylate the CCK receptor (9, 31). To determine whether the PKC effects we observed were downstream of the receptor, we examined the effects of kinase isoform inhibition in a cell-free reconstitution system. Using this preparation, we have shown that both zymogen granule-enriched and microsomal fractions from the pancreas could support cytosol-dependent zymogen activation (34). Furthermore, with chymotrypsin, there was an ATP-dependent component to activation that was sensitive to protein kinase inhibition.
To determine whether the unidentified kinase was an isoform of PKC, zymogen activation was measured in both zymogen granule-enriched and microsomal fractions reconstituted with dialyzed cytosol and ATP, as described (34). Although there is no ATP-dependent increase in trypsin activation, a reduction in cytosol-dependent trypsin activity was observed in the presence of PKC-δ or -ε inhibitor (Fig. 4, A and B), agreeing with our acinar cell data, which demonstrated that trypsin activation was PKC sensitive. A similar effect was seen for cytosol-stimulated chymotrypsin activation in both membrane fractions. In addition, the enhanced activation seen in the presence of both cytosol and ATP was also inhibited (Fig. 4, C and D). A greater effect was seen for chymotrypsin than trypsin with up to a 50% reduction from maximal in chymotrypsin activation in both zymogen granule-enriched and microsomal fractions (Fig. 4, C and D). Finally, the lack of any effect with the scrambled peptide further validated the specificity of the PKC inhibitors.
Fig. 4.

PKC-δ and -ε isoform-specific inhibitors reduce trypsin and chymotrypsin activation in zymogen granule-enriched fraction (ZG) and microsomal pancreatic fractions (MIC) in a reconstituted system. Trypsin (A and B) and chymotrypsin (C and D) activity were enhanced by the addition of a cytosolic fraction (CYT) and, in the case of chymotrypsin, enhanced further by 5 mM ATP (experimental conditions described in MATERIALS AND METHODS). Data were normalized to amylase content and expressed as fold vs. ZG/MIC + CYT + ATP. Cytosol was preincubated for 15 min with relevant PKC inhibitors (1 μM), scrambled peptide (1 μM), or DMSO in controls (see MATERIALS AND METHODS for details). *P < 0.05 vs. ZG/MIC + CYT + ATP (n = 5).
Thus results from the cell-free system demonstrated that effects of PKC were downstream from the CCK receptor and that PKC-sensitive zymogen activation could take place in more than one type of acinar cell compartment.
Translocation and cellular distribution of PKC isoforms
Translocation of PKC isoforms from cytosol to membranes is a characteristic of activated PKCs. In previous studies, activated PKC-δ and -ε have been shown to translocate to intracellular membranes, whereas activated PKC-ζ did not translocate (28). Since PKC-δ and -ε affected zymogen activation in diverse cellular compartments in a cell-free system, we investigated whether they translocated from cytosol to a zymogen granule-enriched or microsomal fraction in cells treated with supraphysiological caerulein for 30 min followed by organelle fractionation.
Using immunoblot analysis, we observed that this treatment lead to reduced PKC-δ content in the cytosol and increased in microsomes (Fig. 5A). When PKC-δ levels were normalized to protein content in each fraction, the average increase of PKC-δ signal in the microsomal fraction after 30 min was 2.0 –2.5-fold (Fig. 5B). PKC-δ inhibitor reduced caerulein-stimulated translocation of PKC-δ to microsomal membranes (Fig. 5C). Similar results were seen for PKC-ε, with a 2.6–3.4-fold increase of PKC-ε signal in microsomal membranes (Fig. 6, A and B). PKC-ε translocation was reduced by PKC-ε inhibitor (Fig. 6C). There was relatively little change in PKC-δ or -ε content in zymogen granules. Thus, in isolated pancreatic acinar cells, supraphysiological caerulein stimulation resulted in PKC-δ and -ε translocating predominantly to microsomal membranes.
Fig. 5.

Subcellular distribution of PKC-δ isoform in pancreatic acinar cells stimulated with supraphysiological caerulein. Dispersed rat pancreatic acinar cells were preincubated for 120 min and then stimulated for 30 min with supraphysiological caerulein (100 nM). Subcellular distribution of PKC-δ was determined in cytosol and two membrane fractions (zymogen granule and microsomal membranes) using a PKC-δ antibody. A: representative Western blots from 3 independent experiments. B: pooled data on the increase of PKC-δ in membrane fractions; data was normalized to total protein content in each fraction and expressed as fold vs. unstimulated condition. *P < 0.05 vs. unstimulated (n = 3). C: representative Western blots from 3 independent experiments showing that PKC translocation inhibitor δV1-1 (10 μM) reduces translocation of PKC-δ to microsomal membranes.
Fig. 6.

Subcellular distribution of PKC-ε isoform in pancreatic acinar cells stimulated with supraphysiological caerulein. Dispersed rat pancreatic acinar cells were preincubated for 120 min and then stimulated for 30 min with supraphysiological caerulein (100 nM). Subcellular distribution of PKC-ε was determined in cytosol and two membrane fractions (zymogen granule and microsomal membranes) using a PKC-ε antibody. A: representative Western blots from 3 independent experiments. B: pooled data on the increase of PKC-ε in membrane fractions; data was normalized to total protein content in each fraction and expressed as fold vs. unstimulated condition. *P < 0.05 vs. unstimulated (n = 3). C: representative Western blots from 3 independent experiments showing that PKC translocation inhibitor εV1-2 (10 μM) reduces translocation of PKC-ε to microsomal membranes.
PKC translocation was confirmed with in vivo studies and immunofluorescence microscopy (see MATERIALS AND METHODS). In tissues from animals treated with or without supraphysiological concentrations of caerulein, the distribution of PKC-δ was not clearly defined, perhaps due to efficacy of the antibody. The distribution of PKC-ε, however, was more obvious. In unstimulated animals, there was intense PKC-ε immunoreactivity at the apical region of the acinar cell. PKC-ε was also diffusely distributed throughout the cytosol, with some punctuate structures being evident (Fig. 7A). Upon supraphysiological caerulein stimulation, redistribution of PKC-ε was observed. Its apical signal decreased, and PKC-ε-positive punctate structures increased in the supranuclear region (Fig. 7B). The redistribution of PKC-ε to a supranuclear region was consistent with its translocation to microsomal membranes as observed in our cell fractionation studies (Fig. 6A).
Fig. 7.

PKC-ε redistributes from an apical region to a supranuclear region of the acinar cell following supraphysiological caerulein stimulation in vivo. Male Sprague-Dawley rats were treated with a supraphysiological concentration of caerulein (given by intraperitoneal injection). After 60 min, the animals were euthanized and pancreatic tissue harvested. The distribution of PKC-ε was detected using isoform-specific antibody (shown in red) in unstimulated (control) cells and supraphysiological caerulein-stimulated cells. Note the redistribution of PKC-ε to the supranuclear region after stimulation (apical region, arrowhead and circle; supranuclear region, arrow). Nuclei are DAPI stained (blue).
PKC-ε codistributes with GRAMP-92-positive compartments
The exact site(s) of zymogen activation in the acinar cell is controversial (11–13, 15, 19, 24, 30). Previous studies from our laboratory have shown that TAP, a small peptide that is cleaved upon trypsinogen activation and marker for zymogen activation, is found in compartments containing GRAMP, a marker of late endosomes/lysosomes (10, 13, 19, 20). Conflicting fixation requirements did not permit colocalization of TAP with PKC-ε antibody, so studies were done with GRAMP-92 as the marker for the site of zymogen activation.
Confocal immunofluorescence demonstrated a predominant localization for PKC-ε in the apical region of the cell (shown in red), with a lesser distribution throughout the cytosol (Fig. 8A). GRAMP-92 was distributed throughout the cell and could be seen in some cells as sharp punctuate structures (shown in green) (Fig. 8A). Upon supraphysiological caerulein stimulation, there was a redistribution of GRAMP-92-positive compartments and PKC-ε to a supranuclear region (colocalization shown in yellow) although some apical signal for PKC-ε remained. The overlap between PKC-ε and GRAMP-92-positive compartments and this codistribution was further verified by the z-section analysis (Fig. 8) in which the tissue was scanned at different levels and the individual sections stacked to give a three-dimensional evaluation. In a previous study (19), we showed that not all GRAMP-92-positive compartments supported zymogen activation, but all TAP-containing compartments were GRAMP-92 positive (19). Although not directly demonstrated, the present study suggests that PKC-ε distribution overlaps with compartments, which may support zymogen activation following supraphysiological caerulein stimulation.
Fig. 8.

PKC-ε and lysosomal/endosomal marker granule membrane protein (GRAMP)-92 codistribution after supraphysiological caerulein stimulation in vivo. Male Sprague-Dawley rats were treated with supraphysiological concentrations of caerulein (given by intraperitoneal injection) for 60 min. The distribution of PKC-ε and GRAMP-92 were detected using specific antibodies (PKC-ε in red; GRAMP-92 in green). In tissue from unstimulated animals, PKC-ε had a predominantly apical signal, and GRAMP-92 was diffusely distributed throughout the cytosol with some punctate structures visible. Following supraphysiological caerulein stimulation, PKC-ε and GRAMP-92 redistributed to the supranuclear region. Codistribution can be seen in yellow, and this is verified by the z-section through the cell shown in the side bar of the figure (apical region, arrowhead and circle; supranuclear region, arrow).
DISCUSSION
In this study, we investigated a potential role for PKC isoforms in premature digestive zymogen activation in acute pancreatitis using PKC activators and inhibitors. Of the four PKC isoforms present in the pancreatic acinar cell, the α, δ, and ε isoforms are sensitive to phorbol esters, known activators of PKC. Pretreatment of isolated pancreatic acinar cells with the phorbol ester TPA led to enhancement of caerulein-induced zymogen activation, both at 0.1 nM and 100 nM (representing “physiological” vs. “supraphysiological” stimulation). In another study, TPA was shown to induce translocation of PKC isoforms in the acinar cell although in our study TPA alone was insufficient by itself to cause zymogen activation; caerulein stimulation was still necessary for PKC-mediated zymogen activation (16).
Using broad spectrum and isoform-specific inhibitors, we have identified two isoforms, PKC-δ and -ε, as the candidates for mediating aberrant zymogen activation. Inhibition of these isoforms did not completely block zymogen activation (Fig. 3). This may be a reflection of the degree and duration of the inhibitors although this is unlikely for a number of reasons. First, previous studies have demonstrated that PKC activity was completely inhibited by these isoform-specific peptide inhibitors at the same concentration used in the present study. Furthermore, these inhibitors have been shown to be effective for at least 4 h, comparable to the time frame of the present investigation, although their efficacy beyond this time point has not been determined. Finally, the inhibitors given in our present study are at concentrations 10–100-fold higher than their Ki to ensure that an adequate final concentration of inhibitor is present in the cell. Thus the incomplete inhibition of zymogen activation we observed suggested that there could be a PKC-independent component to the process. Each inhibitor reduced zymogen activation by a similar degree, an effect also observed for NFκB activation (28). When a combination of inhibitors was applied (δ/ε/GF-109203X), the reduction in zymogen activation was similar to that seen for δ, ε, or GF-109203X inhibition alone, indicating that the effect was not additive but maybe sequential.
In a previous study using a cell-free reconstitution system, we were able to induce zymogen activation in both zymogen-granule enriched and microsomal fractions by adding back cytosol and ATP (34). Furthermore, we found that cytosol/ATP-dependent activation could be reduced by broad spectrum protein kinase inhibitors. In this study, our evidence strongly supported PKC-δ and -ε as candidate kinases for mediating zymogen activation. Both trypsin and chymotrypsin activity were significantly reduced in the presence of PKC-δ or -ε inhibitors. A lower concentration of inhibitor was used in the cell-free system compared with the intact cells studies (1 μM vs. 10 μM), as cell permeability of the inhibitors was not an issue in the cell-free system. Preliminary studies showed that the same degree of inhibition was seen for either concentration of PKC inhibitor in the cell-free environment (data not shown); hence, the lower concentration was used. Reduction from maximal activity for trypsin was much less than that seen for chymotrypsin (10 –20% for trypsin vs. 40 – 60% for chymotrypsin). In the reconstituted cell system, we did not observe a significant ATP-dependent increase in trypsin activity but did find that ATP had a prominent effect on the activation of chymotrypsinogen. Additional data from our previous reconstitution studies also suggested that some trypsinogen and chymotrypsinogen activation may be mediated by independent mechanisms (34). This sensitivity of chymotrypsinogen activation to PKC seen in the reconstituted cell system (Fig. 4) was further supported by our acinar cell studies, which showed that chymotrypsinogen activation was much more sensitive to PKC inhibition than trypsinogen activation. At 1 μM GF-109203X, for example, we saw a reduction in chymotrypsin but not trypsin activity (Fig. 2). The mechanism responsible for the differences in trypsinogen and chymotrypsinogen activation, however, remains unclear.
The reconstituted cell method used unstimulated tissue; therefore, one might expect PKC to be largely in an inactive form located predominantly in the cytosol. However, we and others found that, even in unstimulated tissues, there was a basal level of zymogen activation. Similarly, some activation of PKC was observed in unstimulated acini (28). Finally, we found that about 10% of total PKC-δ and -ε were associated with membranes in unstimulated cells (Figs. 5 and 6). These observations suggested that a fraction of PKC was active in unstimulated tissues.
A further observation from the reconstituted cell studies was that cytosol-induced zymogen activation could be reduced in the zymogen granule-enriched fraction by PKC inhibitors, even when ATP was not present (Fig. 4, A and C). The cytosol used in these assays had been dialyzed to remove endogenous ATP. Therefore, one would not expect protein kinases to be active in the cytosol. However, it was possible that the zymogen granules, which contained ATP, could release endogenous ATP during the incubation with cytosol (32). This might provide sufficient ATP for activation of PKC isoforms, particularly those which may be anchored to the surface of the granule (Figs. 5A and 6A).
From our reconstituted cell studies, we have shown that PKC-δ and -ε can modulate zymogen activation in diverse cellular compartments such as zymogen granules and microsomes. However, in intact cells, PKC translocates preferentially to microsomal membranes upon stimulation. What can account for this difference? First, anchoring sites for PKC isoforms are present on both zymogen granules and microsomes as is evident from their localization in resting cells, i.e., some PKC is endogenously bound to these membranes. Thus PKC has the capacity to bind to target sites on either organelle and mediate zymogen activation but selectively translocate to microsomal compartments in intact cells. The mechanism by which PKC translocates is unknown, but it is possible that the cellular machinery responsible for this phenomenon may not be intact in the cell-free preparation.
Whether the zymogen activation compartments observed in reconstituted cell studies reflect the site of PKC-sensitive zymogen activation in the stimulated intact acinar cell is unclear. In previous studies, PKC-δ and -ε have been shown to translocate to intracellular membrane fractions from the cytosol following caerulein or CCK stimulation (28). We explored this further and showed that these isoforms translocated predominantly to microsomal membranes as opposed to zymogen granule membranes upon supraphysiological caerulein stimulation (Figs. 5 and 6). Studies from a number of laboratories, including ours, indicated that early intracellular zymogen activation was occurring in a non-zymogen granule compartment (19, 30). The fact that PKC-ε and δ mediated zymogen activation and were translocating to microsomal membranes in subcellular fractionation studies supported this idea. Using immunofluorescence, we observed that PKC-ε redistributed in the acinar cell following stimulation to a supranuclear region (Fig. 7) in agreement with that observed in previous studies (29). Our data further showed that it codistributed with GRAMP-92, an endosomal lysosomal marker, which has been associated with zymogen activation compartments (Fig. 8) (19, 21, 26, 35). Although this does not directly demonstrate that PKC translocates to zymogen activation compartments, it demonstrates that PKC-ε is in the correct location to participate in zymogen activation.
This study provides strong evidence for a role of PKC-δ and -ε in zymogen activation, but the targets of the kinases remain unclear. Two candidate processes that mediate zymogen activation are changes in 1) intracellular calcium and 2) intraluminal pH of zymogen-containing compartments (21, 26, 35). Several lines of evidence suggest that PKC can regulate these responses. First, PKC can phosphorylate calcium release channels, such as the inositol 1,4,5-trisphosphate receptor, altering the calcium dynamics of the cell (1, 5, 6, 17, 22). Some of these studies show an increase in calcium, a known effector of premature zymogen activation, but others show a decrease in calcium levels. None of these studies, however, demonstrate which PKC isoform is involved in these processes. Second, vacuolar ATPase (vATPase) is an ATP-dependent proton pump, which acidifies zymogen-containing compartments, optimizing conditions for zymogen activation (35). Little is known about the mechanism of vATPase regulation. It is likely that phosphorylation of various vATPase subunits by kinases including PKC may play a role in its assembly or activation. This would lead to acidification of intracellular compartments, providing a suitable environment for premature zymogen activation (4, 18).
An additional target for PKC isoforms in the pancreatic acinar cell is protein kinase D (PKD). PKD is an enzyme involved in a number of cellular processes including protein secretion, apoptosis, and cell proliferation. Recent evidence suggests that PKD 1 is a downstream target of PKC-δ, and it is possible that PKD may contribute to zymogen activation (3).
In conclusion, the results of the present study provide further insights into the role that the novel PKC isoforms δ and ε play in the mechanism of acute pancreatitis. In addition to activating transcription factors such as NFκB, they also mediate aberrant zymogen activation. Given their profound impact on the early stages of acute pancreatitis, novel PKC isoforms present themselves as attractive therapeutic targets. As future studies reveal more about the dynamics of the novel PKC family in the pancreatic acinar cell, strategies that block PKC-dependent mechanisms may reduce initiation and perpetuation of the disease.
Acknowledgments
We thank Bechien Wu, Courtney Jones, Meghan Kelly, and Davidson Gomes for expert technical assistance.
This work was supported by a National Institutes of Health Grant R21 (DK-69702 to E. Thrower), National Institutes of Health Grants RO1 (DK-33850 to J. Reeve, Jr. and DK-54021 to F. Gorelick), USC-UCLA Research Center for Alcoholic Liver and Pancreatic Injury Grant (5 P60 AA-11999 from the National Institute of Alcohol Abuse and Alcoholism, through H. Tsukamoto and S. Pandol; to S. Pandol), and a Veterans Affairs Merit and Senior Career Development Award (to F. Gorelick).
References
- 1.Arguin G, Regimbald-Dumas Y, Fregeau MO, Caron AZ, Guillemette G. Protein kinase C phosphorylates the inositol 1,4,5-trisphosphate receptor type 2 and decreases the mobilization of Ca2+ in pancreatoma AR4-2J cells. J Endocrinol. 2007;192:659–668. doi: 10.1677/JOE-06-0179. [DOI] [PubMed] [Google Scholar]
- 2.Bastani B, Yang L, Baldassare JJ, Pollo DA, Gardner JD. Cellular distribution of isoforms of protein kinase C (PKC) in pancreatic acini. Biochim Biophys Acta. 1995;1269:307–315. doi: 10.1016/0167-4889(95)00120-0. [DOI] [PubMed] [Google Scholar]
- 3.Berna MJ, Hoffmann KM, Tapia JA, Thill M, Pace A, Mantey SA, Jensen RT. CCK causes PKD1 activation in pancreatic acini by signaling through PKC-delta and PKC-independent pathways. Biochim Biophys Acta. 2007;1773:483–501. doi: 10.1016/j.bbamcr.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brisseau GF, Grinstein S, Hackam DJ, Nordstrom T, Manolson MF, Khine AA, Rotstein OD. Interleukin-1 increases vacuolar-type H+-ATPase activity in murine peritoneal macrophages. J Biol Chem. 1996;271:2005–2011. doi: 10.1074/jbc.271.4.2005. [DOI] [PubMed] [Google Scholar]
- 5.Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH. Calcineurin associated with the inositol 1,4,5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell. 1995;83:463–472. doi: 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 6.Caron AZ, Chaloux B, Arguin G, Guillemette G. Protein kinase C decreases the apparent affinity of the inositol 1,4,5-trisphosphate receptor type 3 in RINm5F cells. Cell Calcium. 2007;42:323–331. doi: 10.1016/j.ceca.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Chaudhuri A, Kolodecik TR, Gorelick FS. Effects of increased intracellular cAMP on carbachol-stimulated zymogen activation, secretion, and injury in the pancreatic acinar cell. Am J Physiol Gastrointest Liver Physiol. 2005;288:G235–G243. doi: 10.1152/ajpgi.00334.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cosen-Binker LI, Lam PP, Binker MG, Reeve J, Pandol S, Gaisano HY. Alcohol/cholecystokinin-evoked pancreatic acinar basolateral exocytosis is mediated by protein kinase C alpha phosphorylation of Munc18c. J Biol Chem. 2007;282:13047–13058. doi: 10.1074/jbc.M611132200. [DOI] [PubMed] [Google Scholar]
- 9.Gates LK, Ulrich CD, Miller LJ. Multiple kinases phosphorylate the pancreatic cholecystokinin receptor in an agonist-dependent manner. Am J Physiol Gastrointest Liver Physiol. 1993;264:G840–G847. doi: 10.1152/ajpgi.1993.264.5.G840. [DOI] [PubMed] [Google Scholar]
- 10.Grady T, Mah’moud M, Otani T, Rhee S, Lerch MM, Gorelick FS. Zymogen proteolysis within the pancreatic acinar cell is associated with cellular injury. Am J Physiol Gastrointest Liver Physiol. 1998;275:G1010–G1017. doi: 10.1152/ajpgi.1998.275.5.G1010. [DOI] [PubMed] [Google Scholar]
- 11.Halangk W, Kruger B, Ruthenburger M, Sturzebecher J, Albrecht E, Lippert H, Lerch MM. Trypsin activity is not involved in premature, intrapancreatic trypsinogen activation. Am J Physiol Gastrointest Liver Physiol. 2002;282:G367–G374. doi: 10.1152/ajpgi.00315.2001. [DOI] [PubMed] [Google Scholar]
- 12.Halangk W, Lerch MM, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, Domschke W, Lippert H, Peters C, Deussing J. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000;106:773–781. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hofbauer B, Saluja AK, Lerch MM, Bhagat L, Bhatia M, Lee HS, Frossard JL, Adler G, Steer ML. Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am J Physiol Gastrointest Liver Physiol. 1998;275:G352–G362. doi: 10.1152/ajpgi.1998.275.2.G352. [DOI] [PubMed] [Google Scholar]
- 14.Kikkawa U, Matsuzaki H, Yamamoto T. Protein kinase C delta (PKC delta): activation mechanisms and functions. J Biochem (Tokyo) 2002;132:831–839. doi: 10.1093/oxfordjournals.jbchem.a003294. [DOI] [PubMed] [Google Scholar]
- 15.Kukor Z, Mayerle J, Kruger B, Toth M, Steed PM, Halangk W, Lerch MM, Sahin-Toth M. Presence of cathepsin B in the human pancreatic secretory pathway and its role in trypsinogen activation during hereditary pancreatitis. J Biol Chem. 2002;277:21389–21396. doi: 10.1074/jbc.M200878200. [DOI] [PubMed] [Google Scholar]
- 16.Li C, Chen X, Williams JA. Regulation of CCK-induced amylase release by PKC-δ in rat pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G764–G771. doi: 10.1152/ajpgi.00111.2004. [DOI] [PubMed] [Google Scholar]
- 17.Matter N, Ritz MF, Freyermuth S, Rogue P, Malviya AN. Stimulation of nuclear protein kinase C leads to phosphorylation of nuclear inositol 1,4,5-trisphosphate receptor and accelerated calcium release by inositol 1,4,5-trisphosphate from isolated rat liver nuclei. J Biol Chem. 1993;268:732–736. [PubMed] [Google Scholar]
- 18.Nordstrom T, Grinstein S, Brisseau GF, Manolson MF, Rotstein OD. Protein kinase C activation accelerates proton extrusion by vacuolar-type H+-ATPases in murine peritoneal macrophages. FEBS Lett. 1994;350:82–86. doi: 10.1016/0014-5793(94)00738-1. [DOI] [PubMed] [Google Scholar]
- 19.Otani T, Chepilko SM, Grendell JH, Gorelick FS. Co-distribution of trypsinogen activation peptide and the granule membrane protein, GRAMP-92, in rat cerulein-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol. 1998;275:G999–G1009. doi: 10.1152/ajpgi.1998.275.5.G999. [DOI] [PubMed] [Google Scholar]
- 20.Otani T, Karne S, Gorelick FS. Generation of trypsinogen activation peptide (TAP) in rat isolated pancreatic acini under secretagogue hyperstimulation. Pancreas. 1998 doi: 10.1097/00006676-200007000-00062. [DOI] [PubMed] [Google Scholar]
- 21.Petersen OH, Sutton R. Ca2+ signalling and pancreatitis: effects of alcohol, bile and coffee. Trends Pharmacol Sci. 2006;27:113–120. doi: 10.1016/j.tips.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 22.Poirier SN, Poitras M, Chorvatova A, Payet MD, Guillemette G. FK506 blocks intracellular Ca2+ oscillations in bovine adrenal glomerulosa cells. Biochemistry. 2001;40:6486–6492. doi: 10.1021/bi010207k. [DOI] [PubMed] [Google Scholar]
- 23.Pollo DA, Baldassare JJ, Honda T, Henderson PA, Talkad VD, Gardner JD. Effects of cholecystokinin (CCK) and other secretagogues on isoforms of protein kinase C (PKC) in pancreatic acini. Biochim Biophys Acta. 1994;1224:127–138. doi: 10.1016/0167-4889(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 24.Raraty M, Ward J, Erdemli G, Vaillant C, Neoptolemos JP, Sutton R, Petersen OH. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci USA. 2000;97:13126–13131. doi: 10.1073/pnas.97.24.13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saluja A, Saito I, Saluja M, Houlihan MJ, Powers RE, Meldolesi J, Steer M. In vivo rat pancreatic acinar cell function during supramaximal stimulation with caerulein. Am J Physiol Gastrointest Liver Physiol. 1985;249:G702–G710. doi: 10.1152/ajpgi.1985.249.6.G702. [DOI] [PubMed] [Google Scholar]
- 26.Saluja AK, Donovan EA, Yamanaka K, Yamaguchi Y, Hofbauer B, Steer ML. Cerulein-induced in vivo activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology. 1997;113:304–311. doi: 10.1016/s0016-5085(97)70108-2. [DOI] [PubMed] [Google Scholar]
- 27.Saluja AK, Saluja M, Printz H, Zavertnik A, Sengupta A, Steer ML. Experimental pancreatitis is mediated by low-affinity cholecystokinin receptors that inhibit digestive enzyme secretion. Proc Natl Acad Sci USA. 1989;86:8968–8971. doi: 10.1073/pnas.86.22.8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Satoh A, Gukovskaya AS, Nieto JM, Cheng JH, Gukovsky I, Reeve JR, Jr, Shimosegawa T, Pandol SJ. PKC-δ and -ε regulate NF-κB activation induced by cholecystokinin and TNF-α in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G582–G591. doi: 10.1152/ajpgi.00087.2004. [DOI] [PubMed] [Google Scholar]
- 29.Satoh A, Gukovskaya AS, Reeve JR, Jr, Shimosegawa T, Pandol SJ. Ethanol sensitizes NF-κB activation in pancreatic acinar cells through effects on protein kinase C-ε. Am J Physiol Gastrointest Liver Physiol. 2006;291:G432–G438. doi: 10.1152/ajpgi.00579.2005. [DOI] [PubMed] [Google Scholar]
- 30.Sherwood MW, Prior IA, Voronina SG, Barrow SL, Woodsmith JD, Gerasimenko OV, Petersen OH, Tepikin AV. Activation of trypsinogen in large endocytic vacuoles of pancreatic acinar cells. Proc Natl Acad Sci USA. 2007;104:5674–5679. doi: 10.1073/pnas.0700951104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smeets RL, Rao RV, van Emst-de Vries SE, De Pont JJ, Miller LJ, Willems PH. Reduced cholecystokinin receptor phosphorylation and restored signalling in protein kinase C down-regulated rat pancreatic acinar cells. Pflügers Arch. 1998;435:422–428. doi: 10.1007/s004240050533. [DOI] [PubMed] [Google Scholar]
- 32.Sorensen CE, Novak I. Visualization of ATP release in pancreatic acini in response to cholinergic stimulus. Use of fluorescent probes and confocal microscopy. J Biol Chem. 2001;276:32925–32932. doi: 10.1074/jbc.M103313200. [DOI] [PubMed] [Google Scholar]
- 33.Tapia JA, Garcia-Marin LJ, Jensen RT. Cholecystokinin-stimulated protein kinase C-delta kinase activation, tyrosine phosphorylation, and translocation are mediated by Src tyrosine kinases in pancreatic acinar cells. J Biol Chem. 2003;278:35220–35230. doi: 10.1074/jbc.M303119200. [DOI] [PubMed] [Google Scholar]
- 34.Thrower EC, Diaz de Villalvilla AP, Kolodecik TR, Gorelick FS. Zymogen activation in a reconstituted pancreatic acinar cell system. Am J Physiol Gastrointest Liver Physiol. 2006;290:G894–G902. doi: 10.1152/ajpgi.00373.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waterford SD, Kolodecik TR, Thrower EC, Gorelick FS. Vacuolar ATPase regulates zymogen activation in pancreatic acini. J Biol Chem. 2005;280:5430–5434. doi: 10.1074/jbc.M413513200. [DOI] [PMC free article] [PubMed] [Google Scholar]