

Abstract
Sustained oxidative stress is a known sequel to focal cerebral ischemia. This study examined the effects of treatment with a single dose or sustained infusion of the redox-modulating MnPorphyrin MnIIITDE-2-ImP5+ on outcome from middle cerebral artery occlusion (MCAO) in the rat. Normothermic rats were subjected to 90 min MCAO followed by 90 min reperfusion and then were treated with a single intracerebroventricular dose of MnIIITDE-2-ImP5+. Neurologic and histologic outcomes were assessed at 1 or 8 weeks post-ischemia. A single dose of MnIIITDE-2-ImP5+ caused a dose-dependent improvement in histologic and neurologic outcome when assessed 1 week post-ischemia. MnIIITDE-2-ImP5+ afforded preservation of brain aconitase activity at 5.5 hrs after reperfusion onset, consistent with its known antioxidant properties. MnIIITDE-2-ImP5+ also attenuated post-ischemic NF-κ B activation. Evidence for effects on cerebral infarct size and neurologic function had completely dissipated when rats were allowed to survive for 8 weeks post-ischemia. In contrast, a 1-week continuous intracerebroventricular MnIIITDE-2-ImP5+ infusion caused persistent and substantive reduction in both cerebral infarct size and neurologic deficit at 8 weeks post-ischemia. Pharmacologic modulation of post-ischemic oxidative stress is likely to require sustained intervention for enduring efficacy in improving neurologic and histologic outcome from a transient focal ischemic insult.
INTRODUCTION
Cerebral ischemia causes oxidative stress. A pro-oxidative state can induce direct tissue damage and also participates in regulation of the brain’s delayed response to injury. Numerous antioxidants have been demonstrated to ameliorate focal ischemic brain injury. Most, but not all [1, 2], preclinical trials have utilized post-ischemic observation intervals of several hours to days to define antioxidant efficacy.
Post-ischemic histologic and neurologic responses to ischemia persist for weeks after perfusion has been restored [3–5]. This is relevant to translation of preclinical studies to clinical trials, which typically assess outcome at intervals of several months post-ictus. Thus, observations made in the first few days after experimental stroke may not predict efficacy in long-term outcome clinical trials.
A major neuroprotective effect of a class of antioxidants, known as MnPorphyrins, has been documented. Post-ischemic treatment of a single dose of either Mn(III) meso-tetrakis (N-ethylpyridinium–2-yl)porphyrin (MnTE-2-PyP5+) or Mn(III) tetrakis(N,N’-diethylimidazolium-2-yl)porphyrin (MnIIITDE-2-ImP5+) [6] decreased cerebral infarct size by up to 50%, even when treatment was begun 6 hours after completion of a 90 min focal ischemic insult [7, 8]. However, there is no information on effects of effects of MnPorphyrins on long-term post-ischemic outcome or in vivo mediators of oxidative stress.
The current studies were designed to: 1) define a dose-response effect for post-ischemic MnIIITDE-2-ImP5+ treatment; 2) measure MnIIITDE-2-ImP5+ effects on post- ischemic aconitase activity and nuclear factor kappa beta (NF-κ B) activation, and 3) examine long-term behavioral and histologic outcome responses to MnIIITDE-2-ImP5+ treatment.
MATERIALS AND METHODS
The following studies were approved by the Duke University Animal Care and Use Committee, Durham, NC, USA.
Surgical Preparation
Male Wistar rats (250–275 gm; Harlan Sprague Dawley, Inc. Indianapolis, IN) were anesthetized with 64-mg/kg intraperitoneal sodium pentobarbital and positioned in a stereotactic head frame. The skin was infiltrated with 1.0% lidocaine and a midline scalp incision was made. A burr hole was drilled over the left hemisphere, 7.2 mm anterior to the interaural line and 1.4 mm lateral to the sagittal suture. An intracerebroventricular (ICV) cannula (33 gauge) was positioned with the tip in the left lateral ventricle and fixed in place with screws and cyanoacrylate. The incision was closed with suture around this assembly. After emergence from anesthesia, the animals were returned to their cages with free access to water and food.
Following 2–3 days recovery, rats were allowed access to water but fasted from food for 12 hours to standardize glycemic state. Rats were then anesthetized with isoflurane in O2. Following tracheal intubation, the lungs were mechanically ventilated to maintain normocapnia. A 22-g needle thermistor was percutaneously placed adjacent to the skull beneath the temporalis. Pericranial temperature was servoregulated at 37.5±0.1 °C by surface heating or cooling. The inspired isoflurane concentration was adjusted to 1.0– 1.5% in 50% O2/balance N2. The tail artery was cannulated. The animals were then prepared for MCAO as previously described [7, 9]. A midline cervical incision was made and the right common carotid artery was identified. The external carotid artery (ECA) was isolated and the occipital, superior thyroid, and external maxillary arteries were ligated and divided. The internal carotid artery (ICA) was dissected distally until the origin of the pterygopalatine artery was visualized. Following surgical preparation, a 20 min interval was allowed for physiologic stabilization.
Five min before MCAO onset, heparin (50 IU intra-arterial) was given to prevent intra-arterial thrombosis. A 0.25-mm diameter nylon monofilament, prepared with a silicone tip, was inserted into the ECA stump and passed distally through the ICA (20 mm from carotid bifurcation) until resistance was felt and the filament was secured. At MCAO onset, isoflurane was reduced to 0.8–1.0%.
After 90 min of MCAO, the occlusive filament was removed. The anesthetic state and pericranial temperature regulation were continued for an additional 100 min. The tail artery catheter was removed and the wounds were closed with suture. Isoflurane was discontinued. Upon recovery of the righting reflex, the tracheas were extubated and the animals were placed in an O2 enriched environment (FIO2=50%) for 1 hour. Animals were then randomized to experimental groups (see below).
Neurologic Evaluation
At completion of the predefined recovery interval, rats underwent a neurologic examination to evaluate sensorimotor function [10]. With the observer blinded to group assignment, this test explored six different functions (spontaneous activity, movement symmetry, forepaw outstretching, climbing, body proprioception and response to vibrissae touch). The performance in each test was rated with a 0–3 point score. The score given to each animal was the sum of all six individual scores, 0 being the minimum (worst) and 18 being the maximum (best) score.
Measurement of Cerebral Infarct Volume
Animals were weighed, anesthetized with isoflurane, and decapitated. The brains were removed, frozen at −40 °C in 2-methylbutane, and stored at −70 °C. Infarct volume was measured by comparing the volume of histologically normal tissue observed in the ischemic hemisphere to the expected volume of normal tissue as derived from measurements of the contralateral, non-ischemic hemisphere [11]. Serial quadruplicate 20-μm thick coronal sections were taken using a cryotome at 660-μm intervals over the rostral-caudal extent of the infarct. The sections were dried and stained with hematoxylin and eosin. A section from each 660-μm interval was digitized with a video camera controlled by an image analyzer. The image of each section was stored as a 1280 x 960 pixel matrix and displayed on a video monitor. With the observer blinded to experimental condition, the following regions of interest (ROI) were cursor outlined: non-infarcted ipsilateral cerebral cortex, non-infarcted ipsilateral subcortex, contralateral cerebral cortex, and contralateral subcortex. The area within each ROI (mm2) was determined by automated counting of calibrated pixels. Ipsilateral non-infarcted cortex and subcortex areas were subtracted from the corresponding contralateral ROI values to estimate the area of ischemic tissue damage to control for brain edema [12, 13]. Infarct volumes (mm3) were computed as running sums of subtracted infarct area multiplied by the known interval (e.g., 660 μm) between sections over the rostral-caudal extent of the infarct calculated as an orthogonal projection [14].
Experimental Designs
The same individuals performed surgical procedures and outcome analyses in all experiments. In each experiment, rats were randomly assigned to respective treatment groups and experimenters were blind to group assignment. An a priori power analysis was conducted using data from the same model reported in prior studies [7, 8], which indicated that a group size of 15 rats would be sufficient to allow detection of a 40% reduction in cerebral infarct size, given β = 0.8 and P<0.05.
Experiment 1: MnIIITDE-2-ImP5+ Dose-Response Analysis/One-Week Recovery
Rats received vehicle (Dulbecco’s phosphate buffered saline, 10 μl, n = 20), 100 ng MnIIITDE-2-ImP5+ (a gift from Incara Pharmaceuticals, Inc, Research Triangle Park, NC) (n = 19), 300 ng MnIIITDE-2-ImP5+ (n = 19), or 900 ng MnIIITDE-2-ImP5+ (n = 20). Drug (or vehicle) was given 90 min after onset of MCAO reperfusion as a 10 μl ICV injection over 5 min. Neurologic function and cerebral infarct size were assessed 1 week post-ischemia.
Experiment 2: Aconitase Activity
Aconitase is selectively inactivated by superoxide [15] and thus provides a marker of early reperfusion oxidative stress [7, 8]. Because 900 ng ICV MnIIITDE-2-ImP5+ provided the largest numeric effect on infarct size in Experiment 1, that dose was used in this and all subsequent studies.
Rats underwent 90 min MCAO and treatment with MnIIITDE-2-ImP5+ (900 ng ICV) or vehicle at 90 min after onset of reperfusion as described in Experiment 2 (n=5 per group). Four hours later rats were re-anesthetized. The cerebral vasculature was perfused with 50 ml cold saline. The brains were removed and bisected into hemispheres. A 5 mm coronal section was cut through the brain to capture tissue within the MCA flow distribution in both hemispheres. Aconitase activity was measured as previously described [16, 17]. The tissue was weighed and homogenized in 1 ml ice-cold mitochondrial isolation buffer (20 mM sucrose, 210 mM mannitol, 5 mM Tris-HCl, 1 mM EDTA, pH 7.4). The suspension was centrifuged (600g) for 5 min at 4 °C. The supernatant was then centrifuged at 17,000 g for 10 min at 4 °C. The mitochondrial fraction was suspended in 0.1 ml buffer containing 50 mM Tris-HCl (pH 7.4) containing 0.6 MnCl2 and sonicated. Protein content was measured using a standard curve derived by using Biorad Protein Assay Dye Reagent concentrate (Hercules, CA). Aconitase activity was spectrophotometrically measured at 340 nm in a 1.0 ml reaction mixture containing 50 mM Tris-HCl (pH 7.4), 5 mM sodium citrate, 0.6 mM MnCl2, 0.2 mM NADP, 2 units isocitrate dehydrogenase, and 100 μg of extracted protein. Values were normalized to the contralateral (non-ischemic) hemisphere in each rat to control for any contamination from intravascular blood). The ratio of aconitase activity in the ischemic and contralateral hemispheres was calculated for each animal.
Experiment 3: NF-κ B Activation
Constitutive overexpression of superoxide dismutase in transgenic mice decreases ischemia-induced superoxide production and NF-κ B activation [18]. MnPorphyrins catalytically dismutate superoxide [6] and prevent in vitro NF-κ B binding to DNA canonical κ B binding motifs [19]. To assess whether these properties persist in the context of post-ischemic oxidative stress, NF-κ B activation was assessed by electrophoretic mobility shift assay (EMSA). Rats were subjected to 90 min of transient MCAO and randomly received either 10 μl of PBS or MnIIITDE-2-ImP5+ (900ng) ICV 90 min after reperfusion (n = 6 per group). Ischemic and contralateral hemisphere brain tissues from within the MCA distribution were harvested at 6 hours after reperfusion and snap frozen in liquid nitrogen. Tissues were stored at −80° C for later analysis. After all samples were collected, nuclear protein extraction was performed. The brain tissues were homogenized in a cold lysis buffer (0.5% Nonidet P 40 Substitute, 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM EDTA, 0.5 mM DTT, 0.12 M sucrose, 1 mM PMSF and 1% protease inhibitor cocktail (Sigma-Aldrich, Co. St. Louis, MO)) using a Dounce homogenizer, incubated on ice for 10 min and centrifuged at 10,000g for 2 min. Nuclear pellets were washed once with lysis buffer without NP40 or sucrose and centrifuged. Supernatants were discarded and nuclear pellets were re-suspended in an equal volume of cold buffer containing 20 mM HEPES, 1.5 mM MgCl2, 0.5 mM DTT, 0.6 M KCl, 25% glycerol, 1 mM PMSF and 1% protease inhibitor, incubated with shaking for 30 min at 4° C and then centrifuged at 12,000g for 20 min at 4° C. The resulting supernatants containing nuclear protein were collected and the protein concentrations were determined using the Bradford protein reagent (Pierce Biochemicals, Inc., Rockford, IL). NF-κ B double – stranded consensus oligonucleotide (5’AGTTGAGGGGACTTTCCCAGGC-3’) was labeled with 32P-ATP using T4 polynucleotide kinase. In a total volume of 15 μl, purified 32P-labeled probe was incubated with 5μg of nuclear protein in a binding reaction mixture containing 10 mM Tris-Hcl pH 7.9, 1 mM EDTA, 50 mM NaCl, 10% glycerol, 0.02% NP40, 0.2 mM PMSF, 1 mM DTT, 0.5μg poly (dI-dC) for 20 min at room temperature. Bound complex was separated from free probe by electrophoresis through a 60 min pre-run 6% non-denaturing acrylamide:bis-acrylamide (19:1) gel in 0.5 X Tris-borate/EDTA (TBE) at 150 V. All gels were vacuum-dried, and then exposed to x-ray film overnight, which was then developed. The NF-κ B bands were analyzed using densitometry. The NF-κ B band used to quantify the effect of drug on NF-κ B DNA binding was confirmed in tissue extracts by supershift using p50 and p65 subunit antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) with 2 μg antibody per 5 μg nuclear protein incubated for 30 min prior to adding the probe. Specificity of probe binding was confirmed by competition with unlabeled probe (NF-κ B consensus oligonucleotide sequence) at a ratio of 100:1.
Experiment 4: Single Dose MnIIITDE-2-ImP5+/Eight-Week Recovery
Vehicle (n = 16) was compared to 900 ng MnIIITDE-2-ImP5+ (n = 15), given ICV 90 min after reperfusion onset. Neurologic function and cerebral infarct volumes were measured at 8 weeks post-ischemia.
Experiment 5: One-Week MnIIITDE-2-ImP5+ Infusion/Eight-Week Recovery
This study examined effects of sustained MnIIITDE-2-ImP5+ treatment on long-term outcome. Surgical procedures were as described above with the following exception. At 90 min post-MCAO, rats were treated with 900 ng MnIIITDE-2-ImP5+ ICV (n = 13) or vehicle (n = 13) bolus. In addition, a primed PE-10 cannula, connected to a precharged osmotic pump, was connected to the ICV cannula to deliver a continuous infusion of MnIIITDE-2-ImP5+ (56 ng/hr) or vehicle to the MnIIITDE-2-ImP5+ and vehicle bolus groups, respectively. The pump assembly was implanted subcutaneously. The animals were then allowed to recover. At 7 days post-MCAO, the rats were anesthetized and the assembly was removed. The rats were allowed to survive an additional 7 weeks.
Statistical Analysis
Parametric data (physiologic values, cerebral infarct volumes, and NF-κ B optical densities, aconitase activities) were compared by 1-way ANOVA and Fischer’s protected least squares difference test when appropriate. Parametric data are expressed as mean ± standard deviation. Neurologic scores were compared by the Kruskal-Wallis H statistic or Mann-Whitney U statistic where appropriate and are expressed as median ± interquartile range.
RESULTS
Experiment 1: MnIIITDE-2-ImP5+ Dose-Response Analysis/One-Week Recovery
Physiologic values are reported in Table 1. No significant differences were present among groups. MnIIITDE-2-ImP5+ improved sensorimotor function in a dose-dependent manner (0 ng = 8±3, 100 ng = 9±2, 300 ng = 10±4, 900 ng = 11±3, P= 0.006). A dose-dependent reduction in total infarct volume was observed for MnIIITDE-2-ImP5+ (0 ng = 172 ± 78, 100 ng = 144 ± 88, 300 ng = 106 ± 91, 900 ng = 95 ± 89, P=0.03). The effect in the cortex did not reach statistical significance (P=0.09), while the effect in subcortex did (P=0.003). See Figure 1.
Table 1.
Physiologic Values (Experiment 1: Dose Response Effects of MnIIITDE-2- ImP5+)
| MnIIITDE-2-ImP5+ | Dose (ng) | |||
|---|---|---|---|---|
| 0 | 100 | 300 | 900 | |
| n | 20 | 19 | 19 | 20 |
| 10 Min Pre-Ischemia | ||||
| MAP (mmHg) | 82±15 | 76±15 | 79±15 | 81±13 |
| Arterial pH | 7.43±.05 | 7.44±.05 | 7.44±.03 | 7.44 ±.04 |
| PaCO2 (mmHg) | 38±4 | 39±4 | 38±3 | 38±3 |
| PaO2 (mmHg) | 166±32 | 165±36 | 186±43 | 174±36 |
| Plasma Glucose (mg/dl) | 115±18 | 117±12 | 112±19 | 116±16 |
| 45 min Post-Ischemia Onset | ||||
| MAP (mmHg) | 87±20 | 90±17 | 87±13 | 90±16 |
| Arterial pH | 7.41±.03 | 7.41±.04 | 7.44±.04 | 7.43±.05 |
| PaCO2 (mmHg) | 39±3 | 42±4 | 39±4 | 39±4 |
| PaO2 (mmHg) | 184±37 | 199±37 | 178±34 | 175±34 |
| 45 min Post-Treatment | ||||
| MAP (mmHg) | 80±15 | 83±16 | 78±8 | 82±13 |
| Arterial pH | 7.43±.04 | 7.44±.06 | 7.45±.04 | 7.45±.04 |
| PaCO2 (mmHg) | 37±4 | 37±5 | 37±5 | 36±5 |
| PaO2 (mmHg) | 184±37 | 199±37 | 178±35 | 175±34 |
Values = mean ± standard deviation. MAP = mean arterial pressure.
Figure 1.
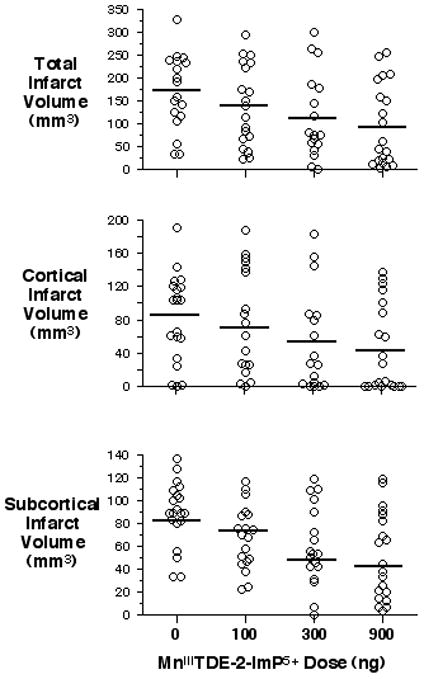
Short-term outcome is dose-dependently improved by a single dose of MnIIITDE-2-ImP5+. Infarct volumes were measured 1 week after 90 min MCAO and treatment with single dose vehicle or MnIIITDE-2-ImP5+ 90 min post-reperfusion. Open circles indicate individual rat values. Horizontal bars indicate group mean. A main effect for MnIIITDE-2-ImP5+ dose was seen in total (P = 0.03) and subcortical (P = 0.003) infarct volumes. Values in the cortex did not reach significance (P = 0.09).
Experiment 2: Aconitase Activity
Aconitase activity in the ischemic hemisphere of vehicle-treated rats was 74±5% of that in the contralateral hemisphere. This ratio (84±10%) was greater in the MnIIITDE-2-ImP5+ group (P=0.03).
Experiment 3: NF-κ B Activation
NF-κ B activation was increased in the ischemic hemisphere at 6 hrs post-ischemia versus the contralateral hemisphere where NF-κ B binding was absent. The NF-κ B band was super-shifted by anti-p65 (Figure 2a). MnIIITDE-2-ImP5+ (900 ng given ICV at 90 min after onset of reperfusion) decreased ipsilateral NF-κ B binding in nuclear extracts at 6 hrs after onset of reperfusion (P = 0.02) (Figure 2b).
Figure 2.

Post-ischemic NF-κ B activation is suppressed by MnIIITDE-2-ImP5+. NF-κ B activation was measured at 6 hrs post-MCAO in rats treated with vehicle or 900 ng MnIIITDE-2-ImP5+ at 90 min after onset of reperfusion. A) Representative EMSA. A=probe with no sample, B=positive control (HeLa), C=positive control with cold probe, D=ischemia hemisphere (MnIIITDE-2-ImP5+), E=ischemia hemisphere with p50 antibody (MnIIITDE-2-ImP5+), F=ischemia hemisphere with p65 antibody (M nIIITDE-2-ImP5+), G=ischemia hemisphere (vehicle), H=ischemia hemisphere (vehicle) with p50 antibody, I=ischemia hemisphere (vehicle) with p65 antibody. A substantially greater supershift was seen for p65 (arrow) than p50. B) Open circles indicate relative optical density values in individual rat NF-κ B bands in the hemisphere ipsilateral to MCAO. Horizontal bars indicate group mean values.
Experiment 4: Single Dose MnIIITDE-2-ImP5+/Eight-Week Recovery
At 8 weeks post-ischemia there were no differences between groups for neurologic score (vehicle = 15 ± 3, MnIIITDE-2-ImP5+ = 15 ± 3, P = 0.37). No differences were present for total (vehicle = 149 ± 73 mm3, MnIIITDE-2-ImP5+ = 149 ± 81 mm3, P = 0.99), subcortical (vehicle = 52 ± 29 mm3, MnIIITDE-2-ImP5+ = 55 ± 24 mm3, P = 0.72), or cortical (vehicle = 97 ± 52 mm3, MnIIITDE-2-ImP5+ = 94 ± 87 mm3, P = 0.87) infarct volumes. See Figure 3.
Figure 3.
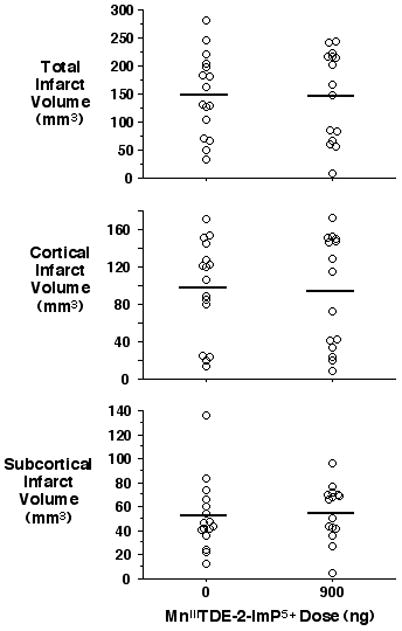
Neuroprotective effect of single dose MnIIITDE-2-ImP5+ is absent at 8 weeks post-MCAO. Infarct volumes were measured 8 weeks after 90 min MCAO and treatment with single dose vehicle or MnIIITDE-2-ImP5+ (900 ng) at 90 min post-reperfusion. Open circles indicate individual rat values. Horizontal bars indicate group mean. There was no statistical difference between groups.
Experiment 5: One-Week MnIIITDE-2-ImP5+ Infusion/Eight-Week Recovery
Sensorimotor function was better in rats treated with MnIIITDE-2-ImP5+ (0 ng = 9±3.75, 900 ng = 10±2.75, P = 0.03). Cortical (vehicle = 79 ± 18 mm3, MnIIITDE-2-ImP5+ = 53 ± 21 mm3, P = 0.003), subcortical (vehicle = 90 ± 44 mm3, MnIIITDE-2-ImP5+ = 38 ± 44 mm3, P = 0.006), and total (vehicle = 168 ± 58 mm3, MnIIITDE-2-ImP5+ = 91 ± 60 mm3, P = 0.003) infarct volumes were smaller in MnIIITDE-2-ImP5+-treated rats (Figure 4).
Figure 4.
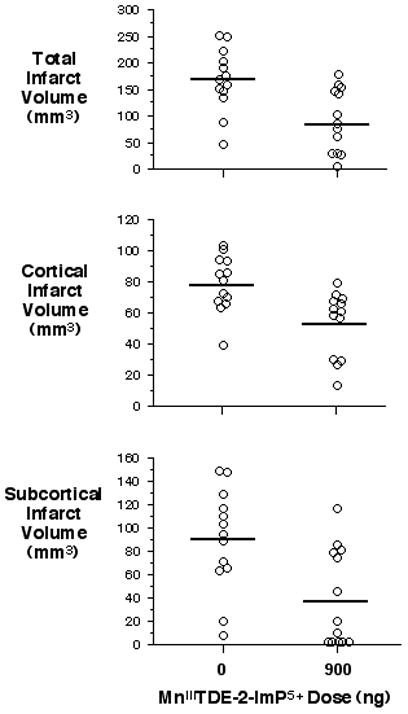
One week MnIIITDE-2-ImP5+ infusion provides protection at 8 weeks post- MCAO. Infarct volumes were measured 8 weeks after 90 min MCAO and treatment with vehicle or MnIIITDE-2-ImP5+ beginning at 90 min post-reperfusion and continued for 7 days. Open circles indicate individual rat values. Horizontal bars indicate group mean. Cortical (P=0.003), subcortical (P=0.006) and total (P=0.003) infarct volumes were reduced by MnIIITDE-2-ImP5+.
DISCUSSION
Experiment 1 confirmed the previously reported observation that post-ischemic treatment with a single bolus of the redox-modulating MnPorphyrin MnIIITDE-2-ImP5+ reduces focal ischemic brain damage when evaluated 1 week post-ischemia [8]. This finding is now extended by demonstration of a dose-response effect. Using the maximally protective dose defined in Experiment 1 (i.e., 900 ng), we next observed an attenuation of post-ischemic aconitase inactivation and NF-κ B nuclear translocation consistent with the known in vitro redox-regulating properties of this compound [6, 19]. Outcome was then examined at 8 weeks post-ischemia. At 8 weeks, no outcome benefit from a single post-ischemic MnIIITDE-2-ImP5+ treatment was present. This indicates deterioration of the protective effect of redox-modulating MnPorphyrins over a sustained recovery interval and the importance of observing effects on long-term outcome when testing pharmacologic interventions in experimental stroke models [20].
In contrast to single bolus treatment, continuous post-ischemic treatment with MnIIITDE-2-ImP5+ for 1 week caused persistent and substantial neurologic and histologic benefit when outcome was evaluated 8 weeks post-ischemia (e.g., 46% reduction in total cerebral infarct size). This is consistent with persistent oxidative stress spanning the first week of recovery that modulates long-term outcome. Previous studies have shown that post-ischemic formation of reactive oxygen and nitrogen species (ROS/RNS) occurs during the first several hours after reperfusion [21, 22]. Classic interpretation is that reperfusion injury occurs rapidly, attributable to renewed availability of oxygen as a substrate. Aconitase is specifically sensitive to superoxide [15]. The expected catalytic consumption of superoxide in the early recirculation interval by MnIIITDE-2-ImP5+ was confirmed by better preservation of aconitase activity when measured at 4-hrs post-MCAO. This is consistent with reported selective effects of the compound on aconitase activity preservation in primary mixed neuronal-glial cell cultures exposed to oxygen-glucose deprivation [8]. However, the importance of this early effect (and thus immediate reperfusion injury) on ischemic outcome can be questioned because several studies have demonstrated substantial benefit with antioxidant treatment given many hours after reperfusion onset when reperfusion-induced formation of ROS/RNS would be expected to have abated [2, 7, 8].
The results of our outcome analyses indicate that persistent oxidative stress modulates long-term ischemic outcome. It is known that oxidative stress modulates post-ischemic inflammatory and apoptotic responses [23, 24]. These responses can be attenuated by either overexpression of endogenous antioxidant enzymes or with pharmacologic treatment [23–26]. Therefore, antioxidants may be pleiotropic acting as scavengers of either diffusely or locally distributed ROS as well as having the potential to modulate redox-regulated transcription factors, such as NF-κ B [27], with the respective roles and impact on outcome being a function of timing of treatment and duration of outcome observation, respectively.
A single bolus of MnIIITDE-2-ImP5+ given at 90 min post-MCAO substantively reduced NF-κ B activation when assessed 4.5 hrs later. This is consistent with the observed amelioration of superoxide-specific aconitase inactivation, as superoxide overproduction participates in NF-κ B activation [28], most likely through increased hydrogen peroxide [29, 30]. NF-κ B activation persists for days post-ischemia and can be inhibited by overexpression of endogenous and pharmacologic antioxidants [18, 31, 32] or inhibition of Akt phosphorylation [33]. Inhibition of NF-κ B activation by MnIIITDE-2-ImP5+ has not been previously evaluated in ischemic brain but has been demonstrated in human pancreatic islet cells [34] and bone marrow-derived macrophages and dendritic cells [19]. Our study demonstrated a suppression of NF-κ B activation at 6 hrs post-ischemia following a single dose of MnIIITDE-2-ImP5+. The MnIIITDE-2-ImP5+ tissue half-life following ICV injection is 8–10 hrs [8]. It is plausible that a single bolus provides an insufficient duration of action to persistently dampen redox-regulated gene transcription stimulated by oxidative stress. This could explain the failure of a single dose of MnIIITDE-2-ImP5+ to improve long-term outcome, which warrants further investigation.
To our knowledge, there have been no other studies comparing acute single dose versus sustained antioxidant therapy on post-ischemic outcome. Marshall et al. [2] reported efficacy at 10 weeks post-MCAO from NXY-059 treatment sustained for 48 hrs post-ischemia onset. Ley et al. [1] demonstrated 30 day protection from stilbazulenyl nitrone in rats subjected to MCAO when treatment was sustained for 48 hrs post-ischemia. In neither study was acute bolus or sustained treatment evaluated. Sustained (days) post-ischemic suppression of pro-inflammatory responses and NF-κ B activation has been observed in mice overexpressing copper/zinc superoxide dismutase (Cu/Zn SOD) [18, 23, 32]. However, because Cu/Zn SOD was constitutively overexpressed, it cannot be stated what duration of increased Cu/Zn SOD activity was essential to persistently inhibit oxidative stress. To pursue the hypothesis that sustained inhibition of cascades stimulated by oxidative stress is required to cause an improved long-term outcome, studies of varied durations of antioxidant therapy and associated relationships with activation of redox-regulated transcription factors and downstream protein expression will be required.
It also is likely that properties of MnPorphyrins other than superoxide dismutation are relevant to long-term outcome. Mn(III) tetrakis(4-benzoic acid)porphyrin, MnIIITBAP, an anionic analogue of MnIIITDE-2-ImP5+, has been shown to inhibit apoptosis-inducing factor (AIF) in post-ischemic murine brain [35], likely through peroxynitrite (but not superoxide) scavenging [36]. It has been shown that the ability of MnPorphyrins to dismute superoxide parallels their ability to reduce peroxynitrite to either nitrogen dioxide or nitrite [37]. MnTDE-2-ImP5+ [6] may also effectively remove peroxynitrite in our MCAO model. The removal of reactive species may then modulate redox-based cellular transcriptional activity, such as inhibition of NF-κ B as shown in this work.
Tauskela et al. [38] have challenged the concept that the principle mechanism by which redox-modulating MnPorphyrins protect against oxidative stress is scavenging of reactive species. They reported that protection of cortical neuronal and PC-12 cultures against oxygen-glucose deprivation correlates with the ability of respective MnPorphyrins to suppress increases intracellular calcium, but not with ROS/RNS-scavenging potential. Whether the multiple proposed neuroprotective mechanisms of antioxidants (e.g., suppression of NF-κ B activation and potentially other redox-regulated transcription factors, inhibition of AIF, and attenuation of intracellular calcium accumulation) play a role in short-term versus long-term protection warrants close inspection.
We believe that our model was valid. Pericranial temperature was monitored and controlled during ischemia and 100 min post-reperfusion. MnIIITDE-2-ImP5+ does not affect body temperature for at least the first 18 hours after treatment [8]. Toxicity studies have demonstrated a species specific arterial hypotensive effect of intravenous MnIIITDE-2-ImP5+ in rats, but not mice, dogs, guinea pigs, or baboons [39]. For that reason, we selected the ICV route for the current rat experiments as the rat remains our best available model for long-term MCAO outcome analysis [40], and this route of administration abrogated any potential interactions between perfusion pressure and ischemic outcome. No effects of MnIIITDE-2-ImP5+ on blood pressure were observed when given ICV. In mice, at least short-term efficacy is provided by intravenous treatment with MnIIITDE-2-ImP5+ [8], leaving it plausible that a similar benefit will be present with intravenous treatment in primates or humans.
A dose of 900 μg ICV yielded maximal benefit in the dose ranges studied (300–900μg, Figure 1). It is likely that this dose approaches maximum efficacy. Prior work using ICV MnIIITDE-2-ImP5+ found no difference between 300 and 4500 ng in a similar MCAO 1-week outcome model [8]. This suggested a U-shaped dose-response, which is in part supported by the current data. It is likely, however, that larger doses would reverse this benefit, but this needs to be determined. ICV MnIIITDE-2-ImP5+ doses exceeding 9 μg (e.g., 10-fold greater doses than the bolus dose used in the current studies) cause central nervous system toxicity (proptosis, ataxia, and sound hypersensitivity) [8]. No evidence of toxicity was observed in the current experiments despite sustained infusions for 1 week.
In summary, treatment with a single dose of MnIIITDE-2-ImP5+ given at 90 min after reperfusion from 90 min MCAO caused a dose-dependent improvement in short-term ischemic outcome. Despite this, complete dissipation of protection was observed at 8 weeks post-ischemia. In contrast, sustained improvement in outcome was produced by continuous infusion of MnIIITDE-2-ImP5+ for 1 week post-ischemia. MnIIITDE-2-ImP5+ was found to inhibit ROS-mediated aconitase enzymatic dysfunction, but also NF-κ B DNA-binding and activation. We conclude that modulation of post-ischemic oxidative stress may require sustained intervention for sustained efficacy.
Acknowledgments
NIH P01HL42444 supported this work.
References
- 1.Ley JJ, Vigdorchik A, Belayev L, Zhao W, Busto R, Khoutorova L, Becker DA, Ginsberg MD. Stilbazulenyl nitrone, a second-generation azulenyl nitrone antioxidant, confers enduring neuroprotection in experimental focal cerebral ischemia in the rat: neurobehavior, histopathology, and pharmacokinetics. J Pharmacol Exp Ther. 2005;313:1090–1100. doi: 10.1124/jpet.105.083386. [DOI] [PubMed] [Google Scholar]
- 2.Marshall JW, Cummings RM, Bowes LJ, Ridley RM, Green AR. Functional and histological evidence for the protective effect of NXY-059 in a primate model of stroke when given 4 hours after occlusion. Stroke. 2003;34:2228–2233. doi: 10.1161/01.STR.0000087790.79851.A8. [DOI] [PubMed] [Google Scholar]
- 3.Colbourne F, Corbett D. Delayed postischemic hypothermia: a six month survival study using behavioral and histological assessments of neuroprotection. J Neurosci. 1995;15:7250–7260. doi: 10.1523/JNEUROSCI.15-11-07250.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Kawaguchi M, Kimbro JR, Drummond JC, Cole DJ, Kelly PJ, Patel PM. Isoflurane delays but does not prevent cerebral infarction in rats subjected to focal ischemia. Anesthesiology. 2000;92:1335–1342. doi: 10.1097/00000542-200005000-00023. [DOI] [PubMed] [Google Scholar]
- 6.Batinic-Haberle I, Spasojevic I, Stevens RD, Hambright P, Neta P, Okado-Matsumoto A, Fridovich I. New class of potent catalysts of O2.-dismutation. Mn(III) ortho-methoxyethylpyridyl- and di-ortho-methoxyethylimidazolylporphyrins. Dalton Trans. 2004:1696–1702. doi: 10.1039/b400818a. [DOI] [PubMed] [Google Scholar]
- 7.Mackensen GB, Patel M, Sheng H, Calvi C, Batinic-Haberle I, Day BJ, Liang LP, Fridovich I, Crapo JD, Pearlstein RD, Warner DS. Neuroprotection from delayed post-ischemic administration of a metalloporphyrin catalytic antioxidant. J Neurosci. 2001;21:4582–4592. doi: 10.1523/JNEUROSCI.21-13-04582.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheng H, Enghild JJ, Bowler R, Patel M, Batinic-Haberle I, Calvi CL, Day BJ, Pearlstein RD, Crapo JD, Warner DS. Effects of metalloporphyrin catalytic antioxidants in experimental brain ischemia. Free Radic Biol Med. 2002;33:947–961. doi: 10.1016/s0891-5849(02)00979-6. [DOI] [PubMed] [Google Scholar]
- 9.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 10.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats: Statistical validation. Stroke. 1995;26:627–635. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- 11.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 12.Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- 13.Gerriets T, Stolz E, Walberer M, Muller C, Kluge A, Bachmann A, Fisher M, Kaps M, Bachmann G. Noninvasive quantification of brain edema and the space-occupying effect in rat stroke models using magnetic resonance imaging. Stroke. 2004;35:566–571. doi: 10.1161/01.STR.0000113692.38574.57. [DOI] [PubMed] [Google Scholar]
- 14.Warner DS, Ludwig PS, Pearlstein R, Brinkhous AD. Halothane reduces focal ischemic injury in the rat when brain temperature is controlled. Anesthesiology. 1995;82:1237–1245. doi: 10.1097/00000542-199505000-00019. discussion 1227A. [DOI] [PubMed] [Google Scholar]
- 15.Gardner PR, Fridovich I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J Biol Chem. 1992;267:8757–8763. [PubMed] [Google Scholar]
- 16.Freiberger JJ, Suliman HB, Sheng H, McAdoo J, Piantadosi CA, Warner DS. A comparison of hyperbaric oxygen versus hypoxic cerebral preconditioning in neonatal rats. Brain Res. 2006;1075:213–222. doi: 10.1016/j.brainres.2005.12.088. [DOI] [PubMed] [Google Scholar]
- 17.Leinenweber SB, Sheng H, Lynch JR, Wang H, Batinic-Haberle I, Laskowitz DT, Crapo JD, Pearlstein RD, Warner DS. Effects of a manganese (III) porphyrin catalytic antioxidant in a mouse closed head injury model. Eur J Pharmacol. 2006;531:126–132. doi: 10.1016/j.ejphar.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 18.Song YS, Lee YS, Chan PH. Oxidative stress transiently decreases the IKK complex (IKKalpha, beta, and gamma), an upstream component of NF-kappaB signaling, after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005;25:1301–1311. doi: 10.1038/sj.jcbfm.9600123. [DOI] [PubMed] [Google Scholar]
- 19.Tse HM, Milton MJ, Piganelli JD. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigen-presenting cells: implication for their use in targeting oxidation-reduction reactions in innate immunity. Free Radic Biol Med. 2004;36:233–247. doi: 10.1016/j.freeradbiomed.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 20.Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30:2752–2758. doi: 10.1161/01.str.30.12.2752. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Piantadosi CA. Prolonged production of hydroxyl radical in rat hippocampus after brain ischemia-reperfusion is decreased by 21-aminosteroids. Neurosci Lett. 1994;177:127–130. doi: 10.1016/0304-3940(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 22.Globus MYT, Busto R, Lin B, Schnippering H, Ginsberg MD. Detection of free radical activity during transient global ischemia and recirculation: Effects of intraischemic brain temperature modulation. J Neurochem. 1995;65:1250–1256. doi: 10.1046/j.1471-4159.1995.65031250.x. [DOI] [PubMed] [Google Scholar]
- 23.Nishi T, Maier CM, Hayashi T, Saito A, Chan PH. Superoxide dismutase 1 overexpression reduces MCP-1 and MIP-1 alpha expression after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:1312–1324. doi: 10.1038/sj.jcbfm.9600124. [DOI] [PubMed] [Google Scholar]
- 24.Saito A, Hayashi T, Okuno S, Ferrand-Drake M, Chan PH. Overexpression of copper/zinc superoxide dismutase in transgenic mice protects against neuronal cell death after transient focal ischemia by blocking activation of the Bad cell death signaling pathway. J Neurosci. 2003;23:1710–1718. doi: 10.1523/JNEUROSCI.23-05-01710.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong KW, Kim KY, Lee JH, Shin HK, Kwak YG, Kim SO, Lim H, Yoo SE. Neuroprotective effect of (2S,3S,4R)-N”-cyano-N-(6-amino-3, 4-dihydro-3-hydroxy-2-methyl-2-dimethoxymethyl-2H-benzopyran-4-yl)-N’-benz ylguanidine (KR-31378), a benzopyran analog, against focal ischemic brain damage in rats. J Pharmacol Exp Ther. 2002;301:210–216. doi: 10.1124/jpet.301.1.210. [DOI] [PubMed] [Google Scholar]
- 26.Bowler RP, Sheng H, Enghild JJ, Pearlstein RD, Warner DS, Crapo JD. A catalytic antioxidant (AEOL 10150) attenuates expression of inflammatory genes in stroke. Free Radic Biol Med. 2002;33:1141–1152. doi: 10.1016/s0891-5849(02)01008-0. [DOI] [PubMed] [Google Scholar]
- 27.Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho YS, Matthews DE, Wouters EF, Janssen-Heininger YM. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang CY, Fujimura M, Noshita N, Chang YY, Chan PH. SOD1 down-regulates NF-kappaB and c-Myc expression in mice after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:163–173. doi: 10.1097/00004647-200102000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt KN, Amstad P, Cerutti P, Baeuerle PA. The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-kappa B. Chem Biol. 1995;2:13–22. doi: 10.1016/1074-5521(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 30.Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J Biol Chem. 2003;278:24233–24241. doi: 10.1074/jbc.M212389200. [DOI] [PubMed] [Google Scholar]
- 31.Clemens JA, Stephenson DT, Yin T, Smalstig EB, Panetta JA, Little SP. Drug-induced neuroprotection from global ischemia is associated with prevention of persistent but not transient activation of nuclear factor-kappaB in rats. Stroke. 1998;29:677–682. doi: 10.1161/01.str.29.3.677. [DOI] [PubMed] [Google Scholar]
- 32.Song YS, Lee YS, Narasimhan P, Chan PH. Reduced oxidative stress promotes NF-kappaB-mediated neuroprotective gene expression after transient focal cerebral ischemia: lymphocytotrophic cytokines and antiapoptotic factors. J Cereb Blood Flow Metab. 2007;27:764–775. doi: 10.1038/sj.jcbfm.9600379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song YS, Narasimhan P, Kim GS, Jung JE, Park EH, Chan PH. The role of Akt signaling in oxidative stress mediates NF-kappaB activation in mild transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2008;28:1917–1926. doi: 10.1038/jcbfm.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bottino R, Balamurugan AN, Tse H, Thirunavukkarasu C, Ge X, Profozich J, Milton M, Ziegenfuss A, Trucco M, Piganelli JD. Response of human islets to isolation stress and the effect of antioxidant treatment. Diabetes. 2004;53:2559–2568. doi: 10.2337/diabetes.53.10.2559. [DOI] [PubMed] [Google Scholar]
- 35.Lee BI, Chan PH, Kim GW. Metalloporphyrin-based superoxide dismutase mimic attenuates the nuclear translocation of apoptosis-inducing factor and the subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. Stroke. 2005;36:2712–2717. doi: 10.1161/01.STR.0000190001.97140.cf. [DOI] [PubMed] [Google Scholar]
- 36.Batinic-Haberle I, Cuzzocrea S, Reboucas JS, Ferrer-Sueta G, Mazzon E, Di Paola R, Radi R, Spasojevic I, Benov L, Salvemini D. Pure MnTBAP selectively scavenges peroxynitrite over superoxide: comparison of pure and commercial MnTBAP samples to MnTE-2-PyP in two models of oxidative stress injury, an SOD-specific Escherichia coli model and carrageenan-induced pleurisy. Free Radic Biol Med. 2009;46:192–201. doi: 10.1016/j.freeradbiomed.2008.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrer-Sueta G, Vitturi D, Batinic-Haberle I, Fridovich I, Goldstein S, Czapski G, Radi R. Reactions of manganese porphyrins with peroxynitrite and carbonate radical anion. J Biol Chem. 2003;278:27432–27438. doi: 10.1074/jbc.M213302200. [DOI] [PubMed] [Google Scholar]
- 38.Tauskela JS, Brunette E, Hewitt M, Mealing G, Morley P. Competing approaches to excitotoxic neuroprotection by inert and catalytic antioxidant porphyrins. Neurosci Lett. 2006;401:236–241. doi: 10.1016/j.neulet.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 39.Ross AD, Sheng H, Warner DS, Piantadosi CA, Batinic-Haberle I, Day BJ, Crapo JD. Hemodynamic effects of metalloporphyrin catalytic antioxidants: structure-activity relationships and species specificity. Free Radic Biol Med. 2002;33:1657–1669. doi: 10.1016/s0891-5849(02)01140-1. [DOI] [PubMed] [Google Scholar]
- 40.Sakai H, Sheng H, Yates RB, Ishida K, Pearlstein RD, Warner DS. Isoflurane provides long-term protection against focal cerebral ischemia in the rat. Anesthesiology. 2007;106:92–99. doi: 10.1097/00000542-200701000-00017. discussion 98–10. [DOI] [PubMed] [Google Scholar]