

Abstract
Discovering DNA motifs from co-expressed or co-regulated genes is an important step towards deciphering complex gene regulatory networks and understanding gene functions. Despite significant improvement in the last decade, it still remains one of the most challenging problems in computational molecular biology. In this work, we propose a novel motif finding algorithm that finds consensus patterns using a population-based stochastic optimisation technique called Particle Swarm Optimisation (PSO), which has been shown to be effective in optimising difficult multidimensional problems in continuous domains. We propose to use a word dissimilarity graph to remap the neighborhood structure of the solution space of DNA motifs, and propose a modification of the naive PSO algorithm to accommodate discrete variables. In order to improve efficiency, we also propose several strategies for escaping from local optima and for automatically determining the termination criteria. Experimental results on simulated challenge problems show that our method is both more efficient and more accurate than several existing algorithms. Applications to several sets of real promoter sequences also show that our approach is able to detect known transcription factor binding sites, and outperforms two of the most popular existing algorithms.
Keywords: DNA motif, optimisation, swarm intelligence, PSO, particle swarm optimisation
1 Introduction
One of the most important mechanisms to regulate gene functions is on the transcription level, where the expression of genes is mediated by the binding of Transcription Factors (TF) to the promoter sequences of genes. Identifying common TFBS from a set of putatively co-regulated genes is an important step towards deciphering the complex gene regulatory networks and understanding the tissue/condition-specific functions of genes.
Although TFBS can be detected experimentally, these experiments are usually expensive, time-consuming, and do not scale well. In practice, many users have relied on a combination of computational and experimental methods. In these approaches, a set of genes that are believed to be co-regulated is first obtained computationally or experimentally (e.g., based on gene expression or Chromatin immunoprecipitation). Given the promoters of these putatively co-regulated genes, one then applies computational motif finding algorithms to identify short DNA sequences (‘motifs’) that are statistically over-represented in these promoters (Roth et al., 1998; Bailey and Elkan, 1994). However, accurate identification of motifs is difficult because they are typically short (8–15 bps) compared to promoter sequences (usually hundreds to thousands of bases long). Furthermore, there is often a great variability among the binding sites of any given TF, and the biological nature of the variability is not yet well understood. Despite these difficulties, in the past decade a large variety of computational methods have been developed, many of which have been proven useful in predicting true binding sites (Tompa et al., 2005; Li and Tompa, 2006; Hu et al., 2005).
The existing motif finding algorithms often differ from one another in their ways of defining motifs, the objective functions for calculating motif significance, and the search techniques used to find the optimal (or near optimal) motifs. Many of these algorithms can be classified into one of two broad categories: stochastic searching algorithms based on Position Specific Weight Matrix (PSWM) motif representations, and combinatorial search algorithms based on consensus sequence motif representations. Examples of the first category include the well-known programs such as MEME (Bailey and Elkan, 1994), AlignACE (Roth et al., 1998), GibbsSampler (Lawrence et al., 1993), BioProspector (Liu et al., 2001),while the second category can be exemplified by Weeder (Pavesi et al., 2001), YMF (Sinha and Tompa, 2002), MultiProfiler (Keich and Pevzner, 2002), and Projection (Buhler and Tompa, 2002). For surveys of the existing methods and assessments of their relative performance, see Tompa et al. (2005), Li and Tompa (2006), Hu et al. (2005). As expected, no single method stands out as the sole best. In fact, assessing the performances of these algorithms is a daunting task itself, and experiments have shown that the overall performance of motif finding algorithms is still quite low (Tompa et al., 2005; Hu et al., 2005). Nevertheless, there seems to be a slight advantage by combinatorial approaches (Tompa et al., 2005; Li and Tompa, 2006).
To test the capacity of various motif finding algorithms, Pevzner and Sze designed a set of challenging cases, the so-called (l, d)-motif, a set of DNA l-mers each of which differ from a common consensus sequence by exactly d mismatches (Pevzner and Sze, 2000). The motifs were then embedded into some random DNA sequences and submitted to various motif finding algorithms. It has been shown that many stochastic searching algorithms fail to recover the embedded motifs even for biologically realistic choices of parameters (e.g., a set of (15, 4)-motifs embedded in 20 sequences each with 600 bases) (Pevzner and Sze, 2000). On the other hand, although combinatorial search algorithms have generally been shown to perform better in these challenging test cases, they typically resort to exhaustive enumeration of all or a large number of variants of consensus sequences, and are therefore limited to small data sets and short motifs only.
In this study, we propose a novel motif finding algorithm based on a population-based stochastic optimisation technique called PSO (Eberhart et al., 2001), which has been shown to be effective in optimising difficult multidimensional problems in many fields. The naive PSO algorithm, however, can only be applied to continuous domains. The motif finding problem, which is essentially a multiple local sequence alignment problem, unfortunately, is discrete, as a slight shift of one sequence completely changes the alignment (see Section 3.2 for more discussions). To circumvent this problem, we break sequences into l-mers and develop a novel mapping scheme to convert the motif finding problem into a semi-continuous domain, and modified the update policy of the original PSO algorithm to solve the motif finding problem. We also propose several strategies for escaping from local optima, and determining the termination criteria automatically.
Compared to previous motif finding methods, our algorithm uses consensus as motif representation, thus avoiding many of the pitfalls associated with PSWM-based methods. On the other hand, PSO does not require exhaustive enumeration of consensus sequences, yet can still quickly converge to an optimal or almost optimal solution. Experimental results on both simulated and real biological data sets have shown that our method is more efficient and more accurate than several existing algorithms.
The remaining sections are organised as follows. In Section 2, we introduce the basic concept and ideas of PSO and the generic PSO algorithm. In Section 3, we discuss the improvement that we made to the generic PSO algorithm in order to accommodate the unique nature of motif finding problems, and other algorithmic issues that we addressed. We present our experimental results in Section 4, and conclude in Section 5 with some possible future improvement.
2 Particle Swarm Optimisation
Particle Swarm Optimisation (PSO) is a population-based stochastic optimisation technique for problem solving that is inspired by the social behaviours of organisms such as bird flocking and fish schooling (Eberhart et al., 2001). The system is initialised with a population of random solutions and searches for the optimal solution by updating iteratively. Although PSO shares many similarities with evolutionary computation techniques such as genetic algorithms, PSO differs from other evolutionary algorithms significantly on how the solutions were updated. In PSO, each potential solution, called particle, is represented by a point in the multiple-dimensional solution space. When searching for the optimal solution, particles fly around the solution space with a certain velocity. During flight, each particle adjusts its position and velocity according to its own experience and the experiences of its neighbours. Specifically, each particle keeps track of the best solution (the position and the fitness value) it has encountered so far. This solution is called pbest, which stands for personal best. Each particle also keeps track of the best solution by any particle in its neighbourhood. This solution is called lbest, which stands for local best. Many types of neighbourhood structures can be implemented by emulating real social networks. In the simplest case, all the particles are directly connected to each other. Then lbest is simply the global optimum of all the particles, hence is also called gbest. Therefore, while each individual particle is performing a local search, the particles also communicate with other particles and learn from them, balancing exploration and exploitation.
Formally, let vectors xi and vi be the current position and velocity of the ith particle (1 ≤ i ≤ n), x̂i be the recorded position of pbest of the ith particle, and ĝ be the position of gbest. The fundamental concept of PSO consists of changing the velocity (vi) of each particle at each time step toward its pbest and gbest locations. Acceleration is weighted by a random number, with separate random numbers generated for acceleration toward pbest and gbest locations. The particles update their positions and velocities based on the two equations below.
where ω is a parameter called the inertial weight, r1 and r2 are vectors of random numbers, usually uniformly distributed within 0 and 1. The operator ○ denotes entry-wise vector multiplication. It is critical to note that different r1 and r2 are generated at each iteration and for each particle. c1 and c2 are positive constants, called the acceleration constant. c1 is a factor determining how much the particle is influenced by its pbest, and c2 is a factor determining how much the particle is influenced by gbest. Although the PSO algorithm is a relatively new technique, it has been developed into many variants and has been successfully applied in many research and application areas (Kennedy and Eberhart, 1995; Eberhart and Shi, 2001; Clerc and Kennedy, 2002; Robinson et al., 2004).
However, as we have mentioned in Section 1, the PSO algorithm cannot be directly applied to the motif finding problem, which is a discrete optimisation problem. In Section 3.2, we will discuss more in detail about the problems in applying PSO to motif finding, and our modifications of the naive algorithm to address these problems.
3 The PSO-motif algorithm
3.1 Motif model and fitness function
In order to apply PSO to the motif finding problem, we need to first define the solution structure and the fitness function. There are several ways to represent a motif. In PSWM-based algorithms, motifs are represented by weight matrices, so a solution can be unambiguously represented by a 3 × l matrix, where l is the length of the motif. Our focus of this paper is to develop a consensus-based algorithm, due to its relative advantage that has been shown recently (Tompa et al., 2005; Li and Tompa, 2006). To represent a motif in consensus-based algorithms, we have two choices. First, we may represent a motif by a consensus sequence, which is a discrete l-dimensional vector. The search space consists of all 4l l-mers. Alternatively, a motif can be precisely defined by the list of motif instances occurred in the input sequences. Therefore, with the motif length fixed, a motif can be represented by a vector of motif starting positions within the input sequences. Between the two representations, we opt for the second representation in this work. The main advantage of the latter is its efficiency in updating. A more detailed discussion of this issue is beyond the scope of this paper.
To evaluate the quality of a motif, we first derive a consensus for the motif by taking the most frequent base at each position, and then measure the total number of mismatches between the individual instances and the consensus. When this fitness function is used, it is assumed that the background sequences have uniform base frequencies, which is usually not true. However, it is straightforward to design a more elaborated fitness function to take into consideration background base frequencies, with slightly increased running time.
3.2 Word dissimilarity graph
The main difficulty associated with applying the PSO algorithm to motif finding problem is that the fitness function is not continuous. In a typical PSO algorithm, one wishes to control the velocity so that at the beginning stage the particles can fly around quickly inside the search space, and when a particle approaches the optimal solution, it should slow down so it can converge. One can achieve this if the fitness function is continuous, since the velocity is updated according to the distances between the current position and the positions of pbest and gbest. However, a solution in our algorithm consists of a vector of positions in the input sequences. Therefore, the distance between two potential solutions has no indication of the difference of their fitness values. For example, as shown in Figure 1, positions 1 and 2 are separated by 6 mismatches, while positions 1 and N have only 1 mismatch.
Figure 1.
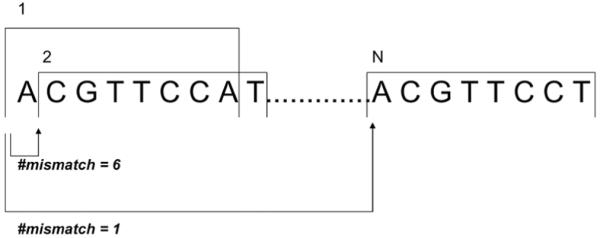
Relationship between motif positions and motif dissimilarity
To solve this problem, we model the neighbourhood information in the solution space by a dissimilarity graph of all l-mers in each sequence. For example, given an input sequence CTCTGCTG and motif length = 3, we can construct a dissimilarity graph, whose adjacency matrix is shown in Table 1. The row index is the current position of a candidate motif in this sequence, the column index is the potential new position of the motif, and the values in the cell are the number of mismatches between the two l-mers starting at the positions represented by the row index and column index, respectively. For example, if the motif start position is updated from 1 to 6, the distance is only 1, meaning CTC and CTG have 1 mismatch. With this graph, we can reformat the sequence into a new order, which is more meaningful and useful. In the matrix below, the ‘neighbourhoods’ order of P2 is (P5), (P4), (P1;P3;P6). This matrix can be pre-computed and stored in the main RAM. As one graph is needed for each sequence, the total space needed is O(nL2), where n is the number of sequences and L is the length of each sequence. For longer sequences, the graph can be made sparse by keeping only the edges whose weights are smaller than a certain threshold, for example, 2d, where d is the maximum number of mismatches expected between the motif consensus and its instances.
Table 1.
Adjacency matrix of an example dissimilarity graph
| P1 | P2 | P3 | P4 | P5 | P6 | |
|---|---|---|---|---|---|---|
| P1 | – | 3 | 1 | 2 | 3 | 1 |
| P2 | 3 | – | 3 | 2 | 1 | 3 |
| P3 | 1 | 3 | – | 3 | 3 | 0 |
| P4 | 2 | 2 | 3 | – | 3 | 3 |
| P5 | 3 | 1 | 3 | 3 | – | 3 |
| P6 | 1 | 3 | 0 | 3 | 3 | – |
3.3 Update policy
In order to decide what new positions should be considered as the motif starting positions, we define the velocity of a particle as the range of the allowed number of mismatches between the new and current motif instances. Let xi and xj represent two potential solutions, and D(xi, xj) be the vector of numbers of mismatches between the motif instances in xi and in xj, we use the following rule to define the velocity of a particle:
Given the upper and lower bounds of the velocity, and , we update xi as follows. Let xi(j) be the starting position of the motif on the jth sequence, and be the new starting position on the same sequence. In order to obtain , we randomly pick a position from sequence j such that the number of mismatches between the motifs started at these two positions are within the upper and lower bound of the velocity, i.e.,
3.4 Occasional full scan and shift check
Inside the algorithm we always keep track a consensus sequence derived from gbest using the most frequent base at each position, and compute the fitness value of gbest. When the fitness value of gbest reaches a certain threshold, we use the consensus sequence to do a full scan on all sequences to check if there is an l-mer in the input sequence that matches the consensus sequence better than the l-mer in gbest.
Second, similar to many motif finding algorithms, the output of PSO algorithm may have a shift issue: the start positions may be one or two positions away from the real positions, and it is difficult for the algorithm to escape from such local optima. To circumvent this problem, we periodically check whether shifting the motif positions by a small number can improve the quality of the solution.
When a particle is almost near the optimal solution, these two tricks can significantly reduce the running time, and make the algorithm converge much more quickly.
3.5 Escape from local optima
If gbest remains stable for a large number of iterations, we consider that the algorithm has reached a local optimum or the global optimum, so we reset the algorithm. There are two ways for reset. The first strategy is to simply discard all the information so far and start the algorithm from scratch. In some cases with this strategy the algorithm might converge to the same local optimum repeatedly. Here we suggest the second strategy, which we call a ‘reset move’. When a local optimum is detected, we move all the current solution, pbest and gbest by a random distance. We have found that this strategy can more effectively help the algorithm escape from local optima.
3.6 Termination
Like many stochastic algorithms, in many cases it is difficult to determine when the algorithm should terminate. For simulated test cases, we typically know the total number of mismatches, so the program can stop when it reaches the threshold. In practice, however, a user typically does not have prior knowledge about the number of mismatches. If the number is given too high, the algorithm may give up the search before it finds the optimal solution. On the other hand, if the number is given too low, the algorithm may spend most of its time trying to improve over an easily detectable global optimum. Here we implemented three methods to determine when the algorithm should terminate: value-based, time-based, and repeat-based. The value-based method is the easiest: when the solution reaches a pre-defined threshold value, the algorithm stops running and returns the solution. The time-based method terminates the algorithm after a certain amount of time has passed. Finally, we recommend the repeat-based method for real test cases. With this method, before resetting the algorithm, we compare the current gbest fitness value with the best gbest fitness value achieved during the course of the algorithm. If the current gbest is better than the history gbest, we update the history gbest; if the two fitness values and the associated motif consensuses are exactly the same, we stop the algorithm and output this result. This method is based on the intuition that, since there are typically many local optima, the probability of repeating one local optimum without an update is very low. This method is very effective when tested on the real test cases (Section 4).
3.7 Post-processing
By default, the quality of a motif is evaluated by the total number of mismatches between the consensus sequence and its instances. There are a number of limitations in this basic strategy when applied to real biological sequences. First, the mismatch based quality function does not consider the background frequency, which is usually not uniform. Second and more importantly, in real TF binding motifs the mutation rates are often not uniformly distributed across every position, and not every site has the same significance in determining the binding strength. For example, many TF binding sites consist of two short conserved regions, separated by a small gap. Therefore, a mismatch in the gapped region should be penalised much less than a mismatch in the conserved region. However, in the consensus-based method, we are forced to select a representative for each position, and the number of mismatches is computed based on that representative.
To address these problems, we apply a post-processing procedure to improve the final motif returned by the PSO algorithm. Given the gbest returned by the algorithm, we construct a position-specific weight matrix. This matrix is then used to scan all input sequences. This scan will likely update some of the motif instances. We then recompute the position specific weight matrix and repeat the scan, until the solution does not vary. With this method, the matching/mismatching score is weighted by the information content contained in each position, and therefore the more conserved positions will have more contribution to the selection of the binding sites. Furthermore, we can also take into account the background base frequencies when scoring a binding site against the weight matrix.
Figure 2 shows the overall structure of the PSO-motif algorithm. The individual steps have been described in the previous subsections.
Figure 2.

Overview of PSO-motif algorithm
4 Experimental results
We have implemented our algorithm in C. To evaluate the performance of our algorithm, we tested it on two types of DNA sequences. The first type of test data consists of simulated data sequences, also known as the (l, d)-motif challenging problem. The second type of data contain real promoter sequences from E. coli and human genes containing known TF binding sites that have been determined experimentally.
4.1 Simulated data sets
To objectively compare with the existing algorithms, we first tested our algorithm on simulated data sets. We synthesised problem instances as follows. First, a motif consensus of length l was generated by randomly picking l bases. Second, we randomly selected d positions from the consensus and mutated the base at each position to a random base, which could be the same as the original base. This generates one instance of the motif. We repeated this process to obtain t instances. Third, we randomly generated t background sequences of length n each. Finally, we assign each motif instance to a random position in a background sequence. This procedure generates t sequences, each containing exactly one instance of the motif. All random choices were made independently and with equal base frequencies.
We first focused on the (15, 4)-motif challenge problem, which is one of the most popular benchmarks for many motif finding programs. We fixed the number of sequences to 20, and varied the length of each sequence from 400 to 1000. Since our algorithm is stochastic, we repeated our algorithm multiple times, and terminated it once the motif was found. Table 2 shows the mean running time of our algorithm and several competing algorithms, including GALF-P (Chan et al., 2008), Projection (Buhler and Tompa, 2002) and Weeder (Pavesi et al., 2001). GALF-P is one of the best genetic algorithm-based motif finding program, while Projection and Weeder are two of the best combinatorial-search motif finding algorithms. Projection is based on random projection. Weeder is a suffix tree-based enumeration algorithm. For each sequence length, the results are based on ten independent runs on five sets of sequences. The running time of Weeder was taken from the original paper published almost seven years ago (Pavesi et al., 2001), and was based on an 89% success probability. We were not able to repeat the experiments ourselves since the original program is not available any more. The new Weeder software only allows motif lengths up to 12 bases. The Projection and GALF-P programs were downloaded from the original author’s websites and were tested on the same data sets as for our algorithm. The number of iterations of Projection is pre-determined by its parameters, so its running time does not vary between runs. PSO-motif and Projection were able to recover all the embedded motifs with 100% accuracy. Motifs found by GALF-P were nearly similar to the embedded motifs, with some tolerable differences, e.g., a shift in the final motifs. Weeder is generally slower than Projection and PSO-motif. The running time of GALF-P is close to PSO-motif on long input sequences, but for shorter sequences PSO-motif is much faster. While PSO-motif and Projection have similar running time in Tables 2 and 3, the running time of Projection increases more rapidly with the length of the input sequences. Furthermore, the number of iterations in Projection has to be estimated from the motif length and the number of mismatches in advance. If a user does not know the number of mismatches, one either has to use a large number of iterations which requires a prolonged running time, or use a small number of iterations with the risk of missing the real motif.
Table 2.
Running time (seconds) on (15, 4)-motif challenge problems
| Sequence length | 400 | 500 | 600 | 800 | 1000 |
|---|---|---|---|---|---|
| Weeder | <60 | 125 | 200 | 450 | 900 |
| Projection | 9 | 23 | 42 | 162 | 418 |
| GALF-P | 99 | 122 | 158 | 205 | 282 |
| PSO | 18 | 34 | 57 | 137 | 288 |
Table 3.
Running time (seconds) on (l, d) motif challenge problems (sequence length = 600)
| (l, d) | (11,2) | (13,3) | (15,4) | (17,5) | (19,6) |
|---|---|---|---|---|---|
| Projection | 4 | 13 | 42 | 94 | 174 |
| MotifEnumerator | 5 | 119 | – | – | – |
| GALF-P | 182 | 164 | 148 | 174 | 174 |
| PSO | 72 | 58 | 57 | 61 | 54 |
It is also worth noting that since PSO is a stochastic algorithm, its running time varies between runs even on the same data set. In rare cases the algorithm may require extremely long running time to converge. As a result, the running time has a long tail distribution, and the mean is often much higher than the median. Figure 3 shows the distribution of the running time of PSO on (15, 4) motifs with a sequence length of 1000. In fact, in 80% of cases the motifs can be found in 335 s, while in one case the program completes after almost 1900 s. A better strategy for detecting local optima may eliminate some of these rare cases and improve the efficiency.
Figure 3.
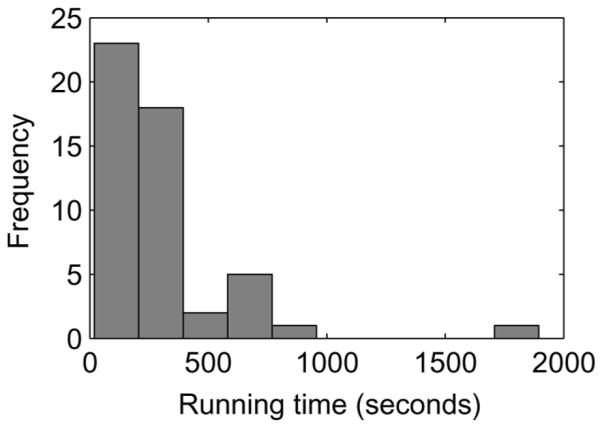
Distribution of running time of PSO on (15,4) motifs with sequence length = 1000
Next, we compared PSO, Projection, GALF-P and MotifEnumerator on a series of challenge problems with varying motif lengths and number of errors. Sequences lengths are fixed at 600. MotifEnumerator is a pattern-driven motif enumeration algorithm (Sze and Zhao, 2006). The program MotifEnumerator was downloaded from the original author’s website (http://faculty.cs.tamu.edu/shsze/motifenumerator/). Again both PSO-motif and Projection were able to recover the embedded motifs with 100% accuracy. As shown in Table 3, although Projection is more efficient than PSO for shorter motifs, its advantage vanishes when motif length increases to about 15, and it is slower than PSO for motifs longer than 17. The running time of PSO and GALF-P is almost independent of motif lengths, with the former being relatively faster. The running time and space requirement of MotifEnumerator are exponential to l. Therefore, for small motif lengths (e.g., l = 11), MotifEnumerator solves the problem very efficiently; however, when l increases to 15, the program aborted with an out of memory exception on our testing computer with 2 GB RAM.
4.2 Real biological sequences
We also tested our algorithm on three sets of biological sequences with known TF binding motifs.
The first sample is the binding sites for the Cyclic AMP Receptor Protein (CRP), which functions as a transcription factor in Escherichia coli. The data set contains 18 sequences, each 105 bp long, which contain 23 sites that have been experimentally determined (Stormo and Hartzell, 1989).
The second test sample is the binding site for the Estrogen Receptor (ER), which is a ligand-activated enhancer protein that binds to Estrogen Response Elements (EREs). The data set includes 25 genomic sequences, each of which is 200 bp long and contains a single known ERE (Klinge, 2001).
The final data set includes 25 mammalian sequences of 200 bp long that are known to contain binding sites for the transcription factors in the E2F family (Frith et al., 2004).
It is important to note the following difference between the above sequences and the simulated ones.
The background base frequencies are not uniform in these real sequences.
The number of mutations is not known.
The mutation rates vary at different positions.
It is interesting to test whether any of these characteristics in real sequences may pose additional difficulty for our consensus-based motif finding algorithm. Furthermore, we chose these three examples because they represent three typical cases in motif finding: ERP is a long and well-conserved prokaryotic TF binding site, which is relatively easy to be identified; ERE consists of two conserved sub-motifs separated by a small gap; E2F is a mammalian TF binding motif present in promoter sequences with high GC contents which may mislead an algorithm into a low complexity region.
Figure 4 shows the sequence logos of the known motifs together with the flanking regions, compared with the motifs predicted by our algorithm. As shown, our algorithm has recovered the consensuses of all three real motifs. On the other hand, the probabilities of observing a given base at some position are different between the predicted and the real motifs, especially for the CRP motif. Further investigation shows that the reason for this partial discrepancy is often due to
each input sequence may contain more than one motif, while our algorithm only allowed one occurrence per sequence
our algorithm does not take into consideration background base frequencies.
In the CRP data set, the 18 sequences contained a total of 24 binding sites. Furthermore, the sequences contain a very high AT content (61%). Our algorithm included several binding sites that match to the consensus sequence equally well as or better than the true sites, but have higher AT contents. Therefore, although the binding sites identified by our algorithm have a smaller number of mismatches to the consensus than the real binding sites, the former may be less biologically interesting. Therefore, we applied to post-processing procedure described in Section 3.7. Figure 4(c) shows that the post-processing did not change the consensus sequence, but fine-tuned the motif instances and the predicted motifs with post-processing are more similar to the real ones than those without post-processing are.
Figure 4.

Results of PSO-motif on real biological sequences: (a) real motifs; (b) predicted motifs and (c) predicted motifs after post-processing (see online version for colours)
As a comparison, we applied two of the most successful motif finding algorithms, MEME (Bailey and Elkan, 1994) and AlignACE (Roth et al., 1998), both of which are based on optimising position specific weight matrices. To our surprise, even for these relatively easy problems, AlignACE failed to find the ERE and E2F motifs. Although it found the consensus sequence of CRP, it missed many of the actual binding sites (Figure 5). MEME correctly identified all the consensus sequences, but still made significant number of errors on the binding site level (Figure 6). These results confirmed the weakness of PSWM-based methods (Tompa et al., 2005; Li and Tompa, 2006).
Figure 5.
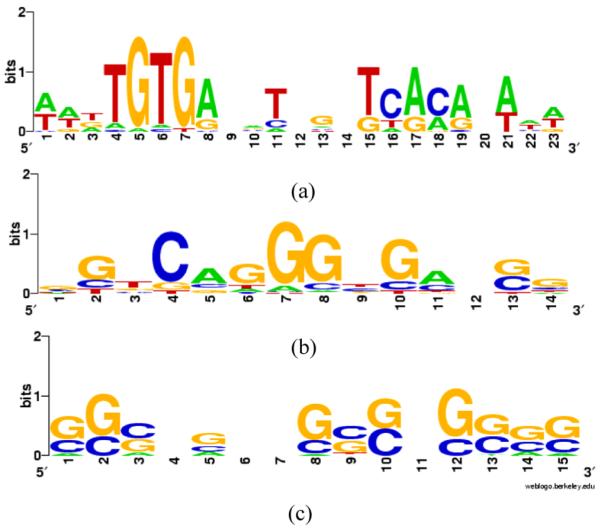
Results of alignACE: (a) CRP; (b) ERE and (c) E2F (see online version for colours)
Figure 6.
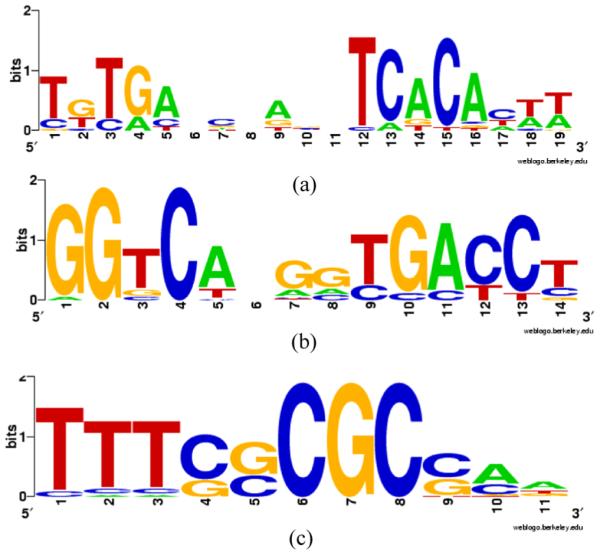
Results of MEME: (a) CRP; (b) ERE and (c) E2F (see online version for colours)
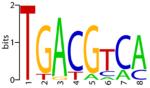
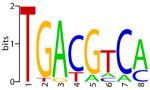
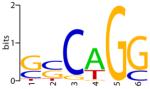
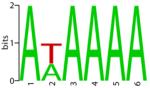
Finally, we tested PSO-motif on five additional real data sets, for vertebrate transcription factors CREB, MEF2, MYOD, SFR and TBF, whose true binding sites are known. The input sequences were obtained from the authors of Chan et al. (2008), and have similar characteristics as the first three test cases. Table 4 shows the sequence logos for the known binding sites, and the motifs predicted by PSO-motif, MEME, and AlignACE. For AlignACE, the results with the default parameter (GC frequency = 0.38) worked poorly on some sequences. We therefore tried several parameters for AlignACE until a satisfactory result was found. For MEME and PSO-motif, motif length is the only parameter varied, and default options were used for all other parameters. As shown in Table 4, the motifs found by PSO are generally in better agreement with the known motifs, while MEME and AlignACE each had some problems in some test cases. AlignACE completely failed to find the MYOD motif, while MEME found a shifted version of MYOD motif. The SRF motif found by MEME only partially resembles the known motif.
Table 4.
Motif results on additional real test cases
| Known | PSO | MEME | AlignACE | |
|---|---|---|---|---|
| CREB |
|
|
|

|
| MEF2 |
|

|

|

|
| MYOD |
|

|

|
|
| SRF |
|

|

|

|
| TBP |
|

|

|
|
5 Conclusions and discussion
In this work, we have proposed a novel algorithm for finding DNA motifs based on a modified version of the PSO algorithm. We have shown that by remapping a promoter sequence into a dissimilarity graph of l-mers, we can successfully apply the PSO algorithm to solve the difficult motif finding problem with high efficiency and high accuracy. Our experimental results on both simulated and real biological data sets are very encouraging. When applied to simulated challenge problems, PSO is slower than Projection in easier cases, while faster in more difficult cases that have longer input sequences or longer motifs and more mutations. Furthermore, our algorithm does not require the number of mismatches to be given as a parameter, which is more useful in practice. For real biological sequences, our method combined with post-processing has successfully identified the known motifs and most of the binding sites. Our studies have shown that PSO is a reliable and efficient technique for solving the difficult motif-finding problem, and we are looking into applying it to other challenging problems in computational biology.
Acknowledgements
This work is supported in part by a University of Texas at San Antonio Faculty Research Award and a NIH grant 1SC3GM086305-01 to JR. The authors would like to thank Zhi Wei for providing real biological sequence data, and UTSA Computational Biology Initiative for providing computing resources. A preliminary version of this paper appeared in the First Workshop on Data Mining in Functional Genomics, in Conjunction with IEEE International Conference on Bioinformatics and Biomedicine 2008, Philadelphia, PA, USA.
Biography
Biographical notes: Chengwei Lei received his Bachelor Degrees in Computer Science and Technology and in Applied Mathematics from Beijing University of Aeronautics and Astronautics in 2005, and his MS Degree in Computer Science from the University of Texas at San Antonio in 2008. He is currently a PhD Student in the Department of Computer Science at the University of Texas at San Antonio, under the supervision of Dr. Jianhua Ruan. His current research focuses on developing fast and accurate algorithms for analysing biological sequences.
Jianhua Ruan received his BS in Biology from the University of Science and Technology of China in 1998, MS in Computer Science from California State University, San Bernardino in 2002, and PhD in Computer Science and Engineering from Washington University in Saint Louis in 2007. He is currently an Assistant Professor in the Department of Computer Science at the University of Texas at San Antonio. His current research interests include functional and structural properties of protein-protein interaction networks, transcriptional and post-transcriptional regulatory networks, microarray data analysis, RNA secondary structure prediction, and data mining algorithms.
References
- Bailey T, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Buhler J, Tompa M. Finding motifs using random projections. J. Comput. Biol. 2002;9:225–242. doi: 10.1089/10665270252935430. [DOI] [PubMed] [Google Scholar]
- Chan T-M, Leung K-S, Lee K-H. TFBS identification based on genetic algorithm with combined representations and adaptive post-processing. Bioinformatics. 2008;24(3):341–349. doi: 10.1093/bioinformatics/btm606. [DOI] [PubMed] [Google Scholar]
- Clerc M, Kennedy J. The particle swarm-explosion, stability, and convergence in a multidimensional complex space. IEEE Transactions on Antennas and Propagation. 2002;6:58–73. [Google Scholar]
- Eberhart R, Shi Y. Particle swarm optimization: developments, applications and resources; Proc. 2001 Congr. Evolutionary Computation, Seoul; South Korea. 2001.pp. 81–86. [Google Scholar]
- Eberhart R, Shi Y, Kennedy J. Swarm Intelligence. Morgan Kaufmann; San Francisco, CA: 2001. [Google Scholar]
- Frith M, Hansen U, Spouge J, Weng Z. Finding functional sequence elements by multiple local alignment. Nucleic Acids Res. 2004;32:189–200. doi: 10.1093/nar/gkh169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Li B, Kihara D. Limitations and potentials of current motif discovery algorithms. Nucleic Acids Res. 2005;33(15):4899–4913. doi: 10.1093/nar/gki791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keich U, Pevzner P. Finding motifs in the twilight zone. Bioinformatics. 2002;18:1374–1381. doi: 10.1093/bioinformatics/18.10.1374. [DOI] [PubMed] [Google Scholar]
- Kennedy J, Eberhart R. Particle swarm optimization; Proc. IEEE Conf. Neural Networks IV; Piscataway, NJ. 1995.pp. 1942–1948. [Google Scholar]
- Klinge C. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence C, Altschul S, Boguski M, Liu J, Neuwald A, Wootton J. Detecting subtle sequence signals: a gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- Li N, Tompa M. Analysis of computational approaches for motif discovery. Algorithms Mol. Biol. 2006;1:8. doi: 10.1186/1748-7188-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Brutlag D, Liu J. Bioprospector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes; Pac. Symp. Biocomput.; Big Island, Hawaii. 2001; pp. 127–138. [PubMed] [Google Scholar]
- Pavesi G, Mauri G, Pesole G. An algorithm for finding signals of unknown length in DNA sequences. Bioinformatics. 2001;17:S207–S214. doi: 10.1093/bioinformatics/17.suppl_1.s207. [DOI] [PubMed] [Google Scholar]
- Pevzner P, Sze S. Combinatorial approaches to finding subtle signals in DNA sequences; Proc. Int. Conf. Intell. Syst. Mol. Biol.; 2000; pp. 269–278. [PubMed] [Google Scholar]
- Robinson J, Sinton S, Rahmat-Samii Y. Particle swarm optimization in electromagnetics. IEEE Transactions on Antennas and Propagation. 2004;52:397–407. [Google Scholar]
- Roth F, Hughes J, Estep P, Church G. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mrna quantitation. Nat. Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- Sinha S, Tompa M. Discovery of novel transcription factor binding sites by statistical overrepresentation. Nucleic Acids Res. 2002;30:5549–5560. doi: 10.1093/nar/gkf669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo G, Hartzell G. Identifying protein-binding sites from unaligned DNA fragments. Proc. Natl. Acad. Sci. 1989;86:1183–1187. doi: 10.1073/pnas.86.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze S-H, Zhao X. Improved pattern-driven algorithms for motif finding in DNA sequences; Proc. 2005 Joint RECOMB Satellite Workshops on Systems Biology and Regulatory Genomics, Lecture Notes in Bioinformatics; La Jolla, CA. 2006.pp. 198–211. [Google Scholar]
- Tompa M, Li N, Bailey T, Church G, De Moor B, Eskin E, Favorov A, Frith M, Fu Y, Kent W, Makeev V, Mironov A, Noble W, Pavesi G, Pesole G, Régnier M, Simonis N, Sinha S, Thijs G, van Helden J, Vandenbogaert M, Weng Z, Workman C, Ye C, Zhu Z. Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]