

Abstract
The M2 protein of influenza A virus performs the crucial function of transporting protons to the interior of virions enclosed in the endosome. Adamantane drugs, amantadine (AMN) and rimantidine (RMN), block the proton conduction in some strains, and have been used for the treatment and prophylaxis of influenza A infections. The structures of the transmembrane (TM) region of M2 that have been solved in micelles using NMR (residues 23-60) [Schnell and Chou (2008)] and by X-ray crystallography (residues 22-46) [Stouffer et al. (2008)] suggest different drug binding sites: external and internal for RMN and AMN, respectively. We have used molecular dynamics (MD) simulations to investigate the nature of the binding site and binding mode of adamantane drugs on the membrane-bound tetrameric M2-TM peptide bundles using as initial conformations the low-pH AMN-bound crystal structure, a high-pH model derived from the drug-free crystal structure, and the high-pH NMR structure. The MD simulations indicate that under both low-and high-pH conditions, AMN is stable inside the tetrameric bundle, spanning the region between residues Val27 to Gly34. At low pH the polar group of AMN is oriented toward the His37 gate while under high-pH conditions its orientation exhibits large fluctuations. The present MD simulations also suggest that AMN and RMN molecules do not show strong affinity to the external binding sites.
Keywords: molecular dynamics, simulations, amantadine, adamantine, transmembrane, ion channel
Introduction
The M2 protein located in the lipid envelope of influenza A virus is activated by the low pH in the endosome, which encloses viral particles following endocytosis [1]. M2 transports protons to the interior of the virion, which then permits the uncoating of the viral RNA and allows the fusion of the viral envelope with the endosomal bilayer: a process essential for the viral RNA release and continuation of the infectious life cycle. The M2 protein is a homo-tetramer with each monomer consisting of three regions: a 24-residue N-terminal domain, a transmembrane (TM) domain of 19 residues, and a 54-residue cytoplasmic domain. Adamantane derivatives, amantadine (AMN) and rimantadine (RMN), block the M2 channel activity, and hence can be used for prophylaxis and treatment of influenza A infections [2]. However, the resistance to adamantane drugs has significantly increased in the last decade [3], leading to the urgent need for developing new and effective drugs targeting M2. Besides the M2 protein, the neuraminidase protein of the virus is the other major target protein of the currently available anti-flu drugs, oseltamivir (Tamiflu) and zanamivir (Relenza). However, the resistance to oseltamivir has also greatly increased in the last two years, while the use of zanamivir suffers from the problem of poor bioavailability of the drug [4, 5]. Thus, the increasing resistance to the currently available drugs calls for the urgent development of new and more potent drugs targeting seasonal and pandemic strains of influenza A virus.
There is a significant body of research that has focused on residues 22-46 of the M2 protein (hereafter referred to as M2-TM), which includes the TM domain with a few hydrophilic residues on either end. M2-TM forms conductive tetrameric bundles, which are able to bind AMN [6-10]. Although model structures of M2-TM were available for some time [9, 11-15], high resolution structures have now been solved using NMR [16] and X-ray crystallography [17]. Specifically, Schnell and Chou reported a RMN-bound ensemble of 15 structures for a 4-helix bundle of M2 residues 23-60 using NMR on micelles samples, under conditions which likely correspond to high pH [16]. The crystal structure of M2-TM with bound AMN, reported by Stouffer et al., represents instead a low-pH conformation of M2-TM [17]. The two structural studies, while capturing conformations of the channel at different pH conditions, propose very different mechanisms of channel inhibition by adamantane drugs. In the crystal structure, AMN is bound in the central cavity of M2-TM, spanning the region containing residues Val27-Gly34 [17]. This binding mode is consistent with a pore-blocking mechanism of proton conduction, and the binding site is located in close proximity to the most relevant mutations found in resistant flu strains, including swine H1N1 and avian H5N1 viruses [18, 19]. On the other hand, the NMR structure contains four RMN molecules bound to the outer surface of the M2 (residues 23-60) tetramer, close to Asp44. In the NMR structure the binding site is located far from principal resistant sites, and an allosteric mechanism has been proposed to rationalize the blocking of proton conduction [16]. Questions about the drug binding site of the M2 protein bundle raised by the two structures were discussed by Miller [20].
Although the low resolution (3.5 Å) of the AMN-bound crystal structure permits the identification of the binding site of AMN in the channel cavity, it does not definitively allow determination of the orientation of the drug polar head group. Stouffer et al. proposed that the most plausible orientation of the AMN amide group is toward the C-terminal end of the bundle, enabling the bulky hydrophobic part of AMN to interact with the hydrophobic internal residues around Val27, while the charged head group can be accommodated in the solvated internal cavity. The location and orientation of AMN in the bundle cavity found by the molecular dynamics (MD) study of Yi et al. [21] agree with the suggestion of Stouffer et al. Chen et al have also shown using MD simulations that AMN binds to the Ala29 region and reduces the proton conductance of M2 by 99.8% [22]. They noted that the primary proton permeation free energy barrier is relocated from His37 to Ala29 region as a result of AMN binding. Moreover, Jing et al. have reported that M2 from influenza A with the mutation Asp44Ala is sensitive to AMN as measured by electrophysiological recordings in Xenopus laevis oocytes and in mammalian cells [23]. Recently, Balannik et al have also reported that mutations of residues that are N-terminal to His37 [Val27, Ala30, Ser31 and Gly34] exhibit significantly lower inhibition by AMN while most mutations of residues which are C-terminal to His37 and far from the internal binding site [Trp41 and Asp44] have little or no effect on AMN inhibition[24]. Thus, these data suggest that Asp44 is not required for AMN sensitivity and the inhibitory binding site is not located outside the cavity as proposed by the NMR structure. However, the functional and structural experiments by Pielak et al. support the external lipid-facing pockets as the primary binding sites [25]. A very recent solid-state NMR study under high pH conditions (pH 7.5) shows that indeed two binding sites (internal and external) do exist in M2 in phospholipid bilayers, though the external binding site is of much lower affinity than the internal site and is bound only when the drug reaches very high concentration [26]. In this paper, we investigate both the binding sites using MD simulations, which provide us molecular level insight into the drug binding in a native-like lipid environment and enable us to build a comprehensive scheme of drug binding under different pH conditions.
We proposed that M2-TM acts like a ‘proton transporter’ rather than a classic ‘proton channel’ based on MD simulations of M2-TM in DMPC bilayers using the drug-free crystal structure and a high-pH model derived from the crystal structure as initial conformations [27]. This view reconciles the known electrophysiological properties of M2, such as the low proton conduction rate, the conductance saturation at low pH, and the strong rectifying behavior [28, 29]. The His37 gate of M2 is involved in the proton selectivity [30, 31], and is closed under external high-pH conditions; while the hydrophobic Val27 gate is open (Openout-Closedin state). Under conditions of external low pH that exist inside the endosome, protons reach the central cavity through the open Val27 gate and the His37 residues get protonated. This leads to the opening of the His37 gate and the closing of the Val27 gate (Closedout-Openin state). Protons can diffuse from His37 to the interior of the virus and the protein goes back to the Openout-Closedin state. Hence, protons are transported to the interior of the virus and the cycle continues until equilibrium is reached [Figure 5 of [27]]. It is interesting to consider the impact of adamantane drugs on the proposed model for proton conduction by M2.
AMN is known to inhibit M2 at both low and high pH [6, 32, 33], and Cross and co-workers showed that AMN binds to M2-TM in spectroscopically distinct manners at low versus high pH [6]. AMN and RMN are very similar structurally sharing the adamantane group and the polar amine group. They affect proton translocation in similar ways, and bind to M2 with a stoichiometry of one drug per tetramer [2, 10]. Although AMN binds to M2 with slightly lower affinity, their mechanism of inhibition is thought to be primarily the same [2, 10, 33].
Herein, we report the results of MD simulations performed to shed light on the nature of the drug-binding mode to the tetrameric M2-TM bundle. For membrane-bound M2-TM peptide bundles we find that for all protonation states of His37 examined (i.e., mimicking both high- and low-pH conditions), AMN binds in the central pore of the bundle, although the preferred orientation of the amine group within the pore changes as local pH conditions change. Moreover, in the membrane environment, adamantane drugs do not show significant affinity for external binding sites located close to Asp44 as proposed by the NMR studies on micelle-bound M2-TM peptide bundles.
Methods
A summary of all the MD simulations including the length of the peptide used, simulation time and backbone rmsd is provided in Table 1. The AMN-bound crystal structure obtained by Stouffer et al. (PDB code: 3C9J; residues 22-46) was used as the initial structure in the +3 (three His37 protonated) [MD1], and +4 (all His37 protonated) [MD2] protonation states to investigate the drug binding mechanism under low-pH conditions [17]. As seen from the rmsd values in Table 1 the structure employed is stable for the ∼20 ns timescale in both MD simulations. The NMR structures reported by Schnell and Chou were used to investigate drug binding under high-pH conditions [16]. MD simulations of the NMR structure (PDB code 2RLF; residues 23-60) with four AMN molecules bound externally [MD3], four RMN molecules bound externally [MD4] and one AMN bound internally [MD5] were also stable on the multi-ns timescale with low rmsd values (Table 1). Finally, the AMN-bound model derived from the replication of chain D of the drug-free crystal structure (D4 model obtained from PDB code: 3BKD; residues 22-46) [MD6] [27] [17] was also used to study drug binding at high-pH conditions. The MD3, MD4, MD5 and MD6 simulations provide clues about drug binding under high-pH conditions.
Table 1.
Summary of all the MD simulations reported in this work.
| MD label | PDB code | Length of peptide | His37 protonation | Unconstrained MD time (ns) | Backbone rmsd (Å) | Drug site |
|---|---|---|---|---|---|---|
| MD1 | 3C9J | 22-46 | +3 | 21.4 | 2.47 ± 0.67 | AMN; internal |
| MD2 | 3C9J | 22-46 | +4 | 24.7 | 1.79 ± 0.51 | AMN; internal |
| MD3 | 2RLF | 23-60 | 0 | 17.0 | 1.53 ± 0.18* | AMN; external |
| MD4 | 2RLF | 23-60 | 0 | 24.8 | 2.09 ± 0.26* | RMN; external |
| MD5 | 2RLF | 23-60 | 0 | 17.9 | 2.17 ± 0.23* | AMN; internal |
| MD6 | 3BKD (D4) | 22-46 | 0 | 29.7 | 2.53 ± 0.65 | AMN; internal |
Backbone rmsd for only the transmembrane part is reported (residues 23-46 of the NMR structure).
In all of the MD simulations, protein structures were inserted in the transmembrane orientation into an equilibrated and hydrated 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) bilayer patch containing 64 lipids in each leaflet to reproduce an in vivo-like environment, which neither the X-ray crystallography nor the NMR micelle sample are able to reproduce. The lipid and water molecules overlapping with the protein were removed. Sodium and chloride ions were added to the MD system at 150 mM concentration to maintain overall charge neutrality using the ‘Autoionize’ plugin of VMD [34]. The system sizes ranged from ∼24,000 to ∼50,000 atoms. All MD simulations were performed using the code NAMD [35] with the CHARMM22 protein force field [36], CHARMM27 lipid force field [37] and TIP3P water model [38]. The amantadine (C10H18N+) molecule was constructed and a gas phase optimization was performed at the B3LYP level with a 6-31G* basis set using the GAUSSIAN package [39]. Partial atomic charges were assigned via the RESP scheme [40] in which they are optimized to best reproduce the quantum electrostatic potential calculated on a set of grid points around the molecule with an additional penalty function. The intermolecular potential parameters were taken directly from the CHARMM force field and the equilibrium bond and angle values were taken from the optimized ab initio structure, which is rigid with respect to torsions. The charges and parameters for rimantadine (C12H22N+) were derived in a similar fashion. The protonation state of the His37 tetrad is zero in all the MD simulations using the NMR structure and the D4 model as the initial structures. Six and two water molecules were inserted randomly in the initial structure in the channel cavity close to His37 in MD3 and MD5 respectively. AMN was docked in the cavity of the NMR structure using VMD with its polar group pointing toward the His37 in the initial structure.
The temperature was maintained in all the simulations at 310 K by coupling to a heat bath using the ‘temperature coupling’ method of NAMD. The energy minimizations of all the systems were followed by equilibration runs. During the equilibration runs for the systems in which AMN was bound internally (MD1, MD2, MD5 and MD6) first the heavy atoms of the protein were constrained for 1 ns; followed by 1 ns of harmonic constraints on the Cα atoms. A force constant of 10 kcal/mol/Å2 was used for the harmonic constraints. During the first 2 ns of equilibration, a time step of 1 fs was used for the integration of the equations of motion. After 2 ns, a time step of 1.5 fs was used, with all the hydrogen atoms constrained using the SHAKE [41] and SETTLE [42] algorithms. Special care was taken to equilibrate the systems in which drug molecules were bound externally (MD3 and MD4). For MD3 (AMN bound externally) the 4 RMN molecules were replaced with AMN in the pdb structure 2RLF. The AMN molecules were placed as close as possible to the original RMN positions while trying to maintain favorable contacts between the AMN amine group and residue Asp44. For MD4, initial positions of RMN molecules as reported in the NMR structure were used directly. For both MD3 and MD4 simulations, the protein and drug were held fixed while the solvent relaxed (mainly lipid) around the protein structure. This was accomplished over a 4 ns simulation. Next the side-chains of the protein were released while the protein backbone and drug were held fixed for a simulation time of 2 ns. Following this, a 2 ns simulation was carried out in which the protein backbone and drug were constrained with weak harmonic springs (1.0 kcal/mol/Å2) while the rest of the system was free to relax. Finally, all constraints were removed from the system.
In all the simulations reported, periodic boundary conditions were applied in three dimensions. The Langevin piston Nosé-Hoover method was used to maintain a pressure of 1 atm, allowing isotropic cell fluctuations. Non-bonded interactions were calculated every time step and full electrostatic interactions were calculated every 2 time steps. Long-range electrostatics was taken into account via the particle mesh Ewald (PME) scheme [43, 44]. The number of grid points in each dimension for PME were decided based on the periodic box size, with roughly one grid point corresponding to 1 Å in each dimension. The default value of PME direct space tolerance in NAMD (10-6) was used.
Results and Discussion
Internal binding site under low-pH conditions
MD simulations using the crystal structure of M2-TM in complex with AMN (PDB code: 3C9J) performed with +3 and +4 protonation states of the His37 tetrad (MD1 and MD2, respectively) are representative of low-pH structures. In both of these simulations, AMN is found to be stable at a binding site spanning the pore region from residues Val27 to Gly34 for the entire length of the trajectories. In both low-pH systems, the hydrophobic core of AMN interacts closely with Val27, while the AMN polar group interacts with water in the cavity and remains mostly oriented toward the His37 gate (Figures 1 and 3). Within this structural arrangement, it is energetically favorable for AMN to point its polar group toward the His37 gate, which is highly solvated. Water molecules cannot go through the closed Val27 gate further blocked by the AMN molecule. These low-pH simulations provide support for the orientation of AMN inside the tetrameric bundle, which was initially inferred from the crystal structure [17].
FIGURE 1. Internal binding at low pH.
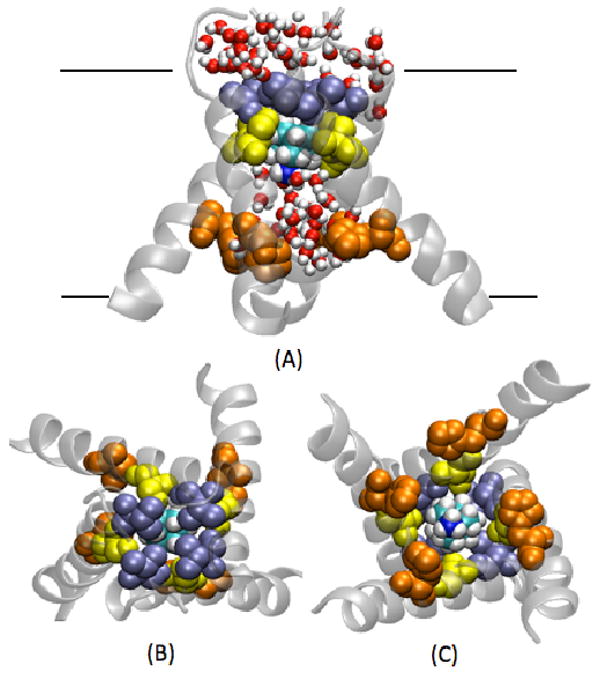
Snapshot taken from the MD2 simulation of the crystal structure with AMN bound in the channel cavity. Val 27 (ice blue), Ser 31(yellow) and His 37 (orange) are indicated. (A) Front view, (B) N-term view, and (C) C-term view.
FIGURE 3. AMN rotation.
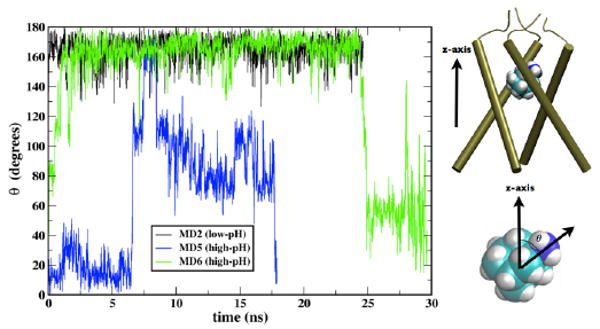
Time evolution of theta, the angle between the C-N bond vector of AMN and the bilayer normal (z-axis) is shown for 3 simulations for the unconstrained runs; MD2 (black), MD5 (blue) and MD6 (green). A value of theta = 180° indicates the AMN polar group is oriented toward the C-terminus (His37 gate).
The simulations clearly indicate that the principal interactions stabilizing the pore binding site of AMN at low-pH conditions are mainly of hydrophobic nature: the adamantane core of AMN strongly interacts with Val27 (Figure 1). This observation also suggests that, sharing the same hydrophobic core, RMN could likely present a similar binding mode at the pore site. It has been commented by Pielak et al [25] that adamantanes could not bind to the pore region in the conformation proposed by the X-ray structure [17] due to the presence of Ser31 residue, which would disrupt favorable hydrophobic interactions with AMN. Our MD simulations show that Ser31, although pointing towards the pore lumen, cannot directly reach the drug; it is a short side-chain and thus does not perturb the Val27/Ala30-AMN interactions, which lock AMN in the pore (Figure 1). The latter observation could also offer a rationale for adamantane resistance emerged from the Ser31Asn point mutation often present in recent influenza strains [18, 19]; the longer side-chain of Asn likely protrudes more into the pore, thus preventing adamantane binding at the site.
External binding sites under high-pH conditions
The coordinates of one of the structures from the NMR ensemble of RMN-bound residues 23-60 (PDB code: 2RLF) reported by Schnell and Chou [16] were used as the initial structure for MD simulations of externally-bound AMN (MD3) and RMN (MD4) molecules near Asp44 residues (Figure 2). In MD3, polar groups of the four AMN molecules drift away from the protein at different rates during the ∼21 ns trajectory (Figure 2C: ∼4 ns constrained run followed by ∼17 ns of unconstrained run). The distance between the AMN nitrogen and Asp44 Cγ atoms for one of the AMN molecules (shown by red in Figure 2(c)) increases immediately after the constrained run (∼4 ns) and becomes > ∼10 Å in ∼7 ns. At this time, the drug does not interact with the protein anymore but shows preferential interactions with the lipid head groups and the surrounding water molecules. On the other hand, two AMN molecules (shown by blue and green in Figure 2 (c)) stay in the vicinity of the external binding site (AMN nitrogen-Asp44 Cγ distance ≤ 6 Å) for a slightly longer time but have moved far away in ∼12 ns. However, the fourth AMN molecule (shown by black in Figure 2 (c)) seems to fluctuate around the binding site (AMN nitrogen-Asp44 Cγ distance varies between ∼ 4 Å and ∼10 Å) for the entire length of the trajectory. Similar results are observed for the RMN-bound system (MD4), in which three RMN molecules diffuse far away from the Asp44 external binding sites during the ∼ 25 ns long simulation (Figure S1). Hence, our results indicate that the external sites do not bind the drug molecules tightly. It should be noted that a very high concentration of the RMN drug was used in the NMR experiment (∼40 mM, comprising 13 mole percent of the micelle in which the protein was embedded) [16]. It is possible that in the NMR micelle experiment, a dynamic equilibrium exists in which the outgoing drug molecules are replaced by the incoming drug molecules and the external sites exhibit binding of the drug under conditions of high drug concentration. Indeed, Cady et al have now shown that amantadine binds to external sites only under high concentration and it shows 40-fold greater affinity for the internal site than the external [26]. Our MD simulations in membranes demonstrate that drug molecules mostly tend to drift away from external sites over tens of nanoseconds, while they are stable at the internal site over this time scale. Hence, the external binding sites reported for the drug molecules at the high concentration of 40 mM likely correspond to secondary binding sites and not the pharmacologically relevant sites. The mutagenesis experiments of Jing et al. also indicate that an external binding site is not the primary site for the pharmacological inhibition of M2 since Asp44Ala is still sensitive to AMN, based on electrophysiological measurements [23].
FIGURE 2. External binding at high pH.
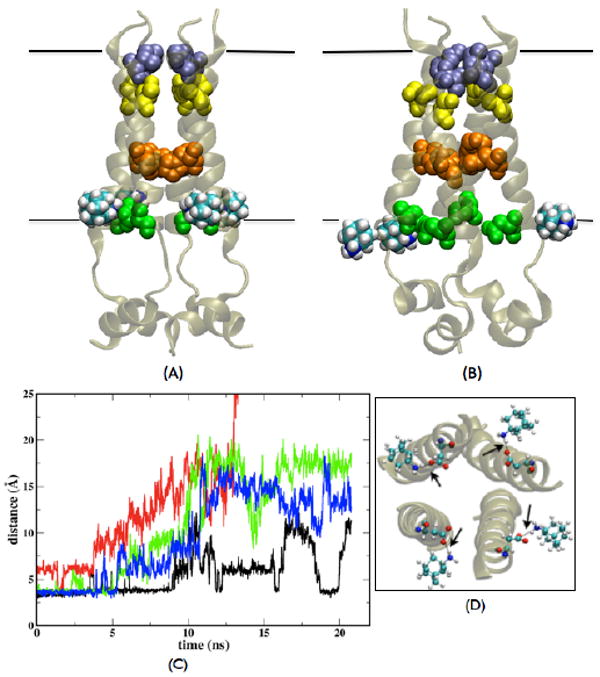
(A) Initial snapshot of the NMR structure [16] with four AMN molecules bound outside. (B) Snapshot after ∼17ns of unconstrained MD3 simulation. Three AMN molecules have disassociated from the external binding sites [one of these (shown by red in (C)) is out of the frame and thus cannot be seen in the image]- Val27 (ice blue), Ser31 (yellow), His37 (orange), Asp44 (green) and transmembrane part (black boundaries) are shown. (C) Time evolution of the distance between AMN nitrogen and Asp44 Cγ atoms of the corresponding helices with which they interacted in the initial conformation [C-term view shown in (D)]; data is shown from a part of the constrained run and entire unconstrained run; different colors are used to indicate the four AMN molecules for clarity. The black arrows in (D) point to the distances plotted in (C).
Internal binding site under high-pH conditions
During the course of the ∼17 ns-long MD3 simulation based on the NMR structure, water molecules were seen to easily pass through the solvated Val27 gate, in agreement with our previous MD simulations representing high-pH conditions [27]. However, no water molecules were detected in the cavity around Val27 in the NMR structure [16]. As discussed by Stouffer et al. [45] perhaps the absence of NOEs with water molecules in this region could be due to the presence of the drug in the cavity, with the possibility that it could not be detected in the NMR experiments due to motional averaging. In order to test the above hypothesis, and to explore the dynamic properties of the drug-binding site in the central cavity under high-pH conditions, an additional MD simulation (MD5) was performed with initial conformation chosen from the ensemble of 15 structures reported by Schnell and Chou (PDB code: 2RLF). The four RMN molecules were removed from the outer surface of the protein and a single AMN was inserted in the cavity between Val27 and Gly34 with its polar group pointing towards His37 residues. The AMN molecule rapidly tumbles during the constrained run, and unlike under low-pH conditions, the orientation of the polar group fluctuates over a wider range during the ∼18 ns unconstrained MD trajectory (Figures 3 and 4). This difference in orientation can be explained by different solvation effects found at the Val27 gate under different pH conditions: at low pH, the His37 gate is open and solvated whereas Val27 is tightly closed, allowing the drug to orient the amine polar group preferentially toward the His37 residues; under high-pH conditions the Val27 gate is much more solvated and water molecules are present in the cavity too (Figures 1 and 4). As a result AMN tumbles much more within the pore with its polar group orientation alternating towards the two gates (Val27 and His37) under high-pH conditions.
FIGURE 4. Internal binding at high pH.
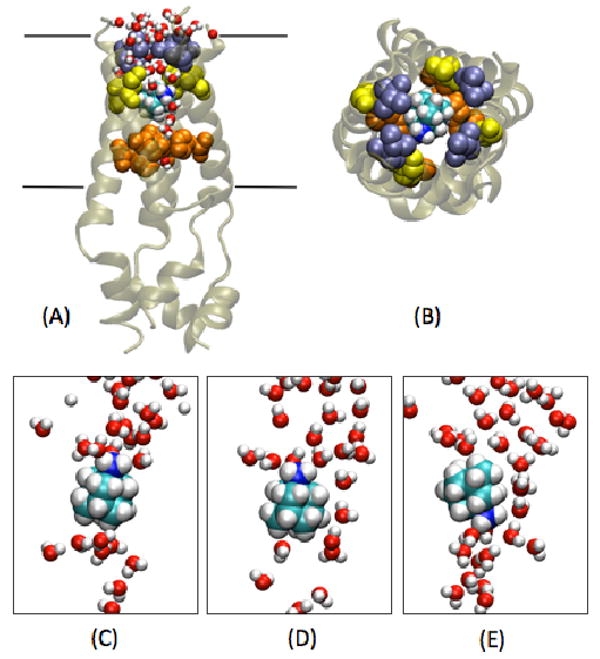
Snapshot of the simulation with AMN in the channel cavity of NMR structure (MD5) with Val27 (ice blue), Ser31 (yellow), His37 (orange) and the transmembrane region (black boundaries) indicated. (A) Front view (B) N-term view. (C), (D) and (E) Three representative snapshots showing intermittent formation of water wires and fluctuation in AMN polar group orientation.
In the drug-free state the Val27 gate is wider in an Openout-Closedin state allowing easy passage of water molecules [27]. In the current MD simulation the presence of AMN impedes the transport of water. However, intermittent formation of water wires allows some water molecules to go through (Figure 4). Rapid spinning of AMN about its molecular symmetry axis is observed with a relaxation time of ∼100 ps. This behavior is also confirmed by an additional simulation of the channel model at high pH derived from the X-ray structure (D4 model) [17, 27]. During the course of ∼30 ns with AMN inserted in the cavity of the D4 model (MD6), we observe that as the pore radius around Val27 increases, the AMN molecule tumbles to point its polar group toward the Val27 gate (Figures 3 and S2). A snapshot from near the end of this MD simulation looks similar to the one in Figure 4A. Thus, when the Val27 pore tends to open and M2 adopts an Openout-Closedin conformation (∼3.5 Å, Figure S2), both the NMR structure and the D4 structure (MD5 and MD6 simulations) show solvation of the Val27 gate and the central cavity which in turn produces similar fluctuations in AMN polar group orientation. In agreement with our simulations, Cady et al have shown recently that the drug binds to the internal pore binding site under high pH conditions and undergoes significant motion [26]. The results of our simulations also agree with those of Yi et al who reported that C-N bond vector of AMN exhibits wide range of orientations inside the M2 cavity under high-pH conditions [46].
Pharmacologically relevant binding site and the development of drug resistance
The MD simulations discussed above support the view that AMN inhibits proton transport through M2 by binding inside the bundle cavity as suggested by the low pH x-ray structure of Stouffer et al. [17] and the very recent high pH solid-state NMR structure of Cady et al [26]. Although the x-ray and solid-state NMR structures are for a shorter peptide (residues 22-46) than the M2 used in the NMR study (residues 23-60) [16], the present MD results yield similar behavior for both structures. Previous studies have shown that M2-TM (residues 22-46) forms tetrameric bundles that bind AMN, providing strong support for using this M2-TM peptide as a minimal model for studying proton conduction and drug binding in the M2 protein [6-9].
In the present MD simulations AMN stays inside the M2 cavity and the molecular details indicate that the polar group of AMN points toward the His37 gate under low-pH conditions. This binding site of AMN also agrees with the previous studies that indicate the site is the hotspot for amantadine resistant mutations [13, 47]. As discussed above, it was noted recently by Pielak et al. that the interactions of the hydrophilic Ser31 side chain and the hydrophobic adamantane group would be unfavorable and thus make the internal binding site difficult to reconcile [25]. They commented that it would be difficult for the hydrophobic drug to replace the water molecules present in the absence of drug [17]. However, in the NMR structure reported by Schnell and Chou, the Ser31 side chain does not face toward the interior of the pore, but rather interacts with the neighboring helices [16, 25]. In the AMN-bound X-ray crystal structure, and in MD simulations based on it, Ser31 does point toward the interior but enough space remains in the cavity that is occupied by water molecules in the absence of AMN. Moreover, the phenomena of a hydrophobic drug (tetrabutylammonium) replacing water molecules present inside a channel cavity has been observed previously in the case of KcsA K+ channel [48]. In the X-ray structure, the hydrophobic interactions of the adamantane group with the other amino acids present at the internal drug binding site, especially Val27, likely lead to the stabilization of the drug at this site. Our MD simulations show that the drug is stable at the internal site in both the X-ray structures (MD1 and MD2) and the NMR structure (MD5).
On the other hand, in MD simulations of the membrane-bound bundle of M2 with residues 23-60, drug molecules do not exhibit strong binding at the external sites observed in the NMR micelle study [16]. However, as discussed above, the high concentration of the drug in the NMR experiments (13% of the micellar components) and the strong partitioning of the drug in the micelle [45] likely explain the observation of NOEs due to drug molecules. Moreover, the notion that the external binding sites are the pharmacologically relevant ones is inconsistent with many previous studies. For example, the drug inhibits proton transport by binding to the channel with a stoichiometry of one drug to one tetramer [32, 33] and not four drugs to one tetramer. A strong evidence in support for the internal binding site being pharmacologically relevant comes from the very recent solid-state NMR experiments that demonstrate that AMN is bound at the internal site at a stoichiometry of one drug to one tetramer and stays bound when the drug concentration is increased [26]. Our results from present MD simulations provide evidence that AMN and RMN interactions with Asp44 are not very stable in the membrane environment and tend to mostly disrupt on the multi-ns timescale.
Stouffer et al. suggested that the absence of NOEs in ∼7 Å wide pore in the NMR structure could be due to the motional averaging of the drug molecule at this site [45]. The present MD simulations show that the drug molecule at this site indeed exhibits dynamic rotation in various orientations under the relevant high-pH conditions. It was proposed by Schnell and Chou that the drug resistant mutation Ser31Asn stabilizes the open conformation. Contrary to this hypothesis, Stouffer et al. have shown that the specific activity of Ser31Asn is very similar to the wild-type [45]. Also, the electrophysiological experiments by Jing et al. show that the mutations of specific residues that interact with the drug in the NMR structure, Asp44Ala and Arg45Ala, exhibit AMN sensitivity [23]. More recently, it has also been shown that mutations of residues that are N-terminal to His37 exhibit significantly lower inhibition by AMN while most mutations of Trp41 and Asp44 had little or no effect on AMN inhibition, indicating that the external binding sites are not the primary sites associated with the pharmacological inhibition of M2 [24]. Thus, preponderance of the experimental evidence along with the present MD simulations support the view that the pharmacologically relevant binding site is located inside the cavity as opposed to the protein bundle external surface.
Conclusions
The present MD simulations are consistent with our recent proposal that the M2 protein shows pH-dependent transporter-like properties modulated by the opening and closing of Val27 and His37 gates [27]. In particular, the simulations suggest that two different binding modes for adamantane drugs are associated with the two distinct states of conduction suggested in a transporter-like mechanism (Figure 5). Under high-pH conditions, when the Val27 gate is open and solvated (Openout-Closedin state), AMN can likely find a favorable entrance and accommodates in the cavity, where the orientation of its polar group fluctuates. In this conformation AMN could severely limit proton conduction into the viral interior by preventing the protonation of His37 residues due to steric hindrance at the Val27 gate, essentially locking M2 in the Openout-Closedin state. Cross and co-workers showed that AMN binding lowers the pKa of the His37 residues by about three orders of magnitude compared to the first pKa of His37 in drug free M2-TM [6]. The change in pKa on AMN binding could also contribute to inhibition of proton transport.
FIGURE 5. Schematic representation of AMN blocking modes at low- and high-pH.
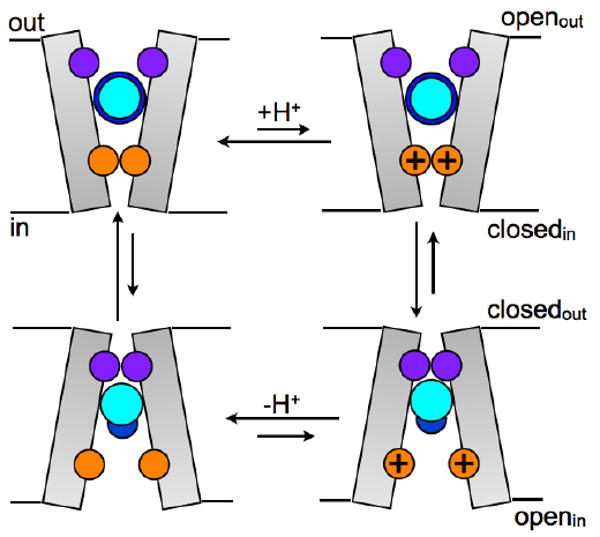
Val27 (purple) and His37 (orange) gates are shown. Under high-pH conditions Openout-Closedin state is dominant, AMN largely prevents proton transport to His37 gate in this state by blocking the Val27 gate and the orientation of AMN polar group (dark blue) fluctuates between the two gates. If, however, protons reach the His37 gate, the Closedout-Openin conformation is favored in which AMN is mostly oriented toward the His37 gate. After the release of protons to the viral interior, the Openout-Closedin state is favored again.
In the event that some protons are able to penetrate inside the M2 cavity via intermittent water wires and the His37 gate becomes charged and opens, the Val27 gate would close (Closedout-Openin state). In this conformation the hydrophobic group of AMN exhibits strong interactions with the closed Val27 gate. Its polar group points toward His37 and is solvated by water molecules in the bundle interior. AMN completely blocks the water (and proton) transport through the Val27 gate under these conditions. AMN could lock the conformation in this Closedout-Openin state or if the equilibrium shifts back to the Openout-Closedin state after release of protons to the viral interior, AMN would still inhibit proton transport due to steric effects and alteration of His37 pKa as discussed above.
Based on the dynamic features obtained from the MD simulations, AMN uptake would likely be more difficult in the Closedout-Openin state, thus AMN may preferentially bind to M2 when the channel is in the Openout-Closedin state at high-pH conditions, which is consistent with the higher drug affinity at higher pH [7, 10]. Because of the structural similarity between AMN and RMN, RMN will likely exhibit the same behavior in the channel pore, hence blocking the proton transport by a similar mechanism.
In conclusion, our MD simulations agree with the prevailing view that adamantane drugs block proton transport through M2 by binding inside the cavity [17, 23, 26]. AMN uptake at the pore binding site likely occurs when the Val27 gate is open and the His37 gate is closed (i.e., mostly in high-pH state). We propose different modes of binding depending on the protonation state of His37 gate, which are consistent with the recently proposed kinetic model for proton conduction through a transporter-like mechanism [27]. The proton transport to the viral interior is inhibited in both the modes via blocking of proton passage through the Val27 gate. The highly persistent Ser31Asn mutation at this site in adamantane-resistant strains of influenza A virus, as present in recent avian and swine flu strains also [18, 19], would then explain the reduction in adamantane drug binding affinity as has been discussed previously [17, 21, 23, 49].
Supplementary Material
Acknowledgments
The authors thank William F. DeGrado for many insightful suggestions, Vincenzo Carnevale for discussions, Axel Kohlmeyer for enabling the computations, and Satyavani Vemparala for providing the amantadine parameters. This work was supported by the National Institutes of Health grant U01-AI-074571.
Footnotes
Supplementary Information: Supplementary Information is available.
Conflict of interest statement: M.L.K. is a founder and member of the Science Advisory Board of InfluMedix.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pinto LH, Lamb RA. Influenza virus proton channels. Photochemical and Photobiological Sciences. 2006;5:629–632. doi: 10.1039/b517734k. [DOI] [PubMed] [Google Scholar]
- 2.Kolocouris N, Kolocouris A, Foscolos GB, Fytas G, Neyts J, Padalko E, Balzarini J, Snoeck R, Andrei G, DeClercq E. Synthesis and antiviral activity evaluation of some new aminoadamantane derivatives .2. Journal of Medicinal Chemistry. 1996;39:3307–3318. doi: 10.1021/jm950891z. [DOI] [PubMed] [Google Scholar]
- 3.Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI. Adamantane resistance among influenza A viruses isolated early during the 2005-2006 influenza season in the United States. Jama-Journal of the American Medical Association. 2006;295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 4.Hurt AC, Ernest J, Deng YM, Iannello P, Besselaar TG, Birch C, Buchy P, Chittaganpitch M, Chiu SC, Dwyer D, Guigon A, Harrower B, Kei IP, Kok T, Lin C, McPhie K, Mohd A, Olveda R, Panayotou T, Rawlinson W, Scott L, Smith D, D'Souza H, Komadina N, Shaw R, Kelso A, Barr IG. Emergence and spread of oseltamivir-resistant A(H1N1) influenza viruses in Oceania, South East Asia and South Africa. Antiviral Res. 2009;83:90–93. doi: 10.1016/j.antiviral.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Hayden F. Developing New Antiviral Agents for Influenza Treatment: What Does the Future Hold? Clinical Infectious Diseases. 2009;48:S3–S13. doi: 10.1086/591851. [DOI] [PubMed] [Google Scholar]
- 6.Hu J, Fu R, Cross TA. The chemical and dynamical influence of the anti-viral drug amantadine on the M2 proton channel transmembrane domain. Biophys J. 2007;93:276–283. doi: 10.1529/biophysj.106.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salom D, Hill BR, Lear JD, DeGrado WF. pH-dependent tetramerization and amantadine binding of the transmembrane helix of M2 from the influenza A virus. Biochemistry. 2000;39:14160–14170. doi: 10.1021/bi001799u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from Influenza A virus. Biophys J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cady SD, Hong M. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1483–1488. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma C, Polishchuk AL, Ohigashi Y, Stouffer AL, Schon A, Magavern E, Jing X, Lear JD, Freire E, Lamb RA, Degrado WF, Pinto LH. Identification of the functional core of the influenza A virus A/M2 proton-selective ion channel. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0905726106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA. Histidines, heart of the hydrogen ion channel from influenza A virus: Toward an understanding of conductance and proton selectivity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6865–6870. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Witter R, Nozirov F, Sternberg U, Cross TA, Ulrich AS, Fu RQ. Solid-state F-19 NMR spectroscopy reveals that Trp(41) participates in the gating mechanism of the M2 proton channel of influenza a virus. Journal of the American Chemical Society. 2008;130:918–924. doi: 10.1021/ja0754305. [DOI] [PubMed] [Google Scholar]
- 13.Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. A functionally defined model for the M-2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Kim S, Kovacs F, Cross TA. Structure of the transmembrane region of the M2 protein H+ channel. Protein Science. 2001;10:2241–2250. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cady SD, Goodman C, Tatko CD, DeGrado WF, Hong M. Determining the Orientation of Uniaxially Rotating Membrane Proteins Using Unoriented Samples: A 2H, 13C, and 15N Solid-State NMR Investigation of the Dynamics and Orientation of a Transmembrane Helical Bundle. J Am Chem Soc. 2007;129:5719–5729. doi: 10.1021/ja070305e. [DOI] [PubMed] [Google Scholar]
- 16.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Update: drug susceptibility of swine-origin influenza A (H1N1) viruses, April 2009. MMWR Morb Mortal Wkly Rep. 2009;58:433–435. [PubMed] [Google Scholar]
- 19.Hurt AC, Selleck P, Komadina N, Shaw R, Brown L, Barr IG. Susceptibility of highly pathogenic A(H5N1) avian influenza viruses to the neuraminidase inhibitors and adamantanes. Antiviral Res. 2007;73:228–231. doi: 10.1016/j.antiviral.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Miller C. Ion channels: Coughing up flu's proton channels. Nature. 2008;451:532–533. doi: 10.1038/451532a. [DOI] [PubMed] [Google Scholar]
- 21.Yi M, Cross TA, Zhou HX. A secondary gate as a mechanism for inhibition of the M2 proton channel by amantadine. J Phys Chem B. 2008;112:7977–7979. doi: 10.1021/jp800171m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen HN, Wu YJ, Voth GA. Proton transport Behavior through the influenza a M2 channel: Insights from molecular simulation. Biophysical Journal. 2007;93:3470–3479. doi: 10.1529/biophysj.107.105742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jing X, Ma C, Ohigashi Y, Oliveira FA, Jardetzky TS, Pinto LH, Lamb RA. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balannik V, Carnevale V, Fiorin G, Levine BG, Lamb RA, Klein ML, Degrado WF, Pinto LH. Functional Studies and Modeling of Pore-Lining Residue Mutants of the Influenza A Virus M2 Ion Channel. Biochemistry. 2010 doi: 10.1021/bi901799k. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pielak RM, Schnell JR, Chou JJ. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc Natl Acad Sci U S A. 2009;106:7379–7384. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khurana E, Dal Peraro M, DeVane R, Vemparala S, DeGrado WF, Klein ML. Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza A virus. Proc Natl Acad Sci U S A. 2009;106:1069–1074. doi: 10.1073/pnas.0811720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mould JA, Li HC, Dudlak CS, Lear JD, Pekosz A, Lamb RA, Pinto LH. Mechanism for proton conduction of the M-2 ion channel of influenza A virus. Journal of Biological Chemistry. 2000;275:8592–8599. doi: 10.1074/jbc.275.12.8592. [DOI] [PubMed] [Google Scholar]
- 29.Chizhmakov IV, Ogden DC, Geraghty FM, Hayhurst A, Skinner A, Betakova T, Hay AJ. Differences in conductance of M2 proton channels of two influenza viruses at low and high pH. J Physiol-London. 2003;546:427–438. doi: 10.1113/jphysiol.2002.028910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gandhi CS, Shuck K, Lear JD, Dieckmann GR, DeGrado WF, Lamb RA, Pinto LH. Cu(II) inhibition of the proton translocation machinery of the influenza A virus M-2 protein. Journal of Biological Chemistry. 1999;274:5474–5482. doi: 10.1074/jbc.274.9.5474. [DOI] [PubMed] [Google Scholar]
- 31.Mould JA, Drury JE, Frings SM, Kaupp UB, Pekosz A, Lamb RA, Pinto LH. Permeation and activation of the M2 ion channel of influenza A virus. J Biol Chem. 2000;275:31038–31050. doi: 10.1074/jbc.M003663200. [DOI] [PubMed] [Google Scholar]
- 32.Wang C, Takeuchi K, Pinto LH, Lamb RA. Ion-Channel Activity of Influenza-a Virus M(2) Protein - Characterization of the Amantadine Block. Journal of Virology. 1993;67:5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Czabotar PE, Martin SR, Hay AJ. Studies of structural changes in the M2 proton channel of influenza A virus by tryptophan fluorescence. Virus Res. 2004;99:57–61. doi: 10.1016/j.virusres.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 34.Humphrey W, Dalke A, Schulten K. VMD- Visual Molecular Dynamics. Journal of Molecular Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 35.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacKerell AD, Bashford D, Jr, Bellott M, Dunbrack RL, Evanseck JD, Jr, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, III, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 37.Feller SE, Yin D, Pastor RW, MacKerell AD. Molecular dynamics simulation of unsaturated lipid bilayers at low hydration: parameterization and comparison with diffraction studies. Biophys J. 1997;73:2269–2279. doi: 10.1016/S0006-3495(97)78259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 39.M.J.T. Frisch, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; and Pople, J. A., Gaussian, Gaussian, Inc., Wallingford, CT, 2004.
- 40.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges-the RESP model. Journal of Physical Chemistry. 1993;97:10269–10280. [Google Scholar]
- 41.Ryckaert JP, Ciccotti G, Berendson HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 42.Miyamoto S, Kollman PA. SETTLE: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. Journal of Computational Chemistry. 1992;13:952–962. [Google Scholar]
- 43.Darden T, York D, Pedersen L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. Journal of Chemical Physics. 1993;98:10089–10092. [Google Scholar]
- 44.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. Journal of Chemical Physics. 1995;103:8577–8593. [Google Scholar]
- 45.Stouffer AL, Ma C, Cristian L, Ohigashi Y, Lamb RA, Lear JD, Pinto LH, DeGrado WF. The interplay of functional tuning, drug resistance, and thermodynamic stability in the evolution of the M2 proton channel from the influenza A virus. Structure. 2008;16:1067–1076. doi: 10.1016/j.str.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yi M, Cross TA, Zhou HX. Conformational heterogeneity of the M2 proton channel and a structural model for channel activation. Proc Natl Acad Sci U S A. 2009;106:13311–13316. doi: 10.1073/pnas.0906553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang YJ, Zaitseva F, Lamb RA, Pinto LH. The gate of the influenza virus M-2 proton channel is formed by a single tryptophan residue. Journal of Biological Chemistry. 2002;277:39880–39886. doi: 10.1074/jbc.M206582200. [DOI] [PubMed] [Google Scholar]
- 48.Yohannan S, Hu Y, Zhou Y. Crystallographic study of the tetrabutylammonium block to the KcsA K+ channel. J Mol Biol. 2007;366:806–814. doi: 10.1016/j.jmb.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 49.Astrahan P, Kass I, Cooper MA, Arkin IT. A novel method of resistance for influenza against a channel-blocking antiviral drug. Proteins-Structure Function and Bioinformatics. 2004;55:251–257. doi: 10.1002/prot.20018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.