

Abstract
The aim of this article is to analyze conformational changes by comparing 10 different structures of Pseudomonas aeruginosa phosphomannomutase/phosphoglucomutase (PMM/PGM), a four-domain enzyme in which both substrate binding and catalysis require substantial movement of the C-terminal domain. We focus on changes in interdomain and active site crevices using a method called computational solvent mapping rather than superimposing the structures. The method places molecular probes (i.e., small organic molecules containing various functional groups) around the protein to find hot spots. One of the most important hot spots is in the active site, consistent with the ability of the enzyme to bind both glucose and mannose phosphosugar substrates. The protein has eight additional hot spots at domain-domain interfaces and hinge regions. The locations and nature of six of these hot spots vary between the open, half-open, and closed conformers of the enzyme, in good agreement with the ligand-induced conformational changes. In the closed structures the number of probe clusters at the hinge region significantly depends on the position of the phosphorylated oxygen in the substrate (e.g., glucose 1-phosphate versus glucose 6-phosphate), but the protein remains almost unchanged in terms of the overall RMSD, indicating that computational solvent mapping is a more sensitive approach to detect changes in binding sites and interdomain crevices. Focusing on multidomain proteins we show that the subresolution conformational differences revealed by the mapping are in fact significant, and present a general statistical method of analysis to determine the significance of rigid body domain movements in X-ray structures.
Keywords: allostery, free energy landscape, hinge bending, ligand binding pocket, computational solvent mapping, X-ray crystallography, conformational change, structure comparison
Introduction
Proteins are dynamic polymers and their conformational flexibility is critical for function, particularly for interactions with other molecules.1 The conformational transitions can be described in terms of the classical induced fit mechanism,2 or the conformational selection model which assumes an ensemble of pre-existing conformations.3 The influence of ligand binding on the conformation of any protein was first demonstrated in 1965 for the enzyme rabbit phosphoglucomutase, a member of the phosphohexomutase superfamily.2 Although no phosphoglucomutase structure was available in 1965, Yankeelov and Koshland were able to show that the binding of the substrate glucose 6-phosphate causes a number of significant changes, including the enhancement of the fluorescence spectrum, an increased rate of inactivation by iodoacetamide, and a decreased reactivity of lysine, methionine, and cysteine residues to alkylation.3 Taking together the data from various experiments, they concluded that a substrate-induced conformational change occurs, and that this conformational change is related to the specificity of the enzyme.
In this article we return to the conformational analysis of a phosphohexomutase, and focus on phosphomannomutase/phosphoglucomutase (PMM/PGM) from Pseudomonas aeruginosa, a human pathogen. PMM/PGM has been the subject of multiple crystallographic and kinetic investigations,4–12 and hence in our analysis we can use 10 X-ray structures of the apoenzyme and its complexes with various substrates. Another novel aspect of this study is that we compare the PMM/PGM conformers in terms of their inter-domain crevices using a very sensitive tool called computational solvent mapping,13–16 originally developed for the identification of “hot spots,” that is, pockets of a protein that bind a variety of small organic molecules. An established experimental approach to finding such hot spots is screening for the binding of fragment-sized organic compounds.17–19 As the binding is very weak, it is usually detected by nuclear magnetic resonance (SAR by NMR17) or by X-ray crystallography18,19 methods. In particular, the multiple solvent crystal structures (MSCS) method involves the soaking of a protein in a series of organic solvents, whereupon binding regions can be determined based on the crystallization of the protein in each solvent and subsequent superimposition of the X-ray structures to identify regions where multiple solvent molecules bind.18,19 The protein mapping algorithm used in this article is a computational analog of the MSCS experiments. The method places molecular probes, small organic molecules containing various functional groups, around the protein surface on a dense grid, finds favorable positions by further search using empirical free energy functions, clusters the low energy conformations, and ranks the clusters on the basis of the average free energy.15 The low energy clusters of different probes are further clustered to identify consensus sites, and the importance of such sites is measured in terms of the probe clusters contained. Since the differences in the number of probe clusters that bind to a particular site highlight even very small conformational changes if those affect the size or surface properties of the pocket, mapping is very useful for comparing different structures of a protein.20–24 In fact, we find pockets that, according to our druggability criteria,15,16 are not likely to bind any ligand beyond the size of the probes, but the changes in their size and shape provide insight and can be verified by other means.
The Pseudomonas aeruginosa PMM/PGM catalyzes the reversible, intramolecular conversion of glucose 6-phosphate (G6P) to glucose 1-phosphate (G1P), or mannose 6-phosphate (M6P) to mannose 1-phosphate (M1P), depending on the biosynthetic pathway in which it is operating. The reaction involves two successive phosphoryl transfer steps (first from enzyme to substrate and second from the intermediate back to the enzyme), and proceeds through a bisphosphorylated sugar intermediate (e.g., glucose 1,6-bisphosphate or G16P). Crystal structures of enzyme-substrate complexes show that the 1- and 6-phosphosugar substrates are accommodated in two distinct, overlapping binding modes in the same binding site via carefully positioned interactions with the O3 and O4 hydroxyl groups.8 These structures showed definitively that the mechanism of the enzyme requires a dramatic 180° reorientation of the intermediate in between phosphoryl transfer steps. In addition, an isotope trapping experiment has shown that the reorientation of G16P occurs without dissociation from the enzyme7 and can, thus, be considered a simple example of processivity, as defined by multiple rounds of catalysis without release of substrate.
The 10 X-ray structures of P. aeruginosa PMM/PGM to be studied are listed in Table I. Figure 1(A) shows four structures that represent four distinct backbone conformations of the protein. PMM/PGM has four domains arranged in an overall “heart” shape [Fig. 1(B)]. The first three domains are bridged by extensive domain-domain interfaces, whereas domain 4 has a less extensive interface with the remainder of the protein. The large active site cleft is formed by residues from all four domains of PMM/PGM. The complexes of different enzyme-substrate complexes are all very similar to each other and are represented by structure 1P5D in Figure 1(A). The superposition of these complexes with the apo-protein (1K35) indicates a rotation of domain 4 relative to the rest of the polypeptide by approximately 9° [Fig. 1(A)], moving some individual residues by as much as 4.5 Å, and changing the active site from a deep pocket which nearly encloses the bound ligand to a relatively open cleft.9 The active site cleft is even more open in 1K2Y, the apo-form of the S108A mutant. The structure 2FKF of the phosphorylated enzyme bound to the reaction intermediate G16P is between the open and closed states, with a half-open active site.11 We note that since in the catalytic cycle the enzyme binding G16P is dephosphorylated, 2FKF is an off-pathway conformation.11 The flexibility of domain 4 relative to the rest of the protein has clear relevance to substrate binding, and is likely to play a role at multiple points during catalysis. In particular, the rotation of domain 4 and its resulting interaction with domain 1 is critical for creating the high-affinity substrate binding site. One would expect that movement of domain 4 would be required upon not only substrate binding and product release, but also upon reorientation of the intermediate during the reaction. However, all closed structures, including the dephosphorylated protein with bound intermediate (2FKM) are very similar, with an overall Cα RMSD of around 0.2 Å. Since this is comparable to the error in the atomic coordinates, these differences are generally considered insignificant.
Table I.
P. aeruginosa PMM/PGM Structures Used for Computational Solvent Mapping
| Protein | PDB ID | Description |
|---|---|---|
| WT complex with G1P | 1P5D | Phosphorylated protein with bound substrate in closed conformation |
| WT complex with G6P | 1P5G | Phosphorylated protein with bound substrate in closed conformation |
| WT complex with M1P | 1PCJ | Phosphorylated protein with bound substrate in closed conformation |
| WT complex with M6P | 1PCM | Phosphorylated protein with bound substrate in closed conformation |
| WT complex with X1P | 2H5A | Phosphorylated protein with bound ligand (inhibitor) in closed conformation |
| WT complex with R1P | 2H4L | Phosphorylated protein with bound ligand (slow substrate) in closed conformation |
| S108D complex with G16P | 2FKM | Dephosphorylated protein with bound intermediate in closed conformation |
| WT complex with G16P | 2FKF | Phosphorylated protein with bound intermediate in half-closed conformation |
| WT apo-enzyme | 1K35 | Phosphorylated protein in open conformation |
| S108A apo-enzyme | 1K2Y | Dephosphorylated protein in open conformation |
Figure 1.
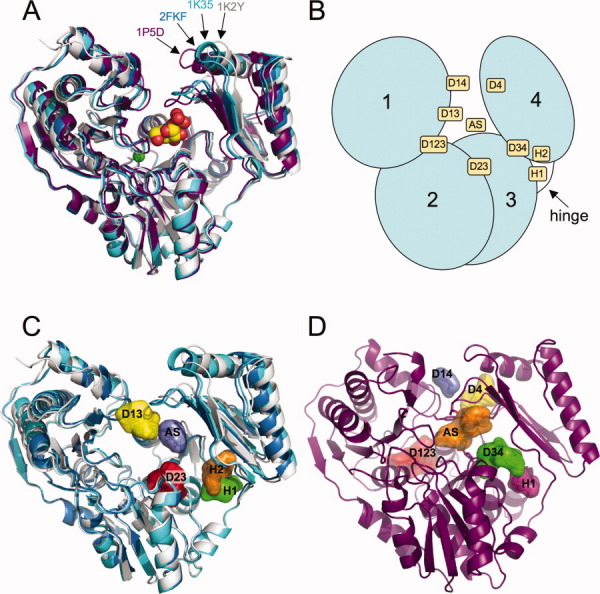
(A) Ribbon diagram showing a superposition of the four P. aeruginosa PMM/PGM conformers. The closed conformer is represented by the 1P5D structure (purple). Three other distinct conformers found in structures 2FKF, 1K35, and 1K2Y are shown in blue, cyan, and light gray, respectively. The substrate glucose 1-phosphate is shown in a space-filling representation as bound in the active site of 1P5D. The metal ion required for phosphoryl transfer is shown as a green sphere. (B) A schematic of PMM/PGM with its four domains (domain 1: residues 1-153; domain 2: residues 154-256; domain 3: residues 257-368; domain 4: residues 369-463) showing the general location of the hot spots from Table II. (C) The three open (or partially open) conformers of PMM/PGM (2FKF, 1K35 and 1K2Y) shown with the top five hot spots for 1K2Y. Hot spots are displayed with a surface surrounding all probes in the site; colors reflect the rank (numbers of probe clusters per site) of the hot spots for 1K2Y, from highest to lowest: red, orange, yellow, green, and blue. (D) The common hot spots for the closed protein conformers, illustrated using 1P5D as an example. Colors reflect the rank of the hot spots for 1P5D, from highest to lowest: red, orange, yellow, green, with blue and magenta for the two hot spots tied for rank 5. An interactive view is available in the electronic version of the article. PRO446 Figure 1
As will be described, the mapping of 10 PMM/PGM structures yields a number of interesting results. First, this multidomain enzyme has a much more complex hot spot structure than the single domain enzymes we have previously studied.14 The simple enzymes usually have a primary hot spot in the main specificity pocket and one or two additional hot spots nearby, accommodating further moieties of the substrate. In contrast, PMM/PGM has nine significant hot spots, one in the active site and the others at domain–domain interfaces and at hinge regions. The locations and nature of these hot spots vary between the open, half-open, and closed conformers of the enzyme, in good agreement with the ligand-induced conformational changes observed in the crystal structures and consistent with the known enzyme mechanism.11 Second, the location of hot spots in the active site suggests an explanation for the ability of this enzyme to bind both glucose and mannose phosphosugar substrates. Finally, the most interesting and somewhat controversial observation is the substantial difference in the importance of a hot spot between the closed structures that bind substrate molecules phosphorylated only at the O1 position (G1P, M1P, ribose 1-phosphate, and xylose 1-phosphate), and the closed structures that bind substrates phosphorylated at the O6 position (G6P, M6P, and G16P), indicating a conformational transition. A follow-up statistical analysis shows that this conformational change is highly significant (P < 10−6), despite the small overall RMSD between the two types of structures. These results further demonstrate the use of computational solvent mapping as a “molecular microscope” which can reveal very small but significant changes in the size and shape of pockets.
Results
Hot spots of PMM/PGM and the effects of ligand binding
Mapping calculations were performed on the 10 crystal structures of P. aeruginosa PMM/PGM listed in Table I. The metal ion (Zn2+) and phosphoserine 108 (when present) were included in the coordinate files. All waters and ligands were removed before mapping. As described in the methods, hot spots were identified by the locations of overlapping probe clusters, also referred to as consensus sites.15 The rank of each hot spot is based on the number of probe clusters it contains. In several cases where two hot spots for the same structure were physically adjacent, either partially overlapping or within a few angstroms of each other, they were combined into one for analysis herein.
The mapping identified nine significant hot spots as described in Table II, which also lists the rank of each hot spot in the four representative structures shown in Figure 1(A), as well as the residues that line each hot spot. A schematic of the enzyme structure showing the approximate location of the nine hot spots is shown in Figure 1(B). One of the most important hot spots coincides with the substrate binding site within the active site cleft, and is denoted AS. As will be discussed, AS is actually the union of two adjacent but distinguishable hot spots that overlap two different moieties of the bound substrates. Finding a few hot spots at the substrate binding site is in good agreement with the results of mapping a number of enzymes.14 However, in contrast to the single-domain enzymes we have previously studied, PMM/PGM has eight additional hot spots, located near domain-domain interfaces and in hinge regions. We have used the locations as descriptive identifiers [Fig. 1(B)]. For example, a hot spot found in a cleft between domains 2 and 3 is called D23. Another structural feature used to identify two hot spots (H1 and H2) is the hinge region, an area involved in substantial conformational change upon ligand binding,9 and located at the juncture of domains 3 and 4 [Fig. 1(B)].
Table II.
Top Ranking Hot Spots Identified by Computational Solvent Mapping for Four Representative Conformers of PMM/PGM
| Rank for conformer |
||||||
|---|---|---|---|---|---|---|
| Hot spot | Location | 1P5D | 2FKF | 1K35 | 1K2Y | Surrounding residues |
| AS | Active site | 2a | 1 | 3 | 5 | Y17, R20, S108, R247, K285, T306, G307, H308, E325, S327, H329, E375, N377, R421, S423, N424, T425, T426, V430 |
| H1 | Hinge | 5 | 4 | 3a | 4 | D261, R262, T287, R289, P368, D370, E434 |
| H2 | Above hinge | 6 | 4 | 2a | D261, K285, T287, S372, T373, E375, Y417, R432, F433, E434 | |
| D123 | Domain 1-2-3 cleft | 1a | 2 | 1 | 5 | Y91, A94, N95, K100, V120, G123, E124, T125, K312, K313, M315, K316, F337, G338, F339 |
| D14 | Domain 1-4 interface | 5a | F14, R15, A16, K385, N424 | |||
| D13 | Domain 1-3 interface | 5 | 5 | 3a | R15, A16, Y17, D18, H308, S309 | |
| D23 | Domain 2-3 cleft | 3 | 2 | 1a | P217, G218, P220, L223, R247, V248, G249, V250, I257, I258, Y259, P260, L263, M326, S327, G328 | |
| D34 | Domain 3-4 cleft | 3a | G218, Y259, P260, K285, M326, S327, T373, P374, E375, R432 | |||
| D4 | Back of domain 4 | 4a | Y17, K305, T306, L310, F386, I389, L406, D407, G408, R421, A422, N424 | |||
Numbers indicate relative ranking of hot spot (based on number of probes in each consensus site). In some cases, two hot spots have the same number of probes and are given equivalent rank. 1P5D is used as a representative structure for all of the closed PMM/PGM conformers. The top five hot spots for each structure were used to compile this table; see Table III for complete listing.
Denotes structure with the highest rank for each hot spot, used for calculating the surrounding residues with CONTACT,33 except for the AS cluster where 1P5D was used to allow comparison with enzyme-substrate contacts.
Table III shows the location and rank of the nine hot spots in the 10 PMM/PGM structures, as well as the number of probe clusters at each location. The hot spot AS located at the substrate binding site is one of the largest in all structures but in 1K2Y, the structure of the dephosphorylated apo-enzyme which has the most open binding site. The site will be further discussed in the next section. The other large consensus site present in all structures is D123, located in a large pocket on the “backside” of the protein [see Fig. 1(C)]. Based on our experience with mapping,13–15 such large and invariant hot spots do not occur without a biological function, but at present no information is available on the potential role of the D123 site. However, some phosphoglucomutases are known to interact with a number of other proteins. For example, according to the database of interacting proteins (http://dip. doe-mbi.ucla.edu/dip/), tandem affinity purifications shows that E. coli phosphoglucomutase interacts with ribonuclease R and acetyl-coenzyme A carboxylase carboxyl transferase. Being on the opposite side of the protein from the active site, D123 is a possible location for such protein–protein interactions.
Table III.
Comprehensive Listing of Hot Spots for All 10 PMM/PGM Structures Used for Mapping
| Structure (Ligand) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Closed conformers |
Open/partially open conformers |
|||||||||
| Hot spot | 1P5Da (G1P) | 1P5Ga (G6P) | 1PCJa (M1P) | 1PCMa (M6P) | 2H4La (R1P) | 2H5Aa (X1P) | 2FKM (G16P) | 2FKFa (G16P) | 1K35a | 1K2Y |
| AS | 2 (32) | 1 (30) | 1 (35) | 2 (26) | 2 (27) | 2 (30) | 3 (25) | 1 (36) | 3 (22) | 5 (12) |
| H1 | 5 (7) | 3 (26) | 6 (10) | 4 (20) | 7 (7) | 2 (27) | 4 (19) | 3 (22) | 4 (14) | |
| H2 | 6 (13) | 4 (19) | 2 (17) | |||||||
| D123 | 1 (42) | 2 (27) | 1 (35) | 1 (39) | 1 (42) | 1 (35) | 1 (38) | 2 (30) | 1 (27) | 5 (12) |
| D13 | 5 (17) | 5 (14) | 3 (16) | |||||||
| D14 | 5 (7) | 7 (5) | 7 (8) | 5 (8) | 5 (9) | |||||
| D23 | 3(20) | 2 (24) | 1 (29) | |||||||
| D34 | 3 (24) | 1 (30) | 3 (18) | 5 (19) | 3 (20) | 3 (27) | 4 (16) | |||
| D4 | 4 (18) | 4 (18) | 2 (20) | 3 (21) | 4 (16) | 4 (14) | 5 (10) | |||
Denotes phosphorylated form of enzyme. Rank of each hot spot is indicated in column below each structure, with the number of probes in the hot spot shown in parentheses. The top three hot spots for each structure are shown in red, orange, and green font, respectively. Hot spots with fewer than five probe clusters are not included. For hot spot definitions, see Table II. For the closed conformers, hot spots found in only one or two structures are omitted for clarity.
The mapping results show substantial differences between the hot spot structures observed in closed [Fig. 1(C)] and open [Fig. 1(D)] conformations. Only three hot spots are present in both conformations: the active site AS, D123 on the opposite side of the protein, and H1 in the hinge region between domains 1-3 and domain 4 which will be discussed in more detail. The closed structures have three additional hot spots, D14 between domains 1 and 4, D34 between domains 3 and 4, and D4 in the back of domain 4. As discussed, in the half-open and open structures domain 4 moves farther from the rest of the protein, and these three pockets cease to be significant. However, we find three new hot spots, D13 and D23 between domains 1 and 3 and between domains 2 and 3, respectively, and H2 in the hinge region close to the conserved pocket H1. The substantial changes in the hot spot structure emphasize that the effects of ligand binding are not constrained to the vicinity of the active site. As will be discussed, some of the conformational changes are due to the rigid body motion of domain 4 upon ligand binding, but the results also reveal significant changes at the interfaces of domain 3 with domains 1 and 2.
In our previous work we have primarily used computational solvent mapping for the identification of druggable subsites of proteins.13–16,20,22 A number of other applications were directed toward the characterization of binding sites,21,23,24 including the comparison of such sites in different structures. Here we also compare different structures, but now all but one of the hot spots are in inter-domain pockets rather than in a binding site. As they are formed by domain-domain interfaces, these pockets were expected to be highly hydrophobic. However, the only pocket with a large fraction of hydrophobic residues is D23, which exists only in the open state (Table II). We emphasize that the existence of hot spots does not imply that the inter-domain pockets serve as binding sites for any compound. In fact, any druggable site (i.e., a site that can bind a drug-size ligand with significant affinity) must have a hot spot with at least 15 probe clusters.15,16 As shown in Table III, only the AS and the D123 sites satisfy this condition. Several pockets have similarly high probe numbers in either open or closed states. While binding of appropriate molecules at these locations might stabilize the particular conformation, at this point we do not have any example of such behavior.
Hot spots in the active site of PMM/PGM
Similarly to the other enzymes we have studied,14 the active site contains one of the most important hot spots in all closed conformers of PMM/PGM. The AS site is typically the second highest in rank for these structures and consists of 26 to 35 probes clusters (Table III). For the closed conformers of PMM/PGM, the location of the AS hot spot coincides almost exactly with the position of bound substrate, as observed in the crystal structures of the PMM/PGM enzyme-ligand complexes. Figure 2(A,B) show a comparison between the AS probe clusters and the position of the bound G1P in the 1P5D enzyme complex.11 As described later, for the closed conformers of PMM/PGM, the interactions of the probe molecules in the AS site recapitulate the direct enzyme-substrate interactions with high fidelity.
Figure 2.
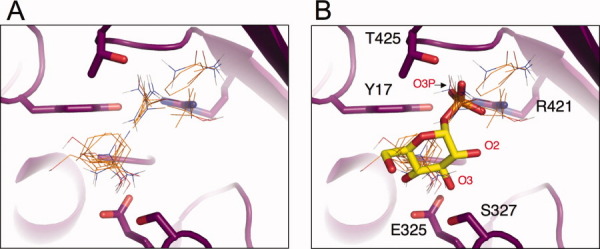
(A) A close-up view of the probes in the active site (AS) hot spots identified in mapping of the 1P5D structure of PMM/PGM. Protein backbone is shown as a magenta ribbon. Probes are shown as thin sticks. (Since each probe site is actually a cluster of the most favorable binding positions of the same probe, only the highest ranked probe from each of the clusters is shown). Two subsites in this hot spot are apparent, which correspond to the sugar and phosphate portions of the substrate (see next panel). (B) A superposition of probes in the AS hot spot and the substrate, with G1P shown in stick model. The side chains of residues involved in enzyme-substrate interactions are shown in stick models; several oxygens of the substrate are indicated with red labels.
Within the AS hot spot, the probes are clearly grouped into two subsites that correspond quite closely to either the sugar moiety or the phosphate group of the substrate. The probes that occupy the sugar subsite include molecules with cyclic structures, such as phenol and benzene, and the rings of these probe molecules closely mimic the position of the sugar ring [Fig. 2(A,B)]. Other probes in the sugar subsite tend to localize to areas that correspond to the sugar hydroxyls of the substrate, especially the O3 hydroxyl [Fig. 2(B)], and make contacts (hydrogen bond or nonbonded) with its interacting residues (E325 and S327), as observed in crystal structures of the enzyme-substrate complexes.11 Probes that occupy the phosphate subsite are generally more polar, and include acetamide, acetone, acetaldehyde, N,N-dimethylformamide, and urea. The common structural feature of these five probes is a carbonyl oxygen positioned to make key contacts with residues in the invariant phosphate-binding site of the enzyme [Fig. 2(B)].11 Superposition of the probes and substrate shows that in each case, the carbonyl oxygen of the probe is virtually superimposable with a specific phosphate oxygen of the substrate: atom O3P. In the enzyme substrate complexes, this oxygen atom participates in a critical three-way contact with the protein, involving residues Y17, R421, and N424.11 In addition to the multiple residues involved, the enzyme contacts to this oxygen span domains 1 and 4 of the protein, essentially “latching the lid” of the closed active site complex. We note that, in contrast to the many contacts made with residues involved in ligand binding, relatively few contacts are found between the probes and residues essential for catalysis (such as the active site phosphoserine S108). This is consistent with the results of previous mapping studies, as the method focuses on binding rather than catalytic properties of the sites.14
An interesting observation from the mapping of the active site is the paucity of probes in the vicinity of the O2 hydroxyl of the substrate [Fig. 2(B)], which is a site of structural variability between the glucose and mannose phosphosugar substrates of PMM/PGM. Unlike the O3 and O4 hydroxyls, which have the same stereochemistry in glucose and mannose and exchange positions in the 1- versus 6-phosphosugar complexes, the O2 hydroxyl occupies a distinct position in each of the four enzyme-substrate complexes.11 In accordance with observations from the crystal structures, the mapping studies show that this region of the site is not very important for binding. In fact, the protein establishes limited contacts with the O2 hydroxyl of its substrates, which enables the enzyme to accommodate its two phosphosugar substrates in their two different binding orientations. Thus, the lack of a hot spot around sites of structural variability in substrates can be considered as an avenue to achieving binding promiscuity restricted to a family of substrates that show conservation in the regions of the hot spots.
Domain motion with subresolution conformational change
As described, the hot spot H1 in the hinge region is present in both closed and open structures (Table III). However, the number of probe clusters at the H1 site substantially varies among different closed structures. The results in Table III suggest that the size of this site (as measured in terms of the number of probe clusters) is directly controlled by the type of substrate or intermediate present in the active site more than 17 Å away. Indeed, the H1 pocket includes few (or none) probe clusters if the ligand in the active site is phosporylated only at the O1 position, that is, in G1P, M1P, R1P, and X1P. However, the number of probe clusters more than doubles when the ligand is phosphorylated at the O6 position as in G6P, M6P, and G16P, clearly indicating a substantial change in the size of the pocket.
It is not difficult to find a plausible explanation for the origin of long-range interactions between the AS and H1 sites. As shown in Figure 3(A,B), the protein has a very large crevice between domains 1-3 and domain 4. This crevice accommodates three hot spots, AS in the active site, D34 in the middle, and H1 in the hinge region. The comparison of open, closed, and half-open conformations reveal that domain 4 can easily move relative to the rest of the protein, and most likely can occupy an ensemble of different conformational states. Binding a ligand phosporylated at the O1 position of the sugar selects for a state with a relatively narrow interdomain crevice and an H1 pocket that binds a relatively small number of probe clusters [Fig. 3(A)]. In contrast, the binding of a ligand phosphorylated at the O6 position of the sugar selects for a slightly more open active site conformation, resulting in a wider crevice and a substantially larger H1 pocket. The slightly wider crevice, highlighted by the mapping studies, may also help explain why the Km of the enzyme is 70-fold lower for G1P than G6P, a fact that even detailed analyses of the high resolution crystal structures failed to illuminate.7 The existence of multiple conformational states is in agreement with the notion of a dynamic free energy landscape with shallow energy wells and transition barriers that allow the protein to sample the conformation appropriate for efficient ligand-binding interactions.
Figure 3.
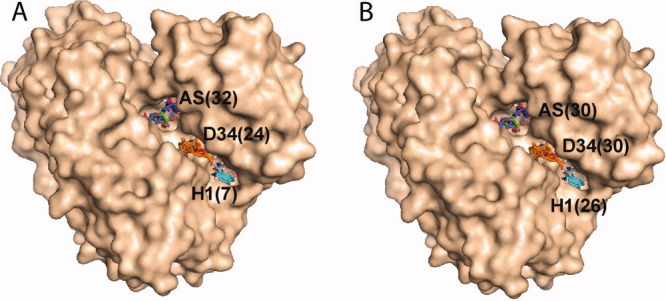
Hot spots AS, D34, and H1 in the interface of domains 3 and 4 of PMM/PGM. Numbers in parentheses show the number of probe clusters in each of the hot spots. A. PMM/PGM structure 1P5D, cocrystallized with G1P. The binding of the substrate, phosporylated at the O1 position, results in a small number of probe clusters at H1. B. PMM/PGM structure 1P5G, cocrystallized with G6P. The binding of the substrate phosporylated at the O6 position results in a much larger number of probe clusters at H1, despite the ∼20 Å distance between H1 and bound ligand.
The assumption that there exists an ensemble of low energy positions for domain 4, ranging from the closed to the open states, helps to explain the changes in the H1 site highlighted by the mapping results. However, all closed structures are very similar to each other, irrespective of the type of the ligand, and hence it is not clear whether the small Cα RMSD—on the order of 0.2 Å—would be compatible with a meaningful conformational change. Can it be that the mapping result is simply an artifact? We show below that this is definitely not the case. In fact, we show that the rigid body translation between two conformations of a protein can be determined with a small standard error which is 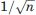 times the standard error in the atomic coordinates, where n is the number of residues in the moveable domain, and thus a relatively small difference between the calculated centers of mass for the two structures may indicate highly significant motion. In more intuitive terms, it is extremely unlikely that random noise in a large number of atomic coordinates would result in significant rigid body motion unless such coordinated motion really exists.
times the standard error in the atomic coordinates, where n is the number of residues in the moveable domain, and thus a relatively small difference between the calculated centers of mass for the two structures may indicate highly significant motion. In more intuitive terms, it is extremely unlikely that random noise in a large number of atomic coordinates would result in significant rigid body motion unless such coordinated motion really exists.
As described in the methods, the above argument can be translated into rigorous statistical evaluation. As an example, we consider the problem of determining the significance of the rigid body motion of domain 4 between 1P5D (with a small H1 pocket) and 1P5G (with a large H1 pocket) that both have the crystallographic resolution of 1.6 Å. We first superimpose domains 1-3 of the two structures. Starting from this common coordinate system, superimposing domains 4 of the two structures requires the translation vector 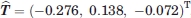 , which represent a shift of
, which represent a shift of 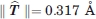 . After the application of this translation and the optimal rotation, the Cα RMSD between the superimposed domains 4 of the two structures is 0.135 Å. As the effects of any rigid body motion have been removed, this residual RMSD is due to the random noise in the data, and can be considered representing the standard error in the atomic coordinates. The value of 0.135 Å is in agreement with the 1.6 Å resolution of the structures. As described in the methods, we calculate the t-statistics defined by
. After the application of this translation and the optimal rotation, the Cα RMSD between the superimposed domains 4 of the two structures is 0.135 Å. As the effects of any rigid body motion have been removed, this residual RMSD is due to the random noise in the data, and can be considered representing the standard error in the atomic coordinates. The value of 0.135 Å is in agreement with the 1.6 Å resolution of the structures. As described in the methods, we calculate the t-statistics defined by  where n = 96 is the number of residues in domain 4, resulting in t = 13.28. This value is very high, and implies that the probability of finding the 0.317 Å translation by chance, rather than due to a rigid body motion, is less than 0.0001%. For comparison, we note that the critical t value from the table of the Student's t-distribution is t* = 3.29 at P = 0.001 and 95 degrees of freedom. The application of the same analysis to all closed structures shows that any of the structures 1P5D, 1PCJ, 2H4L, and 2H5A (all with ligands phosphorylated only at the O1 position) differs from any of the structures 1P5G, 1PCM, and 2FKM (with ligands phosphorylated at the O6 position) with similarly high significance. Thus, we conclude that despite the small conformational change, the position of domain 4 is significantly affected by the position of the phosphorylated oxygen of the ligand bound to the active site of the enzyme. We note that the internal equilibrium constant, that is, the ratio of enzyme-bound G6P and G1P, calculated from the rapid-quench data, is 2.9.7 By contrast, the Keq for the interconversion of α-G1P and α-G6P in solution equals 7. Thus, PMM/PGM provides differential stabilization of G1P relative to its energy free in solution compared to G6P, which is consistent with the mapping results showing the G1P complexes are more closed than the ones with G6P.
where n = 96 is the number of residues in domain 4, resulting in t = 13.28. This value is very high, and implies that the probability of finding the 0.317 Å translation by chance, rather than due to a rigid body motion, is less than 0.0001%. For comparison, we note that the critical t value from the table of the Student's t-distribution is t* = 3.29 at P = 0.001 and 95 degrees of freedom. The application of the same analysis to all closed structures shows that any of the structures 1P5D, 1PCJ, 2H4L, and 2H5A (all with ligands phosphorylated only at the O1 position) differs from any of the structures 1P5G, 1PCM, and 2FKM (with ligands phosphorylated at the O6 position) with similarly high significance. Thus, we conclude that despite the small conformational change, the position of domain 4 is significantly affected by the position of the phosphorylated oxygen of the ligand bound to the active site of the enzyme. We note that the internal equilibrium constant, that is, the ratio of enzyme-bound G6P and G1P, calculated from the rapid-quench data, is 2.9.7 By contrast, the Keq for the interconversion of α-G1P and α-G6P in solution equals 7. Thus, PMM/PGM provides differential stabilization of G1P relative to its energy free in solution compared to G6P, which is consistent with the mapping results showing the G1P complexes are more closed than the ones with G6P.
Discussion
Phosphomannomutase/phosphoglucomutase (PMM/PGM) from P. aeruginosa catalyzes the reversible, intramolecular conversion of glucose 6-phosphate (G6P) to glucose 1-phosphate (G1P), or mannose 6-phosphate (M6P) to mannose 1-phosphate (M1P). Based on crystallographic and kinetic investigations, both substrate binding and catalysis require substantial conformational changes by the enzyme. The changes are largely enabled by the multidomain structure of the protein. Indeed, the C-terminal domain 4 is connected to the other three domains by a flexible hinge, and upon substrate binding rotates by ∼9°, changing the active site from a relatively open cleft to a deep pocket.
One of the most important hot spots identified for the PMM/PGM structures is found in the large active site cleft. Clearly this region contains structural features favorable for binding of small molecules. For the closed conformers of the enzyme, the highly ranked AS hot spot recapitulates the enzyme-substrate interactions with high fidelity: probe interactions mimic both the sugar and phosphate moieties of the ligand. The identification of a “substrate-like” hot spot only for the closed conformer is consistent with the crystallographic studies, which show that key structural features required for substrate binding (e.g., the invariant phosphate-binding site)9 are present only when the protein adopts its characteristic ligand-bound conformation.
The interesting and somewhat unique finding for PMM/PGM is the large number of hot spots, located in the domain–domain interfaces that surround the active site cleft and at the hinge region between domains 3 and 4. All but one of these sites are less hydrophobic than expected for pockets in interfaces,25 but only one of them appears to be a potentially druggable binding site. It is also clear from the mapping studies that most of these hot spots are very sensitive to structural differences in the different conformers of PMM/PGM. Three of the nine hot spots are present in all structures, three others are found only in the closed states, and the remaining three are only in the open and the half-open structures. Most of the changes are associated with the large conformational change of domain 4 upon ligand binding. However, the mapping also reveals that small conformational changes in domain 4 create a more complex conformational landscape for the enzyme than previously appreciated. In particular, in the closed structures the size of a pocket in the hinge region significantly depends on the position of the phosphorylated oxygen in the bound substrate. Although the RMSD between the two types of structures is very small, rigorous statistical analysis shows that the difference in the pocket size is due to a small but statistically very significant rigid body motion of domain 4. We identify only two different states, but this finding is in agreement with the notion of a dynamic free energy landscape with shallow energy wells and transition barriers that allow the protein to sample a variety of conformations. This part of the article makes two interesting and fairly general contributions. First, we show that the mapping is capable of detecting changes in the binding sites that are due to very small conformational changes and hence are generally missed by simple inspection. Second, the method of analysis we have developed confirms that conformational changes can be real and significant even at an overall RMSD that is close to the expected error in the atomic coordinates.
Materials and Methods
Computational solvent mapping
The FTMAP algorithm consists of five steps as follows:
Soft rigid body docking of probe molecules
Protein structures are downloaded from the Protein Data Bank (PDB).26 For each structure, we use 16 small molecules as probes (acetaldehyde, acetamide, acetone, acetonitrile, benzaldehyde, benzene, cyclohexane, dimethyl ether, N,N-dimethylformamide, ethane, ethanol, isobutanol, isopropanol, methylamine, phenol, and urea). For each probe, billions of docked conformations are sampled by soft rigid body docking based on Fast Fourier Transform (FFT) correlation approach.15 The method performs exhaustive evaluation of an energy function in the discretized 6D space of mutual orientations of the protein (receptor) and a small molecular probe (ligand). The center of mass of the receptor is fixed at the origin of the coordinate system. The translational space is represented as a grid of 0.8 Å displacements of the ligand center of mass, and the rotational space is sampled using 500 rotations. The energy function is a linear combination of four different energy terms
representing van der Waals (Evdw), electrostatics (Eelec), cavity-modulated hydrophobic (Ecavity), and structure-based pairwise (Epair) interactions. The details of the energy functions are described in the original FTMAP article.24 Note that mapping requires only the atomic coordinates of the two molecules, that is, no a priori information on the binding site is used. The 2000 best poses for each probe are retained for further processing.
Minimization and rescoring
The free energy of each of the 2000 complexes, generated in Step 1, is minimized using the CHARMM27 potential with the Analytic Continuum Electrostatic (ACE) model representing the electrostatics and solvation terms as implemented in version 27 of CHARMM27 using the parameter set from version 19 of the program. During the minimization by an adopted basis Newton-Raphson method the protein atoms are held fixed while the atoms of the probe molecules are free to move.
Clustering and ranking
The minimized probe conformations from Step 2 are grouped into clusters using 3 Å RMSD as clustering radius.15 Clusters with less than 10 members are excluded from consideration thereby avoiding narrow energy minima with low entropy.15 The retained clusters are ranked on the basis of their Boltzman averaged energies. Ten clusters with the lowest average free energies are retained for each probe.
Determination of consensus sites
To determine the consensus sites, that is, the positions at which several probe clusters overlap, the clusters of different probes are clustered using the distance between the cluster centers as the distance measure. The site with the maximum number of probe clusters (with cluster centers within a 4 Å radius) is selected as consensus site 1 (CS1). The clusters in CS1 are removed from consideration, and the procedure is repeated until all clusters are assigned to a consensus site. The consensus sites are ranked based on the number of the probe clusters they contain. Duplicate clusters of the same type are considered in the count. For each structure the six highest ranking consensus sites are defined as the hot spots shown in the results. The residues lining each consensus site were calculated with CONTACT.28 Figures were prepared using PYMOL.29
Determining the significance of domain translation
To explain the origin of the method to be developed we first consider two random variables x and y, each observed n times, resulting in the samples x1, x2,…, xn, and y1, y2,…, yn, respectively. One can use the two-sample t-test to assess whether the means of the two groups are statistically different from each other. The null hypothesis of the test is that E(x) = E(y), where E denotes the expected value of the variables. Assuming that x and y have the same (unknown) variance σ2, the t statistic to test whether the means  and
and  are different can be calculated by
are different can be calculated by 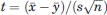 , where s is the estimate of the standard error σ from the sample, n is the sample size, and
, where s is the estimate of the standard error σ from the sample, n is the sample size, and 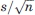 estimates the standard error of the mean. Since can be arbitrarily reduced by increasing n, we emphasize that using a large enough sample the difference
estimates the standard error of the mean. Since can be arbitrarily reduced by increasing n, we emphasize that using a large enough sample the difference  can be significant even when it is substantially smaller than the standard error σ determined by the accuracy of the measurement.
can be significant even when it is substantially smaller than the standard error σ determined by the accuracy of the measurement.
Consider now the problem of determining the significance of the rigid body motion of domain 4 between two structures, say 1P5D (with a small H1 pocket) and 1P5G (with a large H1 pocket). We first superimpose domains 1-3 of the two structures. Let u1, u2,…, un and v1, v2,…, vn denote the coordinates of the Cα atoms in domains 4 in 1P5D and 1P5G, respectively, after the superposition of domains 1-3. We assume that domains 4 in the two structures relate to each other by a rigid body rotation and translation, that is, vi = Rui + T, where R is a 3x3 rotation matrix and T is a translation vector. Considering the measurement errors ɛi in the coordinates of the i-th atom, we adopt the statistical model vi = Rui + T + ɛi, and assume that the coordinate errors ɛi (after accounting for the rigid body movement) are independent are normally distributed. Notice that in this formulation the problem is similar to the ones considered in regression analysis.30 The null hypothesis is that there is no rigid body motion, that is, R = I and T = 0, where I is the 3 × 3 unit matrix. Rejecting any of these two hypotheses implies significant rigid body motion. We focus on the significance of the translation vector which is estimated by 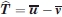 , where
, where 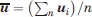 and
and 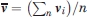 are the centers of mass of domains 4 in the two structures. Thus, the test is very similar to comparing the means of two samples, with the only difference that both
are the centers of mass of domains 4 in the two structures. Thus, the test is very similar to comparing the means of two samples, with the only difference that both  and
and  are vectors. We can apply the t-test componentwise, or to the norm
are vectors. We can apply the t-test componentwise, or to the norm 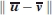 If σ denotes the standard error in the Cα coordinates, then by definition the standard error for each component of the means
If σ denotes the standard error in the Cα coordinates, then by definition the standard error for each component of the means  and
and  is given by
is given by 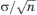 The value of σ can be estimated by the Cα RMSD between domains 4 of the structures 1P5D and 1P5G after the two domains have been superimposed, that is, the effect of the rigid body translation and rotation has been removed, leaving the random “noise” component as the only source of deviation. Thus, the RMSD between the superimposed domains represents the variance in the coordinates, and the estimate of the standard error in the components of and is
The value of σ can be estimated by the Cα RMSD between domains 4 of the structures 1P5D and 1P5G after the two domains have been superimposed, that is, the effect of the rigid body translation and rotation has been removed, leaving the random “noise” component as the only source of deviation. Thus, the RMSD between the superimposed domains represents the variance in the coordinates, and the estimate of the standard error in the components of and is 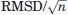 Accounting for the three components of the vector and using the norm
Accounting for the three components of the vector and using the norm 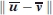 the t-statistics for comparing and is given by
the t-statistics for comparing and is given by 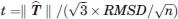 . Thus, as in the case of the t-test for comparing two sample means, the “true” rigid body translation vector T may significantly differ from 0 even when the estimate
. Thus, as in the case of the t-test for comparing two sample means, the “true” rigid body translation vector T may significantly differ from 0 even when the estimate 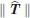 is smaller than the random noise in the atomic coordinates due to the limited crystallographic resolution of the structures. In fact, the error in
is smaller than the random noise in the atomic coordinates due to the limited crystallographic resolution of the structures. In fact, the error in 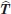 is proportional to
is proportional to 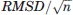 rather than to the RMSD itself.
rather than to the RMSD itself.
Glossary
Abbreviations:
- G16P
glucose 1,6-bisphosphate
- G1P
glucose 1-phosphate
- G6P
glucose 6-phosphate
- M1P
mannose 1-phosphate
- M6P
mannose 6-phosphate
- MSCS
multiple solvent crystal structures
- NMR
nuclear magnetic resonance
- PMM/PGM
phosphomannomutase/phosphoglucomutase
- R1P
ribose 1-phosphate
- RMSD
root mean square deviation
- WT
wild-type
- X1P
xylose 1-phosphate
References
- 1.Hardy MA, Wells JA. Searching for new allosteric sites in enzymes. Curr Opin Struct Biol. 2004;14:1–10. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 2.Yankeelov JA, Jr, Koshland DE., Jr Evidence for conformation changes induced by substrates of phosphoglucomutase. J Biol Chem. 1965;240:1593–602. [PubMed] [Google Scholar]
- 3.Ma B, Shatsky M, Wolfson HJ, Nussinov R. Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci. 2002;11:184–197. doi: 10.1110/ps.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naught LE, Gilbert S, Imhoff R, Snook C, Beamer L, Tipton P. Allosterism and cooperativity in Pseudomonas aeruginosa GDP-mannose dehydrogenase. Biochemistry. 2002;41:9637–9645. doi: 10.1021/bi025862m. 2002. [DOI] [PubMed] [Google Scholar]
- 5.Naught LE, Tipton PA. Kinetic mechanism and pH dependence of the kinetic parameters of Pseudomonas aeruginosa phosphomannomutase/phosphoglucomutase. Arch Biochem Biophys. 2001;396:111–118. doi: 10.1006/abbi.2001.2618. [DOI] [PubMed] [Google Scholar]
- 6.Naught LE, Regni C, Beamer LJ, Tipton PA. Roles of active site residues in P. aeruginosa phosphomannomutase/phosphoglucomutase. Biochemistry. 2003;42:9946–9951. doi: 10.1021/bi034673g. [DOI] [PubMed] [Google Scholar]
- 7.Naught LE, Tipton PA. Formation and reorientation of glucose 1,6-bisphosphate in the PMM/PGM reaction: transient-state kinetic studies. Biochemistry. 2005;44:6831–6836. doi: 10.1021/bi0501380. [DOI] [PubMed] [Google Scholar]
- 8.Regni C, Tipton PA, Beamer LJ. Crystal structure of PMM/PGM: an enzyme in the biosynthetic pathway of P. aeruginosa virulence factors. Structure. 2002;10:269–279. doi: 10.1016/s0969-2126(02)00705-0. [DOI] [PubMed] [Google Scholar]
- 9.Regni C, Naught LE, Tipton PA, Beamer LJ. Structural basis of diverse substrate recognition by the enzyme PMM/PGM from P. aeruginosa. Structure. 2004;12:55–63. doi: 10.1016/j.str.2003.11.015. 2004. [DOI] [PubMed] [Google Scholar]
- 10.Regni C, Shackelford GS, Beamer LJ. Complexes of the enzyme phosphomannomutase/phosphoglucomutase with a slow substrate and an inhibitor. Acta Crystallograph Sect F Struct Biol Cryst Comm. 2006;62:722–726. doi: 10.1107/S1744309106025887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Regni C, Schramm AM, Beamer LJ. The reaction of phosphohexomutase from Pseudomonas aeruginosa: structural insights into a simple processive enzyme. J Biol Chem. 2006;281:15564–15571. doi: 10.1074/jbc.M600590200. [DOI] [PubMed] [Google Scholar]
- 12.Schramm AM, Mehra-Chaudhary R, Furdui CM, Beamer LJ. Backbone flexibility, conformational change, and catalysis in a phosphohexomutase from Pseudomonas aeruginosa. Biochemistry. 2008;47:9154–9162. doi: 10.1021/bi8005219. [DOI] [PubMed] [Google Scholar]
- 13.Dennis S, Kortvelyesi T, Vajda S. Computational mapping identifies the binding sites of organic solvents on proteins. Proc Natl Acad Sci USA. 2002;99:4290–4295. doi: 10.1073/pnas.062398499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silberstein M, Dennis S, Brown L, Kortvelyesi T, Clodfelter K, Vajda S. Identification of substrate binding sites in enzymes by computational solvent mapping. J Mol Biol. 2003;332:1095–1113. doi: 10.1016/j.jmb.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 15.Brenke R, Kozakov D, Chuang GY, Beglov D, Hall D, Landon MR, Mattos C, Vajda S. Fragment-based identification of druggable ‘hot spots’ of proteins using fourier domain correlation techniques. Bioinformatics. 2009;25:621–627. doi: 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landon MR, Lancia DR, Jr, Yu J, Thiel SC, Vajda S. Identification of hot spots within druggable binding regions by computational solvent mapping of proteins. J Med Chem. 2007;50:1231–1240. doi: 10.1021/jm061134b. [DOI] [PubMed] [Google Scholar]
- 17.Hajduk PJ, Huth JR, Fesik SW. Druggability indices for protein targets derived from NMR-based screening data. J Med Chem. 2005;48:2518–2525. doi: 10.1021/jm049131r. [DOI] [PubMed] [Google Scholar]
- 18.Mattos C, Bellamacina CR, Peisach E, Pereira A, Vitkup D, Petsko GA, Ringe D. Multiple solvent crystal structures: probing binding sites, plasticity and hydration. J Mol Biol. 2006;357:1471–1482. doi: 10.1016/j.jmb.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 19.Mattos C, Ringe D. Locating and characterizing binding sites on proteins. Nat Biotechnol. 1996;14:595–599. doi: 10.1038/nbt0596-595. [DOI] [PubMed] [Google Scholar]
- 20.Landon MR, Lieberman RL, Hoang QQ, Ju S, Caaveiro1a HMM, Orwig SD, Kozakov D, Brenke R, Chuang G-Y, Beglov D, Vajda S, Petsko GA, Ringe D. Detection of ligand binding hot spots on protein surfaces via fragment-based methods: application to DJ-1 and glucocerebrosidase. J Comp Aided Mol Des. 2009 doi: 10.1007/s10822-009-9283-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheu SH, Kaya T, Waxman DJ, Vajda S. Exploring the binding site structure of the PPAR-γ ligand binding domain by computational solvent mapping. Biochemistry. 2005;41:1193–1209. doi: 10.1021/bi048032c. [DOI] [PubMed] [Google Scholar]
- 22.Landon MR, Amaro RE, Baron R, Ngan CH, Ozonoff D, McCammon JA, Vajda S. Novel druggable hot spots in avian influenza neuraminidase H5N1 revealed by computational solvent mapping of a reduced and representative receptor ensemble. Chem Biol Drug Des. 2008;71:106–116. doi: 10.1111/j.1747-0285.2007.00614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngan C-H, Beglov D, Rudnitskay AN, Kozakov D, Waxman DJ, Vajda S. The structural basis of pregnane X receptor binding promiscuity. Biochemistry. 2009 doi: 10.1021/bi901578n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chuang G-Y, Kozakov D, Brenke R, Beglov D, Guarnieri F, Vajda S. Binding hot spots and amantadine orientation in the influenza A virus M2 proton channel. Biophys J. 2009;97:2846–2853. doi: 10.1016/j.bpj.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones S, Marin A, Thornton JM. Protein domain interfaces: characterization and comparison with oligomeric protein interfaces. Protein Eng. 2000;13:77–82. doi: 10.1093/protein/13.2.77. [DOI] [PubMed] [Google Scholar]
- 26.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brooks BR, Bruccoleri RE, Olafson BD, States OJ, Swmainathan S, Karplus M. CHARMM: a program for macromolecular energy, minimzation, and dynamics calculations. J Com Chem. 1983;4:187–217. [Google Scholar]
- 28.CCP. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 29.DeLano WL. The PyMOL molecular graphics system. CA: DeLano Scientific; 2002. [Google Scholar]
- 30.Schroeder LD, Sjoquist DP, Stephan PE. Understanding regression analysis: an introductory guide. CA: Sage Publications; 1986. [Google Scholar]