

Abstract
The traditional NMR-based method for determining oligomeric protein structure usually involves distinguishing and assigning intra- and intersubunit NOEs. This task becomes challenging when determining symmetric homo-dimer structures because NOE cross-peaks from a given pair of protons occur at the same position whether intra- or intersubunit in origin. While there are isotope-filtering strategies for distinguishing intra from intermolecular NOE interactions in these cases, they are laborious and often prove ineffectual in cases of weak dimers, where observation of intermolecular NOEs is rare. Here, we present an efficient procedure for weak dimer structure determination based on residual dipolar couplings (RDCs), chemical shift changes upon dilution, and paramagnetic surface perturbations. This procedure is applied to the Northeast Structural Genomics Consortium protein target, SeR13, a negatively charged Staphylococcus epidermidis dimeric protein (Kd 3.4 ± 1.4 mM) composed of 86 amino acids. A structure determination for the monomeric form using traditional NMR methods is presented, followed by a dimer structure determination using docking under orientation constraints from RDCs data, and scoring under residue pair potentials and shape-based predictions of RDCs. Validation using paramagnetic surface perturbation and chemical shift perturbation data acquired on sample dilution is also presented. The general utility of the dimer structure determination procedure and the possible relevance of SeR13 dimer formation are discussed.
Keywords: residual dipolar coupling, homo-oligomer, weak dimer, NMR, paramagnetic relaxation
Introduction
Many proteins function not as monomers, but as multimeric complexes. There are distinct functional advantages implicit in the use of multimeric structures. For example, functional proteins can be produced more efficiently when several short sequences are synthesized, since single disabling transcriptional or translational errors affect a part, as compared to an entire single chain protein.1,2 Lower organisms, with less complex quality control mechanisms, in fact, seem to exploit the use of multimeric assemblies more frequently.1 Likewise, it may be easier to tailor the properties of oligomeric complexes to the demands of evolution by mutation of an oligomeric subunit rather than a domain within a single chain protein. Associations between subunits of an oligomeric complex also provide additional means of regulating different biological functions2 and a means of enhancing affinity in receptor interactions, as they often do in multimeric lectins.3,4
For these reasons, knowledge of the geometry of subunit assembly is important for understanding structure-function relationships and protein surface properties. For tightly associating complexes, X-ray crystallography provides this type of information. However, not all complexes associate tightly. In addition, even for tight complexes, it may be difficult to distinguish biologically relevant interprotein interactions observed in a crystal structure from interactions driven by the energetics of crystallization. Solution methods for characterizing geometry, such as those from nuclear magnetic resonance (NMR), provide an important complement. These methods present their own challenges, including the size limitations of NMR, the lack of NOE constraints across multimer interfaces of weak complexes, and degeneracies of NMR resonances when symmetric homo-oligomers are involved. We have shown previously that residual dipolar coupling (RDC) data can be used to characterize tightly associated homodimeric structures.5 Here we present an NMR methodology that can provide structures of even weakly associating homo-oligomer complexes in the absence of NOE data.
Traditionally, determination of homo-oligomeric protein structures by NMR has relied on distance information derived from intermolecular NOE crosspeaks. In a symmetric homo-oligomer complex, however, NOEs associated with close approach of proton pairs across a multimer interface must be distinguished from those associated with the same pair of protons, but within the same subunit. This distinction is difficult because resonances for protons in equivalent sites of different subunits are degenerate. Isotope filtering strategies have been devised to deal with this problem.6–8 A protein preparation having a uniform high level of enrichment in isotopes such as 13C is mixed with a preparation having only natural abundance in 13C. Selective excitation of protons with covalent bonds to 12C sites followed by NOE transfer and detection through protons bonded to 13C sites allow selective detection of intersubunit NOEs. Sensitivity of this experiment is maximally 50% that of a conventional NOE experiment and it suffers from background generated by the 1% natural abundance of 13C. For weak complexes there are additional problems. For a homodimer with a dissociation constant of 100 μM and at a monomer equivalent concentration of 300 μM, just 66% of the protein is in the dimeric state reducing the sensitivity further. Also, weak complexes tend to have fewer hydrophobic contacts that generate most intersubunit NOEs.
RDCs are easily measured orientation-dependent NMR parameters used most commonly to report the backbone N—H bond orientation for each residue in the protein.9 They are measured as additions to scalar couplings in modified HSQC or TROSY spectra that occur under conditions of partial orientation of the complex in a magnetic field, and require no more prior work other than the assignment of the backbone resonances in these spectra. When a structure for the monomer unit of a homo-oligomeric complex is known (from NMR or X-ray studies), analysis of the RDCs provides information on the orientation of the alignment frame in terms of molecular coordinates. For multimeric complexes, one of the principal axes of the alignment frame must be parallel to the oligomer symmetry axis, and it is possible to use this information to restrict the orientation between subunits of multimeric complexes.10,11
The NOE independent approach described here follows previous work of Wang et al. who used a combination of restraints from RDCs and a residue pair scoring function to generate a dimer model of a high affinity complex.5 The work built on an X-ray crystallography study of the YkuJ protein (Northeast Structural Genomics target SR360) which provided a reliable monomer structure but two alternative dimer structures. Two independent alignment media were used to remove the three-fold ambiguity in identifying the symmetry axis. The initial dimer model was generated by rotating the first monomer unit by 180° about that axis to produce a second monomer unit. A grid search algorithm was then employed to explore translational degrees of freedom for the second monomer unit. The final dimeric models of YkuJ, were selected using scores based on the approach of shape dependent simulated RDCs12 to experimental RDCs, residue pairing potentials for interfacial residues, and van der Waals interaction energies.
There have recently been other excellent examples of NOE independent approaches for determination of oligomeric structures using RDCs as an orientation restraint.5,13,14 A weakly associated hetero-oligomeric complex was solved by Ortega-Roldan et al. using RDC orientational restraints along with chemical shift perturbations.13 Dimer RDCs were extracted by titrating isotopically-enriched monomer A with unlabeled monomer B and the complex was assembled using the alignment tensors calculated from four types of RDC measured in a single medium. An alternate approach for determining the structure of homo- and hetero-oligomer complexes was demonstrated by Wang et al. using one set of RDCs as orientation restraints in combination with small-angle X-ray scattering (SAXS) data as the main shape restraint in a three-dimensional orientation grid search.14
In this work, we expand the RDC-assisted modeling method used in the study of YkuJ to extend applications to proteins that associate weakly. The updated methodology begins with the acquisition of concentration dependent RDC data in two different alignment media. The RDC information for the pure dimeric state is then extrapolated from experimental RDC data sets using the dissociation constant derived from concentration-dependent chemical shift data. The extrapolated RDC values from the two alignment media are then used to determine the symmetry axis of the dimer, allowing the dimer models to be built using the same simple grid search. A paramagnetic perturbation study,15–20 based on shielding of the dimer interface from spin relaxation enhancements has also been added to validate the structures found.
The focus of this study is the Northeast Structural Genomics Consortium target, SeR13, an 86 residue, negatively-charged protein from Staphylococcus epidermidis (UniProt ID Q8CSK1, Gene name SE_1124). The structure of this protein as a monomer was determined by NMR methods and the details of those methods are reported here. The monomeric structure was previously deposited to the Protein Data Bank (PDB) with accession number 2K1H. The determination of its dimer structure provides validation of the monomer structure as well as association characteristics that may have regulatory functions. The protein's highly conserved amino acid sequence suggests that it is related to the nitrogen fixation network family and is homologous to the N-terminal domain of the NifU protein in bacteria, which is necessary for Fe-S cluster biosynthesis.21 A ligand screening study on the homologous protein from Staphylococcus aureus (SAV 1430, NESG ID: ZR18), suggested it preferentially binds a p-Tyr group at its active site, which, by homology, includes the SeR13 residues I6-P10, T14-K16 and I61-V63.22 This suggests that SeR13 may form complexes with other proteins carrying a phosphorylated tyrosine. Additional lines of evidence suggest SeR13 may interact with a protein encoded by the gene SE_0630, which is homologous to the C-terminal domain for NifU and is predicted to act as a reductase in complex with NifS.22,23 The biological function of SeR13 has not been identified experimentally, but based on these results a speculative function could include regulation of SE_0630 activity, possibly mediated by phosphorylation of one of the abundant phosphorylation sites on SE_0630.22
We anticipate that information on the structure of SeR13 and identification of interaction surfaces, even in a homodimer situation, will be useful in deriving and understanding functional characteristics. The dimer structure presented may participate in competitive protein-protein associations that more directly influence its function. Hence, in addition to documentation of a new protocol for the structure determination of weakly associating symmetric homodimers, important structural information on a previously uncharacterized protein is presented.
Results
Determining the monomeric structure of SeR13
A number of screening experiments, pulsed-field-gradient diffusion, dynamic light scattering, and concentration dependent chemical shifts indicated the presence of a monomer-dimer equilibrium for SeR13. Because the equilibrium favored the monomer, structure determination as a monomer was pursued, but with caution. The backbone assignment of SeR13 resonances was obtained using standard triple resonance experiments.24 Out of 86 amino acids, 81 (non-Proline) residues were assigned. The assignments have been deposited under the biological magnetic resonance data bank (BMRB) accession number 15678. The distance restraints for SeR13 were derived from 15N-edited NOESY-HSQC and 13C-edited NOESY-HSQC spectra using a 1 mM uniformly 13C,15N-enriched SeR13 sample.25–28 The locations of the interfacial residues were identified by concentration-dependent chemical shifts and paramagnetic perturbation studies. NOE distances restraints for these residues were carefully assigned to exclude possible intermolecular NOEs. The initial structure for SeR13 was generated by CYANA using a combination of manually assigned and auto-assigned NOE peaks.29 The structures were further refined in XPLOR-NIH using the NOE distance restraints produced by CYANA, dihedral angle restraints, and RDC's measured in DMPC/DHPC bicelle (Bicelle) and negatively charged compressed polyacrylamide gels (Gel).30 Weightings for RDC data were set to generate final Q-factors of 0.10–0.15, which are consistent with the expected accuracy of the RDC data.31 The 10 structures with the smallest number of violations have a backbone RMSD value of 0.5 Å for the secondary structure ordered region (RCSB accession number 2K1H). The ribbon representation of the ensemble is shown in Figure 1 and the structural statistics for SeR13 are shown in Table I.
Figure 1.

The ensemble of 10 structures with the least distance violations showing the monomeric form of SeR13 calculated using XPLOR-NIH. The coordinates for SeR13 have been deposited in the protein data bank with an accession code of 2K1H. The colors in the display progress from blue to red based on sequence.
Table I.
The Structural Statistics of Monomeric SeR13 Solution Structure
| No. of NOE-derived distance restraints | ||
| Intraresidues | 103 | |
| Sequential | 287 | |
| Medium range | 188 | |
| Long range | 331 | |
| Total | 909 | |
| No. of backbone dihedral angle restraints | 135 | |
| RDC | ||
| NH gel | 51 | |
| NH bicelle | 52 | |
| RMSD (Å) | All | Ordered |
| Backbone | 0.7 | 0.5 |
| All heavy atoms | 1.3 | 1.1 |
| Backbone dihedral angle distribution | ||
| % in most favored region | 83.3 | |
| % in additionally allowed region | 15.0 | |
| % in generously allowed region | 1.7 | |
| % in disallowed region | 0 |
The structure determined for SeR13 is a two layered α/β protein with an anti-parallel β-sheet formed by nonsequential segments of the protein, and two anti-parallel α-helices. The chemical shift index analysis and the prediction of TALOS32 indicated that residues D2 to S7 and F28 to Y31 have high β-strand propensities, but the solution structure of SeR13 shows a flexible loop for these regions. This inconsistency is likely due to the lack of sufficient NOE restraints in these regions and the fact that no hydrogen bond restraints were used during the calculation.
Concentration-dependent chemical shift studies
To determine the dissociation constant for the dimer SeR13, concentration-dependent chemical shift studies were carried out. Dilution studies of SeR13 were performed over concentrations ranging from 0.025 to 2.1 mM. 15N-1H HSQC spectra collected over this range are shown in Figure 2(A). The changes in 1H and 15N chemical shifts as a function of protein concentration [Fig. 2(B,C)] were used to calculate the self-dissociation constant for SeR13 using a 1:1 binding model and Eq. (1) of the Materials and Methods section; a Kd of 3.4 ± 1.4 mM was determined. Accordingly, about 30% of the protein is in the dimeric form under conditions used in monomer structure determination. This could partially explain, a failure to observe intersubunit NOEs.
Figure 2.
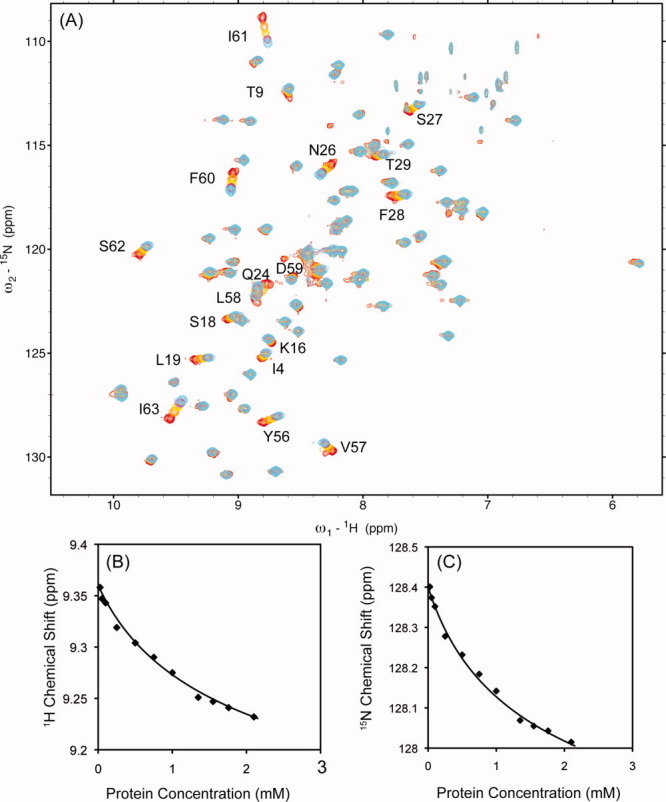
Concentration dependent chemical shift studies of SeR13. (A) The stacked 15N-1H HSQC spectra of SeR13 with protein concentration of 0.025, 0.5, 1.0, 1.5, and 2.1 mM are shown in red, orange, yellow, magenta, and cyan, respectively. The self-dissociation constant of SeR13 was obtained by monitoring the chemical shift changes as a function of protein concentrations. The fitting of chemical shifts assumes a 1:1 protein-protein complex. (B) 1H of L19 and (C) 15N of Y56.
The 1H-15N backbone chemical shifts shown in Figure 2 can also be used to suggest a dimerization interface. The changes in both 15N and 1H chemical shifts observed at dilutions from 2.1 mM to 0.025 mM were normalized and converted into a simplified combined score (Δδppm) as suggested by Farmer et al. and are presented as a function of sequence in Figure 3(A).33 These shifts are color coded on the molecular representation in Figure 3(B). It is clear that the perturbed region is largely on the β-sheet face of the protein and not on the helical region. The loops (I4 and T9) show some perturbation, as depicted in the structure presented. These perturbations could indicate that the loops are involved in the dimerization surface or that some allosteric rearrangement occurs during the dimer formation.
Figure 3.
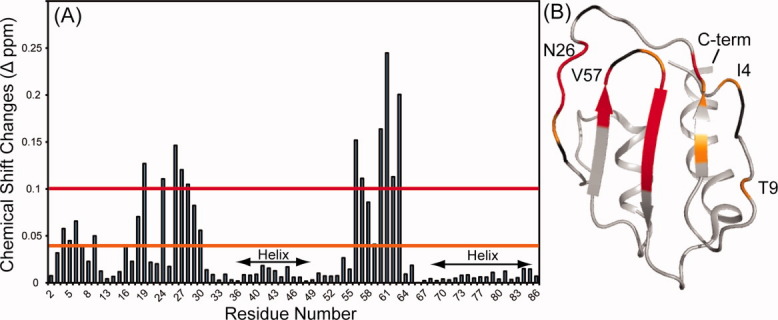
(A) The combined 15N and 1H chemical shifts perturbations of each resonance for SeR13 between 0.1 and 2.1 mM. (B) The backbone of residues with amide chemical shift perturbations > 0.1 Δppm are shown in red (L19, N26, S27, F28, T29, Y56, D59, F60, I61, S62, and I63). The backbone of residues with amide chemical shift perturbations of < 0.1 but greater than 0.04 Δppm are shown in orange (I4, T9, K16, S18, T29 and D59). Residues that show chemical shifts changes but are overlapped with another crosspeaks are shown in black.
Paramagnetic studies using Gd-DTPA
Protection from paramagnetic relaxation enhancement (PRE) can identify residues that comprise the dimerization interface. Paramagnetic agents that weakly associate with protein surfaces can shorten T2 spin relaxation times, leading to loss of signal during coherence transfer and coupling refocusing periods of HSQC experiments. If access to these surfaces is inhibited on dimerization, the loss of signals in the contact region is reversed.16 PRE studies on SeR13 were carried out using two different protein concentrations (0.1 and 1.9 mM) in the presence of 1 and 2.5 mM Gd-DTPA respectively. Amide resonances that experienced a protection from Gd-DTPA-enhanced relaxation are shown in Figure 4(A), where the differences in intensity compared to a standard containing no Gd-DTPA for each condition were normalized and compared to the mean. Residues showing the largest changes in protection are depicted as a function of sequence in Figure 4(A). The positions of these perturbed residues on the structural model are shown in Figure 4(B).
Figure 4.
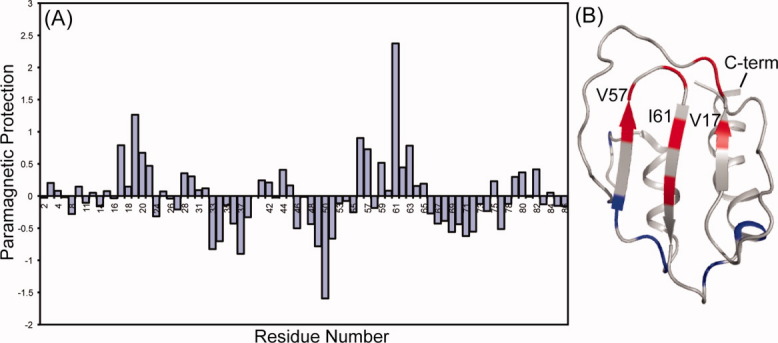
(A) Comparison plot of protection from paramagnetic relaxation enhancement on increasing SeR13 concentration from 0.1 to 1.9 mM in the presence of 1.0 and 2.5 mM Gd-DTPA, respectively. (B) Regions where the protection factor decreased by more than one standard deviation are shown in blue (A33, A34, G37, E49, G50, K52, A69, W71, and N72) and regions where protection increased by more than 1 standard deviation are shown in red (V17, L19, S20, Y56, V57, D59, I61, and I63).
The regions highlighted in red are in general agreement with the chemical shift perturbation data; the primary areas of protection on dimerization are located on the β-sheet face. There are a few regions on the outer loops that show enhanced, as opposed to reduced, relaxation upon dimerization. This enhancement is possibly due to the clustering of some positively charged groups upon dimer formation. Gd-DTPA has a net negative charge and association with these regions may increase in dimer formation. While neither chemical shift perturbation nor PRE protection are unambiguous indicators of a dimerization interface, their common perturbation of residues on the β-sheet face adds confidence to the identification of this surface as the dimerization interface.
Symmetry axis constraints on SeR13 dimerization
Given the qualitative nature of identification of a dimerization interface by chemical shift perturbation and PRE protection, we sought a more quantitative source of geometry restriction to build a model for the SeR13 dimer. It is well known that for a symmetric dimer aligned by any source of molecular interaction, one of the axes of the alignment tensor must lie along the symmetry axis.10,11 RDCs collected on 1H-15N pairs in structured regions of the protein can be used to define the alignment frames in multiple media, and for a tight dimer the symmetry axis can be identified as the alignment frame axis which is preserved among different alignment media.5,34 For weak dimers it is important to realize that the measured RDC values will be the average of the monomer and dimer values. For SeR13 this complication could be addressed by extrapolating RDCs measured at a series of concentrations to RDCs at infinite concentration.
RDCs were measured in negatively charged compressed polyacrylamide gels (7%) using protein concentrations of 0.20, 0.46, 1.00, and 2.10 mM. SeR13 was also aligned in a 4.2% bicelle using protein concentrations of 0.15, 0.60, and 0.90 mM. RDCs for each residue were plotted against the fraction (f) of dimer calculated from the equilibrium constant. The RDCs for dimeric and monomeric populated states were extracted using a linear least squares curve fitting method, where the Y-intercept at f = 0 is the predicted RDC for the monomer and the Y-intercept at f = 1 is the predicted RDC for the dimer [Fig. 5(A)]. To avoid complications in data analysis, only the RDCs from the ordered region of the SeR13 protein were used in this analysis. Representative plots are shown in Figure 5.
Figure 5.
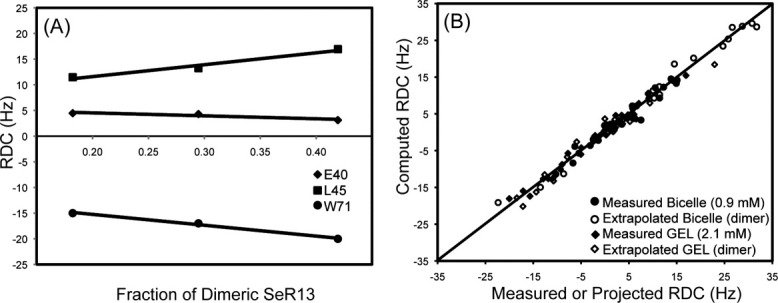
RDCs for the dimer of SeR13 by data extrapolation. (A) The RDCs values were extrapolated using data points collected with different protein concentrations. (B) The RDCs correlation plot between the measured or projected dimer RDCs against the computed RDCs using the monomer NMR structure.
The validity of the RDC analysis and appropriateness of the monomeric model can be assessed by comparison of measured and back-calculated RDCs. A plot of these data for the 0.9 mM Bicelle data, the 2.1 mM Gel data, and the projected dimer values for each is shown in Figure 5(B). Q values corresponding to these sets are 0.19, 0.13, 0.10, 0.23 for the 0.9 mM Bicelle data, the 2.1 mM Gel data, and the projected dimer values for each, respectively. Both the measured and extrapolated RDC values are in good agreement with the solution structure of SeR13.
The orientations of principal order tensor solutions allowed within estimated RDC error limits (10% of the RDC ranges) for both alignment media for SeR13 are plotted on a Sauson-Flamsteed plot in Figure 6(A). The differences in alignments between the Gel and Bicelle media are not large, possibly due to domination of steric alignment terms in both media. However, there are clear differences in the extent to which positions are conserved. The Gel Sxx and the Bicelle Syy axes deviate by no more than 10° while the other pairs of axes deviate by 40–50°. This clearly identifies the Gel Sxx/Bicelle Syy axis as the C2V symmetry axis and this axis is shown as red arrows in the molecular frame of the monomer [Fig. 6(B)]. Identification of the symmetry axis was unchanged when using the upper and lower limits of the dissociation constant as defined by the error estimates, and Euler angles for the alignment frame specification deviated by less than five degrees at these limits. The proper orientation of the second subunit of the dimer was then generated by rotating the monomer unit depicted by 180° about the symmetry axis.
Figure 6.
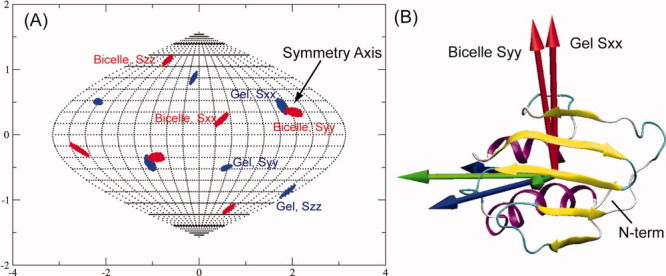
Alignment axis directions for SeR13. (A) The possible principal order tensor solutions of SeR13 for bicelle and Gel are plotted onto the Sauson-Flamsteed projection grid. (B) The directions of the order tensors are plotted onto the molecular frame of SeR13 to illustrate the symmetry axis between the two alignment media.
Constructing the dimeric model of SeR13
After producing a pair of monomers related by a 180° rotation about the identified axis, a dimeric model of SeR13 was constructed based on the grid search algorithm previously used by Wang et al. to model a symmetric homo-oligomer.5 Details of this procedure are given in the methods section. Out of 5107 possible dimeric models (each with different X and Z grid points), 29 models gave both acceptable RDC correlations and residue pair scores. The accepted dimeric models were then subjected to a rigid body energy minimization by XPLOR-NIH with the backbone atomic positions of protein fixed and the sidechains of the interface residues free to move. The 10 structures having the lowest energy from the XPLOR-NIH minimizations were selected and these are presented as ribbon diagrams in Figure 7(A). The RMSD for atomic positions of the 10 structures, not including and including sidechain atoms are 0.87 and 1.00 Å, respectively.
Figure 7.
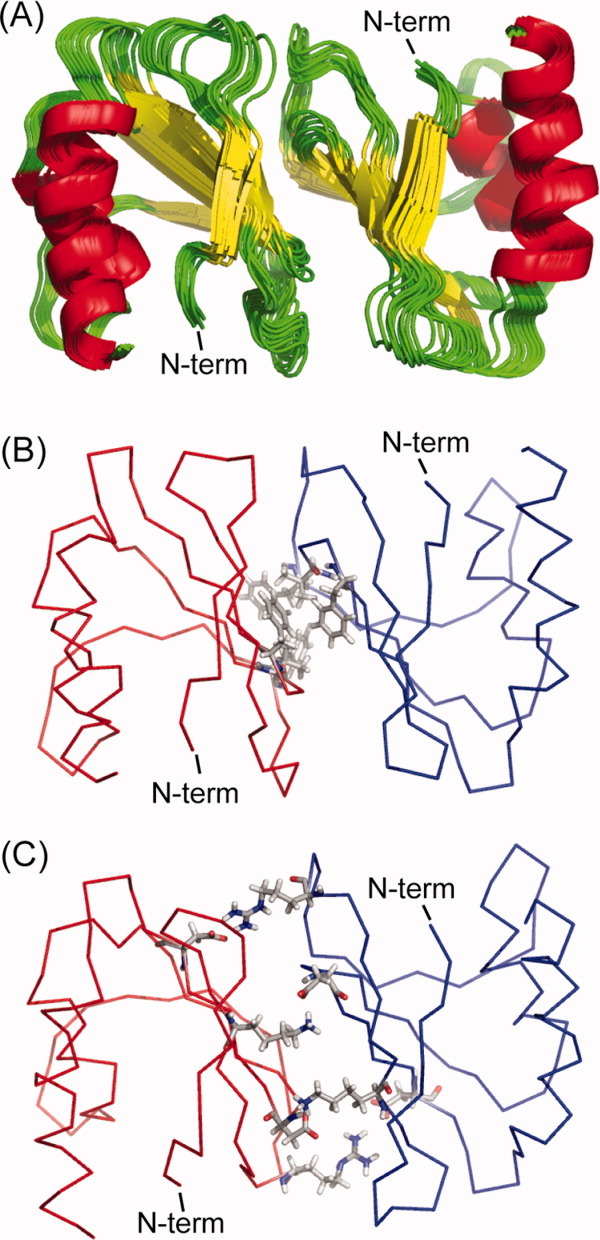
The dimeric models of SeR13. (A) The ensemble of 10 structures with lowest energies. (B) The hydrophobic interactions between the sidechains of F28, V57, and F60 of subunit A (red) and subunit B (blue) are shown in ball and stick representations. (C) The ionic interactions between sidechains of K16 and R23 of one subunit to the sidechain of D59 and D64 of the other subunit are shown in ball and stick representations.
Discussion
Examination of the structure produced suggests that it is indeed a very reasonable structure. One commonly used criterion for identification of an oligomer interface is the size of the interfacial area. In crystallography an interface area of 400 Å2 is commonly used as a minimum value to determine whether contacts represent crystal packing artifacts or the formation of a functional oligomer.35 The interface areas of all ten structures, as determined using VMD (measure solvent accessible surface area functions)36 are well above the cut off values of 400 Å2 with the average interface area being 736 Å2. The residue-residue contacts across the interface are also reasonable. The sidechains of F28, V57, and F60 from subunit A form hydrophobic interactions with the corresponding residues from subunit B [Fig. 7(B)]. The sidechains of K16 and R23 from one subunit also form a pair of salt bridges with sidechains of D59 and D64 from the other subunit, respectively [Fig. 7(C)].
Chemical shift perturbation and PRE protection experiments also support the structure presented. The majority of the residues experiencing chemical shift perturbations are located on the β-strands region of the SeR13. Of the 11 residues showing perturbations above 0.1 ppm, none of these residues lie outside the interface region described earlier. Likewise, of the eight residues showing a PRE protection factor greater than one standard deviation, none of these residues lie outside the interfacial area. Many of the residues experiencing protection from relaxation at higher protein concentration also experienced a chemical shift perturbation.
Of more general interest is the demonstration of an alternate, highly efficient approach to the determination of homodimer structures. The traditional approach involving mixing of differentially isotopically labeled proteins and filtering for NOEs that cross the interfacial boundary has been quite successful,6–8 especially for tightly associating dimers. However, for weakly associating systems, the intermolecular NOEs between the subunits can be difficult to observe for several reasons. One is that the number of possible NOEs is usually smaller because of more limited interfacial areas with hydrophobic contacts. In addition, ion pair interactions seldom bring nonexchangeable protons pairs within the 5 Å NOE limit. A second is that exchange between monomer and dimer species at intermediate rates can lead to exchange broadening of interfacial resonances. A third is that contacts can be mediated by pair wise interaction of equivalent residues, F28 with F28, for example, as occurs in the SeR13 structure. In these cases donating and accepting resonances can be equivalent and unresolved, or they can be degenerate with intra-residue pairs that always give NOEs. A similar situation exists with V57 and F60.
The amount and type of data acquired for the new approach is typical of traditional protein structure determination. The differences are the use of a combination of computational methods and the use of RDC data to restrain the relative orientations of subunits. RDC data, particularly 1H-15N data, are easily acquired. Acquisition builds on the HSQC or TROSY spectra normally used as a platform for acquisition of assignment and NOE data in the course of normal structure determination. RDC data are also increasingly acquired for the purpose of improving the quality of normal NMR structures, and their use in the additional step of determining an oligomeric structure may require little additional sample and little additional acquisition of data. In what we presented, we did acquire additional PRE protection data, which proved to be useful in terms of validating the SeR13 dimer structure, but this may be unnecessary in certain cases.
For the symmetric dimer case presented here we took advantage of the requirement that one axis of the principal alignment frame must coincide with the C2V axis of the dimer. There are similar requirements for other symmetries; for example, a trimer with three-fold rotational symmetry will display axially symmetric alignment with the unique alignment axis along the three fold axis.37 This provides less restriction on subunit geometry, but is still useful. There are some homo-oligomer geometries for which the approach will not work. For example, a complex with tetrahedral symmetry will not align with field induced alignment or the use of nematic liquid crystals. For hetero-oligomeric complexes, of course, one can determine alignment tensors independently for each subunit and assemble a structure by rotating subunits to superimpose principal axis systems.9,10,13,14,38
One of the primary limitations of the methods described is that the structure of the monomeric unit must be available. This structure could come from existing crystal structures that may not show the proper assembly for weak complexes, or it could come from NMR based structure determination. In the latter case, one must be aware that mis-interpretation of intersubunit NOEs as intra-monomer NOEs can lead to distortion of monomer structures.39–43 To a certain extent it is possible to avoid building errors into the dimer structures by restricting RDCs to those in well structured regions and away from regions that show concentration dependent chemical shifts or PRE protection. This was the procedure followed in the case presented. Omitted regions can be left mobile in the course of dimer structure determination and residue contacts that occur across an oligomer interface in derived dimer structures can be used after the fact to screen for possible mis-assignments to intra monomer NOEs. We can illustrate this process with the SeR13 structure by considering all contacts between protons on different subunits that fall within a nominal 4.5 Å cut-off. For SeR13 these include 2 pairs that gave rise to NOE crosspeaks observed and assigned to intra-monomer interactions in the structure determination. But the list of contacts also includes 18 pairs of proton-proton contacts, which would unambiguously give rise to intermonomer crosspeaks in a tight dimer structure. Crosspeaks for these pairs were not initially observed or used in structure determination, and on further examination of NOE data, only a single crosspeak for these pairs could be found. Given the absence of crosspeaks for this unambiguous set of intersubunit contacts, we believe the two observed crosspeaks were intra monomer in origin and were properly used in structure determination. As discussed earlier there are logical reasons for the absence of intermonomer NOEs, however, the absence of significant intermonomer NOEs does illustrate the potential problems in determining structures for weak dimers and underlines the unique value of the approach presented here. In particular, it would not have been possible to determine the dimeric structure of SeR13 using X-filtered NOESY approach,6–8 since the intermolecular NOEs would simply be too weak to observe. Though it may be possible to crystallize such weak dimers, in such circumstances crystal lattice interactions may dominate, resulting in non-native oligomeric interactions in the crystal. Hence, the method outlined in this work is unique in its ability to provide structures of weakly associating dimeric proteins that cannot be reliably determined by any other method.
As pointed out in the introduction, there is reason to believe that the SeR13 protein plays a regulatory role that requires protein-protein interaction. Weak homo-dimerization interfaces may mimic stronger hetero-dimer interfaces and provide guidance in identifying such interfaces. They may also play regulatory roles by competing with hetero-subunits for dimer formation. Clarification of the exact relevance of dimer formation must await further functional characterization of SeR13.
Materials and Methods
Expression and purification of SeR13
NESG target SeR13 was cloned, expressed, and purified based on the standard procedures of NESG to produce a uniformly labeled protein sample for NMR spectroscopy.44,45 The full-length protein was cloned into a pET21b vector, along with a S28F mutation and a short noncleavable C-terminal hexa His tag. The transformed cells were cultured at 37°C in MJ9 minimal medium45 containing (15NH4)2SO4 and U-13C-glucose as the sole nitrogen and carbon sources, respectively. SeR13 was purified using an AKTAexpress FPLC apparatus with a two-step protocol consisting of HisTrap HP affinity and HiLoad 26/60 Superdex 75 gel filtration chromatography. The purity of SeR13 (>98%) was verified with SDS-PAGE and MALDI-TOF mass spectrometry.
NMR experiments
All NMR experiments were performed on a Varian Inova 600 MHz spectrometer equipped with a cryogenically cooled probe. The pulse sequences were supplied by Varian as part of the BioPack distribution. The NMR spectra for resonances and NOE assignments were collected using protein concentrations of 0.9–1.1 mM prepared in 0.02% NaN3, 10 mM DTT, 5 mM CaCl2, 100 mM NaCl, 1x Protease Inhibitors, 20 mM MES pH 6.5, 10% D2O, 50 μM DSS at 25°C. Sequence-specific backbone resonance assignments for SeR13 were determined using HNCO, HNCA, HN(CO)CA, HNCACB, and CBCA(CO)NH experiments. The side chain assignments for SeR13 were carried out using CCONH, HCCH-TOCSY, and 15N-edit TOCSY-HSQC experiments. The NOE distance restraints for structural calculation were derived from 15N-edited NOESY-HSQC and 13C-edited NOESY-HSQC (for both the aliphatic and aromatic regions). NMR data were processed using NMRPipe and analyzed using SPARKY software packages.46,47
The protein concentration was measured with ɛ280 = 8480 M−1 cm−1 for SeR13. NMR samples for concentration dependent studies were prepared by diluting protein samples using buffer with the following composition 0.02% NaN3, 10 mM DTT, 5 mM CaCl2, 100 mM NaCl, 1x Protease Inhibitors, 20 mM MES pH 6.5, 10% D2O, 50 μM DSS. 15N-1H HSQC spectra were acquired for SeR13 at protein concentrations of 0.025, 0.1, 0.25, 0.50, 0.75, 1.0, 1.35, 1.56, 1.76, and 2.1 mM at 25 °C.
Paramagnetic surface mapping studies were carried out using gadolinium complexed with diethylenetriamine penta-acetic acid (Gd-DTPA) as a paramagnetic relaxation agent. 15N-1H HSQC spectra were acquired for SeR13 with protein concentrations of 0.1 and 1.9 mM without Gd-DTPA as diamagnetic references. 15N-1H HSQC spectra were acquired with protein concentrations of 0.1 and 1.9 mM in the presence of 1.0 mM Gd-DTPA for paramagnetic perturbation studies plus an additional point at 2.5 mM Gd-DTPA.
All RDCs for SeR13 were measured from an interleaved TROSY HSQC set of experiments.48 SeR13 was first aligned in a 4.2% DMPC/DHPC bicelle (4.9:1 ratio) medium using protein concentrations of 0.15, 0.6, or 1.0 mM. The deuterium splitting for these samples was 8.63, 5.68, or 8.28 Hz, respectively.49 SeR13 was also aligned in a negatively charged (50% 2-acrylamido-2methyl-1-propanesulfonic acid + 50% acrylamide) compressed gel medium (Gel) using protein concentrations of 0.2, 0.46, 1.0, or 2.1 mM.50 The negatively charged gel was initially cast in a 3.2 mm diameter plastic tube by overnight polymerization. The polymerized gel was washed in deionized water (two cycles over a period of 2 days), followed by washing with protein buffer to equilibrate the pH (two cycles over a period of 2 days). Finally, the gel was washed with deionized water to remove the buffer (two cycles over a period of 2 days). The swelled gel (∼7 mm diameter) was trimmed to a length of 35 mm and dried at room temperature for 2 days. The gel pellet was swollen in a 5 mm Shigemi NMR tube using the protein solution. The plunger of the Shigemi tube was fixed at a height of 14 mm from the bottom of the tube.
Structure calculations
The backbone dihedral angles for SeR13 were predicted using TALOS based on the assigned chemical shifts of HA, CA, CB, CO, and N.32 The structure calculations for SeR13 were initially done using CYANA with 634 manually assigned and 305 CYANA automatically assigned distance restraints.29,51 During the optimization stage for the NOE distance and dihedral angle restraints, 50 structures were calculated by CYANA and 20 structures with the lowest target energies were selected for analysis. For incorporation of RDC data, starting Da and R values for 0.6 mM SeR13 aligned in a bicelle medium were calculated from principle order parameters determined in REDCAT (4.39 and 0.44, respectively).52 Similarly, the Da and R for 0.46 mM SeR13 aligned in a negatively charged compressed gel medium were calculated as −7.54 and 0.32, respectively. The structural refinement was performed using NOE distance, dihedral angle, and orientation restraints based on the protocol described in the gb1_rdc example supplied with XPLOR-NIH and the top 10 structures with the lowest NOE violations of the 50 calculated structures were selected for final structure deposition.30
Concentration dependent dilution study
The resonances for residues with perturbed chemical shifts were assigned by following crosspeak movement as functions of concentration starting with the assigned reference spectrum collected at 1 mM protein concentration. The dissociation constant for SeR13 was calculated using both nitrogen and backbone amide proton chemical shifts fitting to an equation for dimer formation [Eq. (1)].
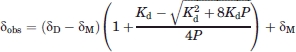 |
(1) |
Here, δD is the chemical shift of the dimer, δM is the chemical shift of the monomer, δobs is the observed chemical shift, Kd is the dissociation constant of the SeR13 dimer, and P is the concentration of total protein. The changes in both 15N and 1H chemical shifts were combined and used as a single variable according to Eq. (2).33
| (2) |
Here, Δδppm represents the combined 15N and 1H chemical shift changes, HΔδppm refers to the 1H chemical shift changes, and NΔδppm refers to the 15N chemical shifts changes. A total of 36 individual nitrogen and backbone amide chemicals shifts were fitted with Eq. (1). The dissociation constant of SeR13 dimer reported is the mean value of the 36 individual fits and the error value reported is the standard deviation of those individual dissociation constants.
Paramagnetic perturbation using Gd-DTPA
The resonances for residues with intensity perturbation were assigned based on the reference spectrum collected at 1 mM protein concentration with no Gd-DTPA. The paramagnetic protection factor (Pp) was determined using Eq. (3):
| (3) |
Here, In refers to the intensity of resonance n observed under the conditions specified in the brackets. These ratios were then compared to the mean for all residues.
RDC analysis
The RDCs for the dimeric populated state of SeR13 (RDCD) were extracted using a linear least square fit of Eq. (4) to experimental RDCs (RDCobs).
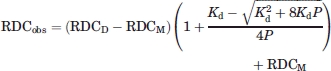 |
(4) |
The equation describes the observed RDCs as a function of protein concentration, P, for a simple homodimer with dissociation constant, Kd. RDCM is the RDC of the monomer.
The program, REDCAT, along with the monomer protein structure (model 1 of 2K1H PDB), was used to determine principal alignment frame axis directions from RDCs at each concentration point and at the dimer limit.52 REDCAT uses singular value decomposition to obtain a set of allowed order tensor solutions based on RDC error estimates. As fixed N—H bond lengths and planar amide groups are assumed in these calculations, the effects of real variations in NH bond lengths and real deviations from peptide planarity53 must be included in the error estimates; 10% of the RDC range was, therefore, used in estimating errors. Experimental errors are well below this limit and are considered not to contribute. The principal axes of the alignment tensor for all order tensor solutions were plotted onto Sauson-Flamsteed plots to depict allowed axis directions, and the C2V axis was identified based on the near overlap of one of the axis directions for the two media. The principal axes of the alignment tensors for both Bicelle and Gel media were placed onto the molecular frame of the PDB using the visual molecular dynamics (VMD) software package with the orient script.36
Grid search algorithm and model evaluations
The dimeric models of SeR13 were constructed using the grid search algorithm previously used by Wang et al.5 First, the monomer of SeR13 was fixed at the center of the grid. A second monomer was then generated by rotating the initial monomer by 180 degrees around the dimer symmetry axis. SeR13 is a fairly spherical molecule with a size of ∼35 Å on each dimension. The grid search was, therefore, performed using 70 grid points for both X and Z axes where 1 Å represents 1 grid point. Models were rejected if any two intermolecular backbone atoms came closer than 4 Å. Models were also rejected if closest intermolecular atomic distance was greater than 2 Å. The side chains of the interfacial residues of the proposed dimeric models from the grid search were subjected to 500 ps molecular dynamic simulation followed by energy minimization (NAMD and CHARMM22 force field, respectively).54
The proposed dimeric models of SeR13 were then evaluated based on the correlation between the measured and simulated RDCs along with a residue-pairing score. The simulated RDCs were calculated using PALES12 in the steric bicelle mode with an rM of 35 Å. The residue-pairing score came from associating the residue pairs in the interfacial area with likelihood statistics observed for those pairs in high resolution X-Ray structures.55 The residue-pairing score compliments well the RDC correlation score as there are multiple minima in the RDC score due to the shape degeneracy of the dimer models The pairing scores ranged from 10.96 for the most favorable pairs to −10.41 for the least favorable. The best dimeric models were selected based on a set of criteria including a Pearson correlation coefficient for RDC comparisons >0.80 and a sum of residue-pairing scores >0. The selected models were then further refined in XPLOR based on the rigid body approach where all of the residues were fixed with the exception of side chains of the interface residues. The simulated annealing processes were initiated at a temperature of 5000 K, cooled to 100 K in 10,000 steps, and finished off with Powell minimizations.
Glossary
Abbreviations:
- NMR
nuclear magnetic resonance
- PDB
protein data bank
- RDC
residual dipolar coupling
- NESG
northeast structural genomics consortium
References
- 1.Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 2.Ali MH, Imperiali B. Protein oligomerization: how and why. Bioorg Med Chem. 2005;13:5013–5020. doi: 10.1016/j.bmc.2005.05.037. [DOI] [PubMed] [Google Scholar]
- 3.Lee RT, Lee YC. Affinity enhancement by multivalent lectin-carbohydrate interaction. Glycoconj J. 2000;17:543–551. doi: 10.1023/a:1011070425430. [DOI] [PubMed] [Google Scholar]
- 4.Drickamer K. C-type lectin-like domains. Curr Opin Struct Biol. 1999;9:585–590. doi: 10.1016/s0959-440x(99)00009-3. [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Bansal S, Jiang M, Prestegard JH. RDC-assisted modeling of symmetric protein homo-oligomers. Protein Sci. 2008;17:899–907. doi: 10.1110/ps.073395108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zwahlen C, Legault P, Vincent SJF, Greenblatt J, Konrat R, Kay LE. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: application to a bacteriophage lambda N-peptide/boxB RNA complex. J Am Chem Soc. 1997;119:6711–6721. [Google Scholar]
- 7.Folkers PJ, Nilges M, Folmer RH, Konings RN, Hilbers CW. The solution structure of the Tyr41–>His mutant of the single-stranded DNA binding protein encoded by gene V of the filamentous bacteriophage M13. J Mol Biol. 1994;236:229–246. doi: 10.1006/jmbi.1994.1132. [DOI] [PubMed] [Google Scholar]
- 8.Ikura M, Bax A. Isotope-filtered 2D NMR of a protein peptide complex—study of a skeletal-muscle myosin light chain kinase fragment bound to calmodulin. J Am Chem Soc. 1992;114:2433–2440. [Google Scholar]
- 9.Prestegard JH, Bougault CM, Kishore AI. Residual dipolar couplings in structure determination of biomolecules. Chem Rev. 2004;104:3519–3540. doi: 10.1021/cr030419i. [DOI] [PubMed] [Google Scholar]
- 10.Al-Hashimi HM, Bolon PJ, Prestegard JH. Molecular symmetry as an aid to geometry determination in ligand protein complexes. J Magn Reson. 2000;142:153–158. doi: 10.1006/jmre.1999.1937. [DOI] [PubMed] [Google Scholar]
- 11.Bewley CA, Clore GM. Determination of the relative orientation of the two halves of the domain-swapped dimer of cyanovirin-N in solution using dipolar couplings and rigid body minimization. J Am Chem Soc. 2000;122:6009–6016. [Google Scholar]
- 12.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 13.Ortega-Roldan JL, Jensen MR, Brutscher B, Azuaga AI, Blackledge M, van Nuland NA. Accurate characterization of weak macromolecular interactions by titration of NMR residual dipolar couplings: application to the CD2AP SH3-C:ubiquitin complex. Nucleic Acids Res. 2009;37:e70. doi: 10.1093/nar/gkp211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Zuo X, Yu P, Byeon IJ, Jung J, Wang X, Dyba M, Seifert S, Schwieters CD, Qin J, Gronenborn AM, Wang YX. Determination of multicomponent protein structures in solution using global orientation and shape restraints. J Am Chem Soc. 2009;131:10507–10515. doi: 10.1021/ja902528f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petros AM, Mueller L, Kopple KD. NMR identification of protein surfaces using paramagnetic probes. Biochemistry. 1990;29:10041–10048. doi: 10.1021/bi00495a005. [DOI] [PubMed] [Google Scholar]
- 16.Bernini A, Spiga O, Ciutti A, Venditti V, Prischi F, Governatori M, Bracci L, Lelli B, Pileri S, Botta M, Barge A, Laschi F, Niccolai N. NMR studies of BPTI aggregation by using paramagnetic relaxation reagents. Biochim Biophys Acta. 2006;1764:856–862. doi: 10.1016/j.bbapap.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Pintacuda G, Otting G. Identification of protein surfaces by NMR measurements with a pramagnetic Gd(III) chelate. J Am Chem Soc. 2002;124:372–373. doi: 10.1021/ja016985h. [DOI] [PubMed] [Google Scholar]
- 18.Bernini A, Venditti V, Spiga O, Ciutti A, Prischi F, Consonni R, Zetta L, Arosio I, Fusi P, Guagliardi A, Niccolai N. NMR studies on the surface accessibility of the archaeal protein Sso7d by using TEMPOL and Gd(III)(DTPA-BMA) as paramagnetic probes. Biophys Chem. 2008;137:71–75. doi: 10.1016/j.bpc.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Bernini A, Venditti V, Spiga O, Niccolai N. Probing protein surface accessibility with solvent and paramagnetic molecules. Prog Nucl Mag Res Sep. 2009;54:278–289. [Google Scholar]
- 20.Otting G. Protein NMR using paramagnetic ions. Annu Rev Biophys. 2010;39:387–405. doi: 10.1146/annurev.biophys.093008.131321. [DOI] [PubMed] [Google Scholar]
- 21.Schilke B, Voisine C, Beinert H, Craig E. Evidence for a conserved system for iron metabolism in the mitochondria of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1999;96:10206–10211. doi: 10.1073/pnas.96.18.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mercier KA, Baran M, Ramanathan V, Revesz P, Xiao R, Montelione GT, Powers R. FAST-NMR: functional annotation screening technology using NMR spectroscopy. J Am Chem Soc. 2006;128:15292–15299. doi: 10.1021/ja0651759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Qi W, Cowan JA. Iron-sulfur cluster biosynthesis: functional characterization of the N- and C-terminal domains of human NFU. Biochemistry. 2009;48:973–980. doi: 10.1021/bi801645z. [DOI] [PubMed] [Google Scholar]
- 24.Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Mag Res Sep. 1999;34:93–158. [Google Scholar]
- 25.Silver MS, Joseph RI, Hoult DI. Highly selective Pi/2 and Pi-pulse generation. J Magn Reson. 1984;59:347–351. [Google Scholar]
- 26.Kay LE, Keifer P, Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. JACS. 1992;114:10663–10665. [Google Scholar]
- 27.Zhang O, Kay LE, Olivier JP, Forman-Kay JD. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J Biomol NMR. 1994;4:845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- 28.Bendall MR. Heteronuclear J coupling precession during spin-lock and adiabatic pulses. Use of adiabatic inversion pulses in high-resolution NMR. J Magn Reson Series A. 1995;116:46–58. [Google Scholar]
- 29.Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 30.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 31.Cornilescu G, Bax A. Measurement of proton, nitrogen, and carbonyl chemical shielding anisotropies in a protein dissolved in a dilute liquid crystalline phase. J Am Chem Soc. 2000;122:10143–10154. [Google Scholar]
- 32.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 33.Farmer BT, 2nd, Constantine KL, Goldfarb V, Friedrichs MS, Wittekind M, Yanchunas J, Jr., Robertson JG, Mueller L. Localizing the NADP+ binding site on the MurB enzyme by NMR. Nat Struct Biol. 1996;3:995–997. doi: 10.1038/nsb1296-995. [DOI] [PubMed] [Google Scholar]
- 34.Al-Hashimi HM, Valafar H, Terrell M, Zartler ER, Eidsness MK, Prestegard JH. Variation of molecular alignment as a means of resolving orientational ambiguities in protein structures from dipolar couplings. J Magn Reson. 2000;143:402–406. doi: 10.1006/jmre.2000.2049. [DOI] [PubMed] [Google Scholar]
- 35.Henrick K, Thornton JM. PQS: a protein quaternary structure file server. Trends Biochem Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 36.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graphics. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 37.Jain NU, Noble S, Prestegard JH. Structural characterization of a mannose-binding protein-trimannoside complex using residual dipolar couplings. J Mol Biol. 2003;328:451–462. doi: 10.1016/s0022-2836(03)00268-7. [DOI] [PubMed] [Google Scholar]
- 38.Fischer MW, Losonczi JA, Weaver JL, Prestegard JH. Domain orientation and dynamics in multidomain proteins from residual dipolar couplings. Biochemistry. 1999;38:9013–9022. doi: 10.1021/bi9905213. [DOI] [PubMed] [Google Scholar]
- 39.Clore GM, Omichinski JG, Sakaguchi K, Zambrano N, Sakamoto H, Appella E, Gronenborn AM. Interhelical angles in the solution structure of the oligomerization domain of p53: correction. Science. 1995;267:1515–1516. doi: 10.1126/science.7878474. [DOI] [PubMed] [Google Scholar]
- 40.Lambert LJ, Schirf V, Demeler B, Cadene M, Werner MH. Flipping a genetic switch by subunit exchange. EMBO J. 2004;23:3186–3186. doi: 10.1093/emboj/20.24.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nabuurs SB, Spronk CAEM, Vuister GW, Vriend G. Traditional biomolecular structure determination by NMR spectroscopy allows for major errors. PLOS Comput Biol. 2006;2:71–79. doi: 10.1371/journal.pcbi.0020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Prot Struct Funct Bioinform. 2007;66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 43.Bardiaux B, Bernard A, Rieping W, Habeck M, Malliavin TE, Nilges M. Influence of different assignment conditions on the determination of symmetric homodimeric structures with ARIA. Prot Struct Funct Bioinformatics. 2009;75:569–585. doi: 10.1002/prot.22268. [DOI] [PubMed] [Google Scholar]
- 44.Acton TB, Gunsalus KC, Xiao R, Ma LC, Aramini J, Baran MC, Chiang YW, Climent T, Cooper B, Denissova NG, Douglas SM, Everett JK, Ho CK, Macapagal D, Rajan PK, Shastry R, Shih LY, Swapna GV, Wilson M, Wu M, Gerstein M, Inouye M, Hunt JF, Montelione GT. Robotic cloning and protein production platform of the northeast structural genomics consortium. Methods Enzymol. 2005;394:210–243. doi: 10.1016/S0076-6879(05)94008-1. [DOI] [PubMed] [Google Scholar]
- 45.Jansson M, Li YC, Jendeberg L, Anderson S, Montelione GT, Nilsson B. High-level production of uniformly N-15- and C-13-enriched fusion proteins in Escherichia coli. J Biomol NMR. 1996;7:131–141. doi: 10.1007/BF00203823. [DOI] [PubMed] [Google Scholar]
- 46.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 47.Goddard TD, Keneller DG. Sparky 3. San Francisco: University of California; 2006. [Google Scholar]
- 48.Kontaxis G, Clore GM, Bax A. Evaluation of cross-correlation effects and measurement of one-bond couplings in proteins with short transverse relaxation times. J Magn Reson. 2000;143:184–196. doi: 10.1006/jmre.1999.1979. [DOI] [PubMed] [Google Scholar]
- 49.Bax A, Tjandra N. High-resolution heteronuclear NMR of human ubiquitin in an aqueous liquid crystalline medium. J Biomol NMR. 1997;10:289–292. doi: 10.1023/a:1018308717741. [DOI] [PubMed] [Google Scholar]
- 50.Cierpicki T, Bushweller JH. Charged gels as orienting media for measurement of residual dipolar couplings in soluble and integral membrane proteins. J Am Chem Soc. 2004;126:16259–16266. doi: 10.1021/ja046054g. [DOI] [PubMed] [Google Scholar]
- 51.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 52.Valafar H, Prestegard JH. REDCAT: a residual dipolar coupling analysis tool. J Magn Reson. 2004;167:228–241. doi: 10.1016/j.jmr.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 53.Ulmer TS, Ramirez BE, Delaglio F, Bax A. Evaluation of backbone proton positions and dynamics in a small protein by liquid crystal NMR spectroscopy. J Am Chem Soc. 2003;125:9179–9191. doi: 10.1021/ja0350684. [DOI] [PubMed] [Google Scholar]
- 54.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moont G, Gabb HA, Sternberg MJ. Use of pair potentials across protein interfaces in screening predicted docked complexes. Proteins. 1999;35:364–373. [PubMed] [Google Scholar]