

Abstract
Mycobacterium tuberculosis encodes five gene clusters (ESX-1 to ESX-5) for Type VII protein secretion systems that are implicated in mycobacterial pathogenicity. Substrates for the secretion apparatus are encoded within the gene clusters and in additional loci that lack the components of the secretion apparatus. The best characterized substrates are the ESX complexes, 1:1 heterodimers of ESAT-6 and CFP-10, the prototypical member that has been shown to be essential for Mycobacterium tuberculosis pathogenesis. We have determined the structure of EsxRS, a homolog of EsxGH of the ESX-3 gene cluster, at 1.91 Å resolution. The EsxRS structure is composed of two four-helix bundles resulting from the 3D domain swapping of the C-terminal domain of EsxS, the CFP-10 homolog. The four-helix bundles at the extremities of the complex have a similar architecture to the structure of ESAT-6·CFP-10 (EsxAB) of ESX-1, but in EsxRS a hinge loop linking the α-helical domains of EsxS undergoes a loop-to-helix transition that creates the domain swapped EsxRS tetramer. Based on the atomic structure of EsxRS and existing biochemical data on ESX complexes, we propose that higher order ESX oligomers may increase avidity of ESX binding to host receptor molecules or, alternatively, the conformational change that creates the domain swapped structure may be the basis of ESX complex dissociation that would free ESAT-6 to exert a cytotoxic effect.
Keywords: X-ray crystallography, structural genomics, protein complex, Mycobacterium tuberculosis, bacterial pathogenesis
Introduction
Mycobacterium tuberculosis is the causative agent of tuberculosis (TB) and a threat to world public health. One third of the world population (2 billion people) is infected with M. tuberculosis, 9.27 million new cases of TB were reported in 2007, and it is estimated that 1.75 million people died from TB in 2007.1 Global incidence rates have peaked, but the overall number of new cases continues to grow with population growth. Incomplete or inadequate antimicrobial therapy has led to multidrug resistant and extensively-drug resistant M. tuberculosis strains, which require the development of new vaccines or antimicrobial therapies.2
ESX complexes, heterodimeric protein complexes of ESAT-6 (early secreted antigenic target of 6 kDa), and CFP-10 (culture filtrate protein of 10 kDa), are secreted by mycobacterial species and are important mediators of host-pathogen interactions. The canonical ESX complex, EsxAB, is encoded within the 9.5 kb region of difference 1 (RD1), which is the only common genetic deletion found in two attenuated mycobacterial vaccine strains [M. bovis bacillus Calmette-Guérin (BCG) and M. microti]. In vivo studies have determined that the RD1 locus is partially responsible for the virulence of M. tuberculosis, M. bovis, M. marinum, and M. microti3–8 and complementation of BCG or the avirulent M. microti vaccine strain with RD1 increased the virulence of these strains.6,9 Biochemical and bioinformatic analyses have determined that the RD1 locus encodes, in addition to EsxAB, the components of a protein secretion system, ESX-1, which is the prototypical member of the Type VII secretion system family (T7S system; reviewed in10). There are four homologous systems present within the M. tuberculosis genome (ESX-2 to ESX-5) and similar systems have been indentified in other mycobacterial species (M. marinum and M. smegmatis) and in some Gram-positive bacteria.11–13 For some ESX systems additional T7S system components and substrates for the secretion system are encoded in isolated loci that lack the secretion apparatus.14,15 Since not all systems have been implicated in bacterial pathogenicity—the sole M. smegmatis ESX system plays a role in bacterial conjugation16—it appears that the original function of Type VII secretion systems did not involve export of virulence factors.
Structural and biochemical studies have determined that ESX complexes form tightly-bound 1:1 heterodimers17–20 and that a novel C-terminal CFP-10 signal sequence is required for secretion of the complex.21 The function of the secreted complexes is unclear but there are two possible mechanisms of action. The first scenario involves ESAT-6·CFP-10 binding to cell surface receptors on antigen presenting cells (APCs) enabling bacterial mediated modulation of APC responses to the pathogen.22 Evidence supporting this hypothesis is that the binding of ESAT-6·CFP-10 to the cell surface of monocytes or macrophages has been shown17 and specific binding of the C-terminal region of ESAT-6 alone to Toll-like receptor 2 (TLR2) of macrophages and inhibition of TLR signaling has been demonstrated.23 In the second scenario, the secreted complex dissociates in response to an environmental stimulus and ESAT-6 alone exerts a cytotoxic effect, possibly through disruption of membranes of the cell or intracellular compartments such as the phagosome. The evidence for this proposal is that the complex dissociates at low pH,5,24 that ESAT-6 alone has been shown to undergo a conformational change in the presence of phospholipids,25 that ESAT-6 can disrupt artificial lipid bilayers,5,24 and that CFP-10 lacks membrane lytic activity.24 The ESAT-6 homolog from the ESX-1 system of M. marinum has also been implicated in disrupting host cell membranes and a genetic deletion of the ESAT-6 gene has been shown to eliminate the pore-forming ability of M. marinum.8 To date only the ESAT-6·CFP-10 complex of ESX-1 has been demonstrated to be essential for virulence. However, there is evidence that the ESX complexes of the ESX-3 and ESX-5 gene clusters, and their homologs encoded in isolated loci, are secreted although their role in M. tuberculosis virulence is unclear.26–28 Expression of genes in the ESX-3 gene cluster has been shown to be regulated by iron and zinc29,30 and mutants that impair ESX-3 gene expression have confirmed that ESX-3 gene products are involved in iron and zinc uptake.31,32
We have determined the structure of EsxRS, an ESAT-6·CFP-10 homolog encoded by the Rv3019c and Rv3020c genes, in a heterotetrameric form created by a domain swap of the C-terminal domain of EsxS, the CFP-10 homolog. The domain swap results from a loop to helix transition of the hinge loop connecting the two α-helical domains of EsxS. As a result EsxS adopts a long helical conformation that pairs with a second EsxS monomer in an antiparallel manner that allows the N- and C-termini of two EsxS subunits to interact with an EsxR subunit at each end of the EsxS dimer thus creating a molecule with four-helix bundles at its extremities. The structure of a domain-swapped tetramer of an ESX complex may help in unraveling the function of these enigmatic protein complexes.
Results
EsxRS exists in heterodimeric and heterotetrameric forms
The EsxR and EsxS subunits were coexpressed and the EsxRS complex purified by Ni-NTA affinity chromatography. Size exclusion chromatography (SEC) was used for additional purification and two peaks containing EsxRS were eluted from the column [Fig. 1(A)]. Comparison with standards of known molecular mass is consistent with the first peak being a tetramer of EsxRS while the retention time for the second peak was consistent with a dimer of EsxRS. The relative ratio of tetramer to dimer was approximately 1:15 and was consistently observed in all protein preparations. Analysis of the two peaks by SDS-PAGE confirmed that both subunits were present in equimolar amounts in the two peaks [Fig. 1(B)]. Native PAGE results indicated that the first peak contains a higher order oligomer with reduced electrophoretic mobility compared to the second peak (data not shown). Mass spectrometric analysis of the two peaks immediately following purification determined that both subunits were present in the purified complexes and that there was no proteolytic degradation of either subunit. When the dimeric and tetrameric forms of EsxRS were reanalyzed by SEC four days after the preparative gel filtration, two populations of EsxRS were obtained for each previously homogeneous sample that corresponded to the dimeric and tetrameric states. The rate of interconversion between the two species was low with approximately 95% of the sample remaining in the original oligomeric state.
Figure 1.

Purification of dimeric and tetrameric forms of EsxRS. (A) Typical chromatographic profile for preparative SEC of EsxRS. Peak 1 contains an EsxRS complex with a molecular mass consistent with a tetramer while peak 2 contains an EsxRS complex with a molecular mass consistent with a dimer. (B) SDS—PAGE (20% gel) analysis of the two peaks from SEC purification. Lane M, Broad-range molecular weight markers; lane 1, peak 1 from SEC purification; lane 2, peak 2 from SEC purification. EsxRS complexes exist in two distinct oligomeric states, dimer and tetramer, that contain the EsxR and EsxS subunits in equal amounts.
The crystal structure of EsxRS
Both oligomeric forms of EsxRS were subjected to crystallization screening and crystals were obtained for the tetramer. The structure of the heterotetrameric M. tuberculosis EsxRS complex was determined by single wavelength anomalous diffraction at 3.0 Å resolution. SOLVE determined the positions of the six selenium sites that were present in the substructure. A partial model was built using RESOLVE, which was subsequently refined against the 1.91 Å resolution dataset and then rebuilt using ARP/wARP, which built ∼75% percent of the residues present in the final model. The remaining parts of the model were built using Coot and the model was refined using Refmac and TLS (translation/libration/screw) refinement with one TLS group per chain to a Rwork and Rfree of 19.6 and 23.8%, respectively. The model has excellent geometry with no residues in the disallowed region of the Ramachandran plot. Refinement statistics and model contents are detailed in Table I.
Table I.
Mycobacterium tuberculosis EsxRS Refinement Statistics (Numbers in Parentheses Refer to the High Resolution Data Shell)
| Crystal Parameters | Native | SeMet | |
|---|---|---|---|
| Space group | P212121 | P212121 | |
| Wavelength | 0.97849 | 0.97849 | |
| Cell constants at 100K | 40.3, 54.6, 103.1 Å | 40.1, 54.8, 102.9 | (90° 90° 90°) |
| Mean redundancy | 13.7 | 10.1 | |
| Resolution range [Å] | 51.6–1.91 (1.96–1.91) | 80.0–3.0 (3.11––3.0) | |
| Number of unique reflections | 18085 | 8778 | |
| Completeness of data [%] | 99.0 (96.7) | 99.1 (99.4) | |
| Mean I/σ(I) | 25.5 (6.1) | 21.7 (8.0) | |
| Number of residues (4 chains / asymmetric unit) | 238 | ||
| Number of protein atoms | 1776 | ||
| Number of glycerol atoms | 1 | ||
| Number of waters | 142 | ||
| Rfree [%] | 23.8 (28.2) | ||
| Rwork [%[ | 19.6 (21.2) | ||
| Test set size [%], selection | 5.0, random | ||
| RMS deviations from target values | |||
| Bond lengths [Å] | 0.011 | ||
| Bond angles [°] | 1.054 | ||
| Ramachandran plot | 98.1 | Most favored | |
| 1.9 | Additionally allowed | ||
| Errat Overall Quality Factor[%] | 98.5 | ||
| Verify3D residues with score >0.2[%] | 95.0 | ||
| PDB accession code | 3H6P |
The coordinates and molecular structure factors for Rv3020c-Rv3019c have been deposited in the Protein Data Bank (http://www.rcsb.org) under the accession code 3H6P.
The structure of EsxRS reveals a heterotetramer that contains two molecules of EsxR and two molecules of EsxS [Fig. 2 (A)]. EsxS is comprised of 97 residues excluding the affinity tag and the asymmetric unit contains two EsxS molecules with residues 17–76 observed for Chain A and residues 14–80 for Chain B. EsxR is 96 residues in length and two molecules are present in the asymmetric unit with Chain C containing residues 20 through 75 and Chain D containing residues 20 through 74. While mass spectrometric analysis of the complex immediately after purification indicated that the full length EsxR and EsxS polypeptides were present, we observed significant degradation of the complex in the protein stock solution (stored at 4°C) after a week. Analysis of the EsxRS crystals by MALDI-TOF mass spectrometry determined that the crystals contained degraded EsxR and EsxS subunits with two dominant species present with molecular weights of 7.1 and 7.4 kDa.
Figure 2.

The structure of the domain-swapped EsxRS complex. (A) Ribbon representation of the EsxRS structure. EsxS subunits are colored in red and orange and the EsxR subunits are in purple and teal. (B) Superposition of the two four-helix bundles of the EsxRS complex shows strong structural similarity. The orientation of the four-helix bundle at the top of the panel is in the same orientation as in Panel A. Subunit colors are the same. (C) Electrostatic surface potential of the EsxRS complex calculated with an ionic strength of 100 mM with red and blue representing negative and positive potentials, respectively. Fully saturated colors indicate a potential of magnitude ≥ ±3 kT. The first view in panel C is the same representation as in Panel A. The surface of EsxRS lacks the intense accumulation of charge associated with nucleic acid binding proteins implying that the EsxRS complex binds to specific target molecules as has been proposed for the EsxAB homolog. The structural similarity and symmetrical charge distribution between the four-helix bundles suggests that both extremities of EsxRS are capable of binding to receptor proteins, which has implications for inhibiting host cell processes that involve protein dimerization. An interactive view is available in the electronic version of the article.PRO451 Figure 2
As expected from secondary structure predictions using the PredictProtein server33 the complex has significant α-helical character. The EsxR subunit is an α-hairpin composed of two α-helical domains (N-terminal helix, Chain C, residues 21–38, Chain D, residues 21–39; and C-terminal domain: Chain C, residues 51–73, Chain D, residues 51–73) that are connected by a loop. The EsxS subunits form long antiparallel α-helices (Chain A, residues 18–74; Chain B, residues 15–76). The N-terminus of one EsxS chain and the C-terminus of the other EsxS chain interact with an EsxR subunit to form a four-helix bundle at each end of the molecule, which we will refer to as Bundle A (EsxR Chain C and the N-terminal segment of EsxS chain A and the C-terminal segment of EsxS Chain B) and Bundle B (EsxR Chain D and the N-terminal segment of EsxS Chain B and the C-terminal segment of EsxS Chain A). A structural superposition of the extended EsxS α-helices shows that there is conformational flexibility in the molecules with a root mean square deviation (r.m.s.d.) of 0.9 Å over 60 amino acids. The superposition of the two EsxR subuits gives an r.m.s.d. of 1.2 Å over 55 aligned amino acid residues with significant structural deviation in the loop that connects the two helical domains. The conformational flexibility of the loop is apparent from the high B-factors in the loop as well as the lower quality electron density seen for this region of the structure. A superposition of the four-helix bundles by truncating the EsxS chains at Gln44 and superimposing the resulting four-helix bundles demonstrates strong structural similarity between the two four-helix bundles with an r.m.s.d. of 1.1 Å over 115 amino acids [Fig. 2(B)]. Examination of the electrostatic potential of the surface of the complex reveals an even distribution of positive and negative charge with no highly charged regions [Fig. 2(C)].
The largest interface in the EsxRS structure, between the EsxS helices, buries ∼1130 Å2 of each subunit, or 20% of the surface of each subunit, in the interface. The four-helix bundles of the EsxRS structure have similar interfaces in terms of buried surface area, surface complementarity, and residues involved in the interface. EsxR Chain C buries 930 Å2 in the interface of Bundle A while the N-terminal segment of EsxS Chain A and the C-terminal segment of EsxS Chain B bury 1050 Å2 in the interface. Likewise, EsxR Chain D buries 870 Å2 in the interface of Bundle B with the C-terminal segment of EsxS Chain A and N-terminal segment of EsxS Chain B burying 1060 Å2 in the interface. The protein-protein interfaces were further compared by calculating the geometric shape complementarity (Sc) of the interfaces using the SC program.34 Theoretical Sc values for perfectly correlated and uncorrelated surfaces are 1 and 0, respectively, whereas the highest values seen for protein-protein interfaces, other than amyloids, are in the range of 0.70–0.76.34 The Sc values for the two EsxRS bundles are similar, 0.76 and 0.78 for Bundles A and B, respectively, and reveal that the interfaces are highly complementary. The charge distribution between Bundle A and Bundle B is highly symmetrical [Fig. 2(C)] owing to the structural similarity of the bundles.
The interface between EsxS and EsxR in the four-helix bundles is primarily hydrophobic and the amino acids in the helical segments of the bundles that contribute to the interface are the same in the two bundles. Variability in the bundle interfaces is because of the presence of additional amino acid residues at the termini of the helices for which there is density for one subunit but not the other. There is also variability in the interface due to different conformations of the loop that connects the helical elements of EsxR as Ser40 of EsxR Chain D contributes to the bundle interface while Ser40 of Chain C does not. There are seven hydrogen bonds in Bundle A between EsxR (Chain C) and the EsxS subunits and eight in Bundle B between EsxR (Chain D) and the EsxS subunits. Four of these are absolutely conserved and two others are between the same amino acids. The discrepancy in the other bonds is due to interactions at the termini of the EsxS subunits. In addition to the hydrogen bonds in the four-helix bundles there are also hydrogen bonds between the EsxS subunits with the following bonding pattern: Chain A Gln44-O to Chain B Gln44-NE2 with Gln40-OE1 making a hydrogen bond to Ser47-OG in both subunits.
Additional stabilization of subunit interactions is through intra- and intermolecular salt bridges. There are five salt bridges between the EsxS subunits: Chain A Glu33-OE2 to Chain B His55-NE2; Chain B Glu33-OE2 to Chain A His55-NE2; Chain A Arg26-NH1 to Chain B Asp70-OD1; Chain B Arg26-NH1 to Chain A Asp70-OD1; and one additional salt bridge between Chain A Asp70-OD1 to Chain B Arg26-NH2. There is one conserved intermolecular salt bridge that occurs between EsxR-Glu35 and EsxS-Lys21 in both of the four-helix bundles.
Comparison with homologous structures
The ESAT-6/CFP-10 proteins that comprise ESX complexes belong to the WXG100 family based on two common characteristics, the presence of an amino acid motif (Trp-Xaa-Gly; WXG) and a size of 100 residues per polypeptide chain.13 The structure of the prototypical member of the ESX family (ESAT-6·CFP-10; EsxAB) has been determined by NMR.17 EsxR and ESAT-6 share 27.1% sequence identity and 42.1% similarity while EsxS and CFP-10 have 31% sequence identity and 40% similarity. Consistent with the high degree of sequence identity there is obvious structural homology between EsxRS and ESAT-6·CFP-10 [Fig. 3(A)]. The four-helix bundle of ESAT-6·CFP-10 and the four-helix bundles at the extremities of the EsxRS structure superimpose well with an overall r.m.s.d. of 2.2 Å for 113 amino acids with a Z-score of 12.2 and sequence identity of 32% in the superimposed region. Our EsxRS structure has truncations at the N- and C-termini of all four chains; secondary structure predictions for EsxRS and comparison with EsxAB suggest that two to three turns of α-helix are missing from each subunit as are the unstructured regions present at the C-termini of ESAT-6 and CFP-10. However, the core component of both complexes is well conserved. Superimposing the EsxAB structure on the two four-helix bundles of EsxRS shows that it is possible for the intact EsxR subunits to exist in close proximity within the tetramer. The EsxR and ESAT-6 subunits have the same domain architecture with two α-helical domains linked by a loop that contains the WXG motif. The structural homology between EsxS and CFP-10 is evident with the exception of the six amino acid segment that contains the WXG motif (residues Gly40-Ala45 in CFP-10). In EsxS, this region adopts an α-helical conformation that results in the fusion of the α-helical N- and C-terminal domains into a single long α-helix whereas in CFP-10 this region is a loop that connects the N- and C-terminal helical domains.
Figure 3.
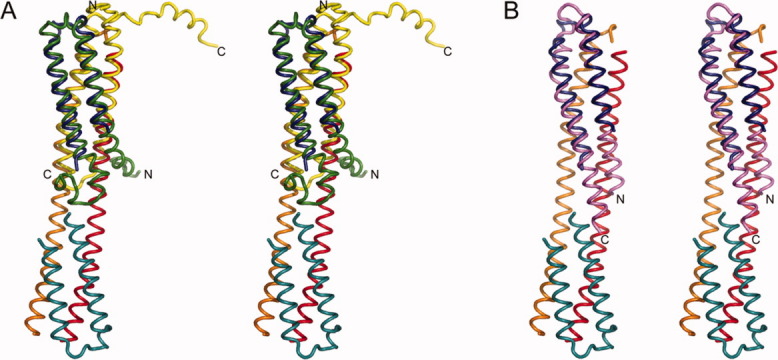
Structural similarity of EsxRS and related ESX molecules. (A) Stereoview of the superposition of EsxRS with M. tuberculosis EsxAB (ESAT-6·CFP-10) in worm representation with EsxRS oriented and colored as in Figure 2. The EsxA (ESAT-6) subunit is colored in green and EsxB (CFP-10) is colored in yellow. (B) Stereoview of the superposition of Staphylococcus aureus EsxA (SaEsxA) on the EsxR subunit of the EsxRS complex. EsxRS is colored as in Panel A and SaEsxA is colored pink. For clarity only the termini of EsxAB and SaEsxA are labeled. Strong structural similarity between the EsxRS and EsxAB complexes suggests that mycobacterial ESX complexes have related functions whereas the divergence in the SaEsxA structure suggests that more distantly related ESX homologs may have evolved new functions. An interactive view is available in the electronic version of the article.PRO451 Figure 3
An analysis of the stability of the EsxAB and EsxGH complexes concluded that the primary feature governing the stability of ESX complexes is van der Waals interactions and that additional hydrogen bonds or salt bridges increase complex stability.19 Analysis of the intermolecular interactions in the EsxRS bundles supports these observations as the EsxRS intermolecular interfaces are primarily hydrophobic with additional stabilization mediated by salt bridges and hydrogen bonds. Sequence alignments of ESAT-6 and CFP-10 homologs combined with data from the EsxRS and EsxAB structures reveals that the residues that form the hydrophobic interface are poorly conserved and that the intermolecular salt bridges that stabilize the complexes are not conserved between these homologs (Fig. 4). The sequence alignment suggests that other ESX complexes are stabilized by salt bridges as the residues that participate in the intermolecular EsxRS salt bridge are substituted by charged residues in equivalent positions in some ESX homologs (Fig. 4).
Figure 4.

Sequence alignment of CFP-10 (bottom) and ESAT-6 (bottom) homologs. The alignment was prepared with ClustalW and colored using BOXSHADE. Secondary structure elements are represented by sinusoidal lines representing α-helices with 310-helices represented by the lighter color. Disordered or missing regions of the structure are represented by black lines. The position of the hinge loop in EsxS (residues 41–46) is indicated as are the salt bridges formed between EsxS domains (red triangles, Arg26-Asp70; black triangles, Glu33-His55) and the intermolecular salt bridge formed between EsxS Lys21 and EsxR Glu35 (blue triangles). Residues involved in the intermolecular interface are designated by black circles. From the structure presented here and from the high conservation in residues 41–46 (this figure) we can speculate that the EsxGH complex will also be capable of domain swapping.
The secondary structure of the M. tuberculosis EsxGH complex, which is closely related to EsxRS, has recently been determined.35 The EsxGH complex is a 1:1 heterodimer of the Rv0287 (EsxG) and Rv0288 (EsxH) gene products and each subunit contains two α-helices (EsxG: α1, residues F18–F42 and α2, residues A49–L76; EsxH: α1, residues A19–A38, and α2, residues Y51–M81). The high degree of sequence homology between EsxGH and EsxRS, EsxG and EsxH are 91.8 and 84.4 percent identical to EsxS and EsxR, respectively, is reflected in the conservation and position of α-helical elements in the two structures (Fig. 4). The only significant variation between the secondary structure elements of the EsxRS and EsxGH complexes is that residues His43-Ser48 of EsxG lack ordered secondary structure while the corresponding region in EsxS adopts an α-helical conformation. A recent study of the EsxGH complex concluded that it was more stable than EsxAB, most likely as the result of increased surface area involved in protein-protein interactions or the presence of additional salt bridges.19 The authors predicted that EsxGH is stabilized by two intermolecular bridges: EsxG-Lys21 to EsxH-Glu31 and EsxG Lys64 to EsxH-Asp64. The EsxRS structure is stabilized by a single salt bridge (EsxR-Lys21 to EsxS-Glu35) although the residues predicted to form salt bridges in EsxGH are conserved in EsxRS. While these amino acid pairs do not form salt bridges in EsxRS, they are in close proximity and subtle variations between the EsxRS and EsxGH structures may allow these residues to form salt bridges in EsxGH. Completion of the EsxGH NMR structure and a detailed comparison of these highly homologous structures will allow these questions to be answered.
The X-ray structure of EsxA, an ESAT-6 or CFP-10 homolog, from Staphylococcus aureus has also been determined.36 In contrast to the Mycobacterial ESX complexes SaEsxA has been found to be a homodimer. The architecture of SaEsxA is similar to the Mycobacterial ESX proteins in that two helical domains are connected by a loop that contains the WXG motif. There is a slight bend in the C-terminal α-helical domain of SaEsxA due to the presence of a proline residue. The bend in this helix combined with the lower overall sequence identity between SaEsxA and EsxR and EsxS, 11.4% and 17.1% identity, respectively, results in a lower degree of structural homology between these less related ESX subunits. A structural superposition of SaEsxA on EsxR [Fig. 3(B)] has an r.m.s.d. of 3.3 Å over 49 Cα atoms. The superposition of SaEsxA on the N-terminal segment of EsxR Chain A (amino acids Ala17-Gln44) and the C-terminal segment of EsxR Chain D (amino acids Gly45-Ala80) has an r.m.s.d. of 2.5 Å over 45 Cα atoms.
The EsxRS heterotetramer is a result of 3D domain swapping
The similarity of the interface in the EsxRS four-helix bundles with the ESAT-6·CFP-10 four-helix bundle indicates that the EsxRS tetramer is the result of 3D domain swapping. 3D domain swapping is a mechanism by which higher order oligomers are formed through the exchange of identical domains between protein molecules. The molecule in which no domains are swapped is the closed reference structure, which constitutes a functional unit. A domain swap requires that the interactions in the reference and domain swapped complex interfaces must be the same and that the only significant difference between the closed reference structure and the domain swapped complex structure occurs in the hinge loop that mediates the domain swap. While the dimeric EsxRS reference structure has yet to be determined it has been purified in solution and we can postulate that it would be structurally similar to the EsxAB dimer.17
The domain swap that generates the tetrameric EsxRS structure is mediated by a conformational change in a short amino acid segment of EsxS that is predicted to lack regular secondary structure. In the ESAT-6·CFP-10 structure, this polypeptide segment (residues Gly40-Ala45), which contains the WXG motif, forms a loop that connects the two helical domains of CFP-10. EsxS and its closest family member, EsxG (Rv0287), contain a modified WXG motif with the tryptophan replaced by a histidine (His43) and in contrast to CFP-10 the hinge loop of EsxS (amino acids Ala41-Glu46) adopts a helical conformation that results in the fusion of the N- and C-terminal helical domains of EsxS into a single continuous α-helix. Two EsxS molecules pair in an antiparallel fashion to create a tetramer with two EsxR molecules. The region surrounding the hinge is stabilized by hydrogen bonds between Gln40 of one EsxS molecule and Ser47 of the other EsxS molecule. An additional hydrogen bond is formed between the main-chain oxygen of Gln44 of Chain A and Gln44-NE2 of Chain B. Gln44 is the variable residue in the WXG motif and the Gln44 sidechain in subunit A has two distinct conformations and could be involved in the interconversion between dimeric and tetrameric forms. The domain swap creates two functional units at each end of the complex composed of a single EsxR molecule and the N-terminal domain of one EsxS subunit and the C-terminal domain of the second EsxS subunit.
The amount of buried surface area in the interfaces of the closed reference structure and the domain swapped tetramer are different with the ESAT-6 and CFP-10 subunits having 1550 Å2 and 1880 Å2, respectively, buried in the interface compared to an average of 900 Å2 and 1050 Å2 for EsxR (ESAT-6 homolog) and EsxS (CFP-10 homolog). Deletion of the additional structural elements in the ESAT-6·CFP-10 complex to match the core molecule seen in our EsxRS structure results in a similar amount of buried surface area with 890 Å2 for ESAT-6 and 1000 Å2 for CFP-10. Thus the overall structural similarity and other characteristics confirm that the tetramer is the result of 3D domain-swapping.
Discussion
Biochemical studies and the NMR structure of the EsxAB complex have established that complexes of ESAT-6 and CFP-10 form 1:1 heterodimers. Our results demonstrate the existence of a higher order oligomer of an ESX complex that results from 3D domain swapping. Domain swapping involves a change in oligomeric state that occurs when a structural element, or domain, is exchanged between two or more protein molecules.37 In our structure of EsxRS, a heterotetrameric complex composed of two molecules of EsxR and EsxS is created through the exchange of the C-terminal domains of the CFP-10 (EsxS) subunits. The existence of an alternate oligomeric form of an ESX complex raises questions about a role for higher order oligomers in ESX function.
The creation of higher order oligomers through domain swapping has been found to play multiple roles in vivo. Domain-swapped proteins have been shown to form amyloid-like fibrils,38 which has led to the proposal that deposition diseases that involve protein aggregation, e.g. Alzheimer's and Parkinson's, may involve runaway domain swapping.39 Domain swapping can increase the enzymatic activity of a protein or increase the avidity of protein-protein interactions. Indeed it has been shown that diphtheria toxin forms a domain-swapped dimer upon binding its cellular receptor.40 As the interaction of the C-terminus of ESAT-6 (EsxA) with a macrophage cell surface receptor has been demonstrated to downregulate the host immune response,23 it would be of interest to determine if higher order ESX oligomers increase the avidity of protein-protein interactions. However, the lack of information on the function of ESX complexes will complicate this effort.
An alternate possibility by which a domain swapped ESX complex may be involved in ESX function can be inferred from studies that have shown that ESAT-6 alone has a destabilizing effect on membranes. The ESAT-6 subunit from M. tuberculosis and M. marinum has been shown to be capable of disrupting artificial membranes and host cell membranes, respectively,5,24,25,41 and it has been proposed that the low pH of the phagosomal compartments leads to complex dissociation allowing ESAT-6 to exert a cytotoxic effect.24 We also have seen pH induced dissociation of EsxRS (our unpublished results) although there is contradictory evidence about pH induced complex dissociation as a recent study of the stability of the EsxAB and EsxGH complexes concluded that they are not destabilized by low pH.19 The conformational change responsible for the domain-swap in EsxRS could be the basis for dissociation of a 1:1 ESAT-6·CFP-10 complex. Under this scenario in which pH or another stimulus, results in complex dissociation our domain-swapped tetramer may represent a non-functional dead-end in which the high concentrations of protein resulting from overexpression could lead to the tetramer by virtue of the complementarity of the subunit interfaces and the unfavorable interaction between the exposed hydrophobic faces of the unpaired ESX domains with aqueous solvent. More study is required to determine whether ESX complexes dissociate in response to environmental conditions and if the conformational change that creates our domain-swapped molecule is the basis of complex disassembly.
The EsxRS crystal structure suggests that domain swapping could occur in the closely related EsxGH complex and potentially in other ESX complexes. While the integrity of the WXG motif in the ESAT-6 subunit of the complex has been shown to be important for ESAT-6 folding and complex assembly42 a tryptophan to arginine mutation in the WXG motif of CFP-10 had no effect on complex assembly43 suggesting that the WXG motif in CFP-10 is not a critical structural element. Furthermore, the WXG motif in ESAT-6 contributes to a small hydrophobic core that stabilizes the ESAT-6 hairpin while the same region in CFP-10 lacks a comparable network of stabilizing interactions.17,19 The lack of a tight interface in CFP-10 suggests that other EsxS/CFP-10 homologs could undergo a similar conformational change to form domain swapped ESX complexes. While NMR studies have reported that EsxGH and EsxAB form 1:1 heterodimers17,35 with no evidence of higher order oligomers (Mark Carr, personal communication) we have seen evidence of higher order oligomers formed by another ESX complex (our unpublished results). As EsxAB has been demonstrated to interact with cell surface receptors of host APCs it would be of interest to explore whether alternate oligomeric forms of EsxAB exist and, if they do, whether these alternate oligomeric states have different effects on cell signaling.
In summary, we have determined the structure of an unpredicted higher order oligomer of an M. tuberculosis ESX complex. Our structure raises questions as to whether the domain-swapped ESX complex is formed by other ESX homologs and the role of domain swapping in ESX function. Future studies that provide insight into the function of ESX complexes will be necessary to determine the function, cellular targets, and role of domain-swapped ESX molecules in M. tuberculosis pathogenicity.
Materials and Methods
Cloning of the EsxRS complex
Rv3020c (EsxS) and Rv3019c (EsxR) were cloned into the pET46Ek-LIC expression vector in the same context as they are found in the M. tuberculosis H37Rv genome with Rv3020c upstream of Rv3019c. The Rv3020c-Rv3019c gene pair was PCR-amplified from M. tuberculosis H37Rv genomic DNA using KOD Hot Start polymerase (Novagen) and the following primers: Rv3020cFor (5′- GACGACGACAAGATGAGTTTGTTGGATGCCCATATTCC) and Rv3019cRev (5′- GAGGAGAAGCCCGGttaCTAGCCGCCCCACTTGGC). The PCR product was treated with T4 polymerase using the procedure supplied with the pET-46 Ek/LIC Vector Kit (Novagen). Following incubation of the insert and vector the ligation mixture was transformed into chemically competent E. coli DH5α cells and plated on LB plates supplemented with 100 μg/mL of ampicillin. Putative positive plasmids were isolated using a Qiaprep miniprep kit (Qiagen) and sequences were confirmed by DNA sequencing (Davis Sequencing).
Protein expression and purification
The EsxRS expression plasmid was transformed into E. coli BL21-Gold (DE3) (Stratagene) and grown overnight in LB broth supplemented with ampicillin (100 μg/mL) at 37°C. The following day the overnight culture was used to inoculate fresh cultures, which were grown at 37°C until an OD600nm of 0.6 was reached. At this point protein expression was induced with 0.75 mM IPTG and the cultures were grown for an additional 4 hours at 37°C. The cells were harvested by centrifugation and the cell pellet was resuspended in lysis buffer (20 mM HEPES pH 7.8, 150 mM NaCl, 10 mM imidazole pH 7.8, 10 mM β-mercaptoethanol). Protease inhibitor cocktail (Sigma-Aldrich), DNase I, and PMSF were added. Cells were lysed by sonication and the lysate was then centrifuged at 39,800 × G for 30 minutes at 4°C. The supernatant was then rocked with 2 mL of Ni-NTA agarose (Qiagen) for one hour at 4°C. The mixture was poured into a gravity column and the beads were washed with 20 column volumes of wash buffer (20 mM HEPES pH 7.8, 150 mM NaCl, 10 mM imidazole pH 7.8), followed by four column volumes of imidazole buffer (20 mM HEPES pH 7.8, 150 mM NaCl, 50 mM imidazole pH 7.8), and then the complex was eluted with four column volumes of elution buffer (20 mM HEPES pH 7.8, 150 mM NaCl, 300 mM imidazole pH 7.8). The imidazole wash and elution fractions were pooled and the hexahistidine tag was removed by proteolytic treatment with enterokinase (Invitrogen) at room temperature for 6 hours and then at 4°C overnight using conditions supplied in the manufacturers protocol. The following day the reaction was centrifuged (14,000 × G) for 5 minutes at 4°C, the supernatant was concentrated to 1 mL, and the complex was further purified by gel filtration using a Superdex75 column (GE Healthcare) equilibrated in gel filtration buffer (20 mM HEPES pH 7.8, and 150 mM NaCl). SDS-PAGE and native gels were used to determine, which peaks contained the complex and the appropriate elution fractions, were pooled and concentrated to 8–12 mg/mL for crystallization.
The selenomethionine derivative of EsxRS was prepared by growing 6 L of E. coli BL21-Gold (DE3) cells harboring the EsxRS expression plasmid in LB media supplemented with ampicillin at 37°C with shaking. Upon reaching an OD600nm of 0.6, the cells were harvested by centrifugation at 3000 × G for 20 minutes at 4°C. The cell pellet was resuspended in 50 mL of M9 buffer (supplemented with: 50 mg leucine, 50 mg isoleucine, 50 mg valine 100 mg phenylalanine, 100 mg lysine, 100 mg threonine, 5% glycerol, 0.002 M MgSO4, 0.0001 M CaCl2, and 10 mg thiamine per liter) and then recentrifuged. Each pellet was resuspended in 1 L of the same supplemented M9 media containing ampicillin and grown with shaking at 37°C. After 30 minutes, 0.75 mg selenomethionine was added to each liter and the culture was induced with 0.75 mM IPTG. Induced cultures were grown for 5 hours at 37°C. Cells were harvested and the EsxRS complex was purified using the same protocol as for native protein. The selenomethionine-labeled complex was concentrated to 8–10 mg/mL for crystallization.
Crystallization of the EsxRS complex
Crystallization was carried out at the UCLA Crystallization Facility using a Mosquito-TTP nanoliter dispenser. Hanging drop crystallization trials that mixed protein and reservoir solution at 1:1, 1:2, and 2:1 ratios were set up using commercially available crystallization screens from Hampton Research and Qiagen. Crystals of the native preparation of EsxRS that grew in condition D4 [0.1 M MMT (Malic acid, MES, Tris buffer system) pH 7, 25% PEG1500] of the PACT screen (Qiagen) were optimized with grid screens that varied buffer, salt, and precipitant concentrations. Optimization of crystallization conditions resulted in crystals that grew in 4–8 weeks when mixed 2:1 with precipitant (0.1 M MMT pH 7.0, 25% PEG1500). SeMet-labeled protein did not initially crystallize in the conditions used for native protein so heterogeneous streak seeding was used to transfer crystal nuclei from native crystals into the SeMet crystallization reactions. After seeding SeMet crystals grew in 0.1 M MMT pH 7.0, 26% PEG1500 using a 2:1 protein to precipitant ratio. Data from single crystals was collected on beamline 24ID-C at the Advanced Photon Source of Argonne National Lab.
EsxRS structure determination
The data for both native and selenomethionine-labeled crystals were processed with Denzo and Scalepack.44 The program SOLVE45 was used to locate selenium sites and density modification and initial model-building was performed with RESOLVE46. Automated model-building employing the higher resolution native data was performed with ARP/wARP47 using the CCP4 crystallography suite.48 Manual building of additional structural elements was performed with Coot49 interspersed with refinement cycles using Refmac,50 also part of the CCP4 crystallography suite. Final refinement used TLS parameters determined with the TLSmd server.51,52 The correctness of the structure was verified using the NIH-MBI SAVS validation server (http://nihserver.mbi.ucla.edu/SAVS/) for analysis with the programs ERRAT,53 PROCHECK,54 and WHAT_ CHECK.55 Ribbon and cartoon representations were prepared with Pymol,56 structural superpositions with DALI,57 electrostatic surface potentials with UHBD,58 and subunit interfaces were analyzed with PISA,59 AreaiMol,60 CONTACT/ACT,48 and SC.34
Acknowledgments
We thank M. Capel, K. Rajashankar, N. Sukumar, Jonathan Schuermann, and I. Kourinov of Northeastern Collaborative Access Team beamline ID-24 at the Advanced Photon Source of Argonne National Laboratory for assistance with X-ray data collection. We thank Robert Riley, Art Langanowsky, Andrew Min, Heungbok Kim, and the ISFI Protein Production Facility at the Los Alamos National Laboratory for technical assistance. Marcin Apostol, Mike Sawaya, Chang-Yub Kim, and Jeff Cox provided helpful insights into the project.
References
- 1.WHO. Global tuberculosis control–epidemiology, strategy, financing. Geneva, Switzerland: WHO; 2009. [Google Scholar]
- 2.Nguyen L, Pieters J. Mycobacterial subversion of chemotherapeutic reagents and host defense tactics: challenges in tuberculosis drug development. Annu Rev Pharmacol Toxicol. 2009;49:427–453. doi: 10.1146/annurev-pharmtox-061008-103123. [DOI] [PubMed] [Google Scholar]
- 3.Pym AS, Brodin P, Majlessi L, Brosch R, Demangel C, Williams A, Griffiths KE, Marchal G, Leclerc C, Cole ST. Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat Med. 2003;9:533–539. doi: 10.1038/nm859. [DOI] [PubMed] [Google Scholar]
- 4.Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, Sherman DR. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J Infect Dis. 2003;187:117–123. doi: 10.1086/345862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR., Jr The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci USA. 2003;100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol. 2002;46:709–717. doi: 10.1046/j.1365-2958.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- 7.Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, Brown EJ. ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol. 2008;10:1866–1878. doi: 10.1111/j.1462-5822.2008.01177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith J, Manoranjan J, Pan M, Bohsali A, Xu J, Liu J, McDonald KL, Szyk A, LaRonde-LeBlanc N, Gao LY. Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect Immun. 2008;76:5478–5487. doi: 10.1128/IAI.00614-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majlessi L, Brodin P, Brosch R, Rojas MJ, Khun H, Huerre M, Cole ST, Leclerc C. Influence of ESAT-6 secretion system 1 (RD1) of Mycobacterium tuberculosis on the interaction between mycobacteria and the host immune system. J Immunol. 2005;174:3570–3579. doi: 10.4049/jimmunol.174.6.3570. [DOI] [PubMed] [Google Scholar]
- 10.Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CM, Appelmelk BJ, Bitter W. Type VII secretion--mycobacteria show the way. Nat Rev Microbiol. 2007;5:883–891. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- 11.Gey Van Pittius NC, Gamieldien J, Hide W, Brown GD, Siezen RJ, Beyers AD. The ESAT-6 gene cluster of Mycobacterium tuberculosis and other high G+C Gram-positive bacteria. Genome Biol. 2001;2:RESEARCH 0044. doi: 10.1186/gb-2001-2-10-research0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tekaia F, Gordon SV, Garnier T, Brosch R, Barrell BG, Cole ST. Analysis of the proteome of Mycobacterium tuberculosis in silico. Tuber Lung Dis. 1999;79:329–342. doi: 10.1054/tuld.1999.0220. [DOI] [PubMed] [Google Scholar]
- 13.Pallen MJ. The ESAT-6/WXG100 superfamily – and a new Gram-positive secretion system? Trends Microbiol. 2002;10:209–212. doi: 10.1016/s0966-842x(02)02345-4. [DOI] [PubMed] [Google Scholar]
- 14.Fortune SM, Jaeger A, Sarracino DA, Chase MR, Sassetti CM, Sherman DR, Bloom BR, Rubin EJ. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc Natl Acad Sci USA. 2005;102:10676–10681. doi: 10.1073/pnas.0504922102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacGurn JA, Raghavan S, Stanley SA, Cox JS. A non-RD1 gene cluster is required for Snm secretion in Mycobacterium tuberculosis. Mol Microbiol. 2005;57:1653–1663. doi: 10.1111/j.1365-2958.2005.04800.x. [DOI] [PubMed] [Google Scholar]
- 16.Coros A, Callahan B, Battaglioli E, Derbyshire KM. The specialized secretory apparatus ESX-1 is essential for DNA transfer in Mycobacterium smegmatis. Mol Microbiol. 2008;69:794–808. doi: 10.1111/j.1365-2958.2008.06299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Renshaw PS, Lightbody KL, Veverka V, Muskett FW, Kelly G, Frenkiel TA, Gordon SV, Hewinson RG, Burke B, Norman J, Williamson RA, Carr MD. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J. 2005;24:2491–2498. doi: 10.1038/sj.emboj.7600732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Renshaw PS, Panagiotidou P, Whelan A, Gordon SV, Hewinson RG, Williamson RA, Carr MD. Conclusive evidence that the major T-cell antigens of the Mycobacterium tuberculosis complex ESAT-6 and CFP-10 form a tight, 1:1 complex and characterization of the structural properties of ESAT-6, CFP-10, and the ESAT-6*CFP-10 complex. Implications for pathogenesis and virulence. J Biol Chem. 2002;277:21598–21603. doi: 10.1074/jbc.M201625200. [DOI] [PubMed] [Google Scholar]
- 19.Lightbody KL, Ilghari D, Waters LC, Carey G, Bailey MA, Williamson RA, Renshaw PS, Carr MD. Molecular features governing the stability and specificity of functional complex formation by Mycobacterium tuberculosis CFP-10/ESAT-6 family proteins. J Biol Chem. 2008;283:17681–17690. doi: 10.1074/jbc.M800123200. [DOI] [PubMed] [Google Scholar]
- 20.Lightbody KL, Renshaw PS, Collins ML, Wright RL, Hunt DM, Gordon SV, Hewinson RG, Buxton RS, Williamson RA, Carr MD. Characterisation of complex formation between members of the Mycobacterium tuberculosis complex CFP-10/ESAT-6 protein family: towards an understanding of the rules governing complex formation and thereby functional flexibility. FEMS Microbiol Lett. 2004;238:255–262. doi: 10.1016/j.femsle.2004.07.043. [DOI] [PubMed] [Google Scholar]
- 21.Champion PA, Stanley SA, Champion MM, Brown EJ, Cox JS. C-terminal signal sequence promotes virulence factor secretion in Mycobacterium tuberculosis. Science. 2006;313:1632–1636. doi: 10.1126/science.1131167. [DOI] [PubMed] [Google Scholar]
- 22.Stanley SA, Raghavan S, Hwang WW, Cox JS. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci USA. 2003;100:13001–13006. doi: 10.1073/pnas.2235593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pathak SK, Basu S, Basu KK, Banerjee A, Pathak S, Bhattacharyya A, Kaisho T, Kundu M, Basu J. Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat Immunol. 2007;8:610–618. doi: 10.1038/ni1468. [DOI] [PubMed] [Google Scholar]
- 24.de Jonge MI, Pehau-Arnaudet G, Fretz MM, Romain F, Bottai D, Brodin P, Honore N, Marchal G, Jiskoot W, England P, Cole ST, Brosch R. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J Bacteriol. 2007;189:6028–6034. doi: 10.1128/JB.00469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meher AK, Bal NC, Chary KV, Arora A. Mycobacterium tuberculosis H37Rv ESAT-6-CFP-10 complex formation confers thermodynamic and biochemical stability. FEBS J. 2006;273:1445–1462. doi: 10.1111/j.1742-4658.2006.05166.x. [DOI] [PubMed] [Google Scholar]
- 26.Skjot RL, Brock I, Arend SM, Munk ME, Theisen M, Ottenhoff TH, Andersen P. Epitope mapping of the immunodominant antigen TB10.4 and the two homologous proteins TB10.3 and TB12.9, which constitute a subfamily of the esat-6 gene family. Infect Immun. 2002;70:5446–5453. doi: 10.1128/IAI.70.10.5446-5453.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenkrands I, Weldingh K, Jacobsen S, Hansen CV, Florio W, Gianetri I, Andersen P. Mapping and identification of Mycobacterium tuberculosis proteins by two-dimensional gel electrophoresis, microsequencing and immunodetection. Electrophoresis. 2000;21:935–948. doi: 10.1002/(SICI)1522-2683(20000301)21:5<935::AID-ELPS935>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 28.Skjot RL, Oettinger T, Rosenkrands I, Ravn P, Brock I, Jacobsen S, Andersen P. Comparative evaluation of low-molecular-mass proteins from Mycobacterium tuberculosis identifies members of the ESAT-6 family as immunodominant T-cell antigens. Infect Immun. 2000;68:214–220. doi: 10.1128/iai.68.1.214-220.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maciag A, Dainese E, Rodriguez GM, Milano A, Provvedi R, Pasca MR, Smith I, Palu G, Riccardi G, Manganelli R. Global analysis of the Mycobacterium tuberculosis Zur (FurB) regulon. J Bacteriol. 2007;189:730–740. doi: 10.1128/JB.01190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez GM, Voskuil MI, Gold B, Schoolnik GK, Smith I. ideR, An essential gene in mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect Immun. 2002;70:3371–3381. doi: 10.1128/IAI.70.7.3371-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serafini A, Boldrin F, Palu G, Manganelli R. Characterization of a Mycobacterium tuberculosis ESX-3 conditional mutant: essentiality and rescue by iron and zinc. J Bacteriol. 2009;191:6340–6344. doi: 10.1128/JB.00756-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siegrist MS, Unnikrishnan M, McConnell MJ, Borowsky M, Cheng TY, Siddiqi N, Fortune SM, Moody DB, Rubin EJ. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci USA. 2009;106:18792–18797. doi: 10.1073/pnas.0900589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32:W321–326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 35.Ilghari D, Waters LC, Veverka V, Muskett FW, Carr MD. 15N, 13C and 1H resonance assignments and secondary structure determination of the Mycobacterium tuberculosis Rv0287–Rv0288 protein complex. Biomol NMR Assign. 2009;3:171–174. doi: 10.1007/s12104-009-9167-3. [DOI] [PubMed] [Google Scholar]
- 36.Sundaramoorthy R, Fyfe PK, Hunter WN. Structure of Staphylococcus aureus EsxA suggests a contribution to virulence by action as a transport chaperone and/or adaptor protein. J Mol Biol. 2008;383:603–614. doi: 10.1016/j.jmb.2008.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Eisenberg D. 3D domain swapping: as domains continue to swap. Protein Sci. 2002;11:1285–1299. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Z, Eisenberg D. Runaway domain swapping in amyloid-like fibrils of T7 endonuclease I. Proc Natl Acad Sci USA. 2006;103:8042–8047. doi: 10.1073/pnas.0602607103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bennett MJ, Sawaya MR, Eisenberg D. Deposition diseases and 3D domain swapping. Structure. 2006;14:811–824. doi: 10.1016/j.str.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 40.Louie GV, Yang W, Bowman ME, Choe S. Crystal structure of the complex of diphtheria toxin with an extracellular fragment of its receptor. Mol Cell. 1997;1:67–78. doi: 10.1016/s1097-2765(00)80008-8. [DOI] [PubMed] [Google Scholar]
- 41.Johnson PD, Stuart RL, Grayson ML, Olden D, Clancy A, Ravn P, Andersen P, Britton WJ, Rothel JS. Tuberculin-purified protein derivative-, MPT-64-, and ESAT-6-stimulated gamma interferon responses in medical students before and after Mycobacterium bovis BCG vaccination and in patients with tuberculosis. Clin Diagn Lab Immunol. 1999;6:934–937. doi: 10.1128/cdli.6.6.934-937.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brodin P, de Jonge MI, Majlessi L, Leclerc C, Nilges M, Cole ST, Brosch R. Functional analysis of early secreted antigenic target-6, the dominant T-cell antigen of Mycobacterium tuberculosis, reveals key residues involved in secretion, complex formation, virulence, and immunogenicity. J Biol Chem. 2005;280:33953–33959. doi: 10.1074/jbc.M503515200. [DOI] [PubMed] [Google Scholar]
- 43.Meher AK, Lella RK, Sharma C, Arora A. Analysis of complex formation and immune response of CFP-10 and ESAT-6 mutants. Vaccine. 2007;25:6098–6106. doi: 10.1016/j.vaccine.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 45.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terwilliger TC. Maximum-likelihood density modification using pattern recognition of structural motifs. Acta Crystallogr D Biol Crystallogr. 2001;57:1755–1762. doi: 10.1107/S0907444901013737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 48.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 49.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 50.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 51.Painter J, Merritt E. TLSMD web server for the generation of multi-group TLS models. J Appl Crystallogr. 2006;39:109–111. [Google Scholar]
- 52.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 53.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 55.Vriend G, Sander C. Quality control of protein models: directional atomic contact analysis. J Appl Crystallogr. 1993;26:47–60. [Google Scholar]
- 56.DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]
- 57.Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics. 2000;16:566–567. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 58.Davis ME, Madura JD, Luty BA, McCammon JA. Electrostatics and diffusion of molecules in solution: simulations with the University of Houston Brownian dynamics program. Comput Phys Commun. 1991;62:187–197. [Google Scholar]
- 59.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 60.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]