

Abstract
l-Arginine hydrochloride (l-ArgHCl) was found to be an effective enhancer for in vitro protein refolding more than two decades ago. A detailed understanding of the mechanism of action, by which l-ArgHCl as co-solvent is capable to effectively suppress protein aggregation, while protein stability is preserved, has remained elusive. Concepts for the effects of co-solvents, which have been established over the last decades, were found to be insufficient to completely explain the effects of l-ArgHCl on protein refolding. In this article, we present data, which clearly establish that l-ArgHCl acts on the equilibrium solubility of the native model protein recombinant plasminogen activator (rPA), while for S-carboxymethylated rPA (IAA-rPA) that served as a model protein for denatured protein states, equilibrium solubilities could not be obtained. Solid to solute free transfer energies for native rPA were lowered by up to 14 kJ mol-1 under the tested conditions. This finding is in marked contrast to a previously proposed model in which l-ArgHCl acts as a neutral crowder which exclusively has an influence on the stability of the transition state of aggregation. The effects on the apparent solubility of IAA-rPA, as well as on the aggregation kinetics of all studied protein species, that were observed in the present work could tentatively be explained within the framework of a nucleation-aggregation scheme, in which l-ArgHCl exerts a strong effect on the pre-equilibria leading to formation of the aggregation seed.
Keywords: l-arginine, rPA, IAA-rPA, aggregation, equilibrium solubility
Introduction
Production of recombinant proteins is a well-established process. One of the most commonly used prokaryotic expression systems is Escherichia coli. Unfortunately, production of proteins of pharmaceutical interest in this system frequently leads to formation of insoluble inactive protein aggregates, the inclusion bodies. These inclusion bodies have to be solubilized with a denaturing agent such as urea or guanidine hydrochloride (GuHCl) and refolded afterwards to obtain correctly folded native protein.1–4
One of the most commonly occurring undesirable side reactions of protein refolding is unspecific aggregation.5 The extent of protein aggregation can be minimized by optimization of process parameters like temperature, protein concentration, pH value, ionic strength, catalysis of disulfide bridge formation, and by additives, which enhance protein refolding in a specific manner.1
The positive effects of some additives like sugar and polyalcohols are based on the stabilization of the most compactly folded protein species which is—in most cases—the native state.6,7 The effect of l-arginine, which is the proteinogenic amino acid with the most basic pKa value, differs from the rest of the natural amino-acids.8 l-Arginine hydrochloride (l-ArgHCl) has no significant effect on protein stability but has been found to increase the solubility of aggregate-prone, unfolded protein species, and folding intermediates.9
The positive effect of l-ArgHCl on protein refolding has been demonstrated for an extensive series of proteins, including lysozyme,9,10 the HA1-domain of hemagglutinin,11 human glucose 6-phosphate dehydrogenase,12 recombinant Fab-fragments,13 and tissue type plasminogen activator (t-PA).14 Although an impressive body of work describing the effects of l-ArgHCl has been accumulated, the mechanisms behind the action of l-ArgHCl are still not fully understood.9,15
The effects of co-solvents on proteins in solution may in principle be divided into kinetic and equilibrium effects. Observation of either type of effect puts constraints on possible mechanistic explanations. Equilibrium effects of co-solvents are based on a relative shift between the free energy of the accessible protein states. If this shift affects the dissolved and/or the macroscopically aggregated state, it would be equivalent to a difference in the equilibrium solubility of the protein. A large body of work exists, in which the effects of co-solvents on the solubility of proteins have been studied (see e.g., Refs. 16–20). As early as 120 years ago, Hofmeister21 ordered a series of cations and anions according to their ability to precipitate (salt out) proteins.
Apart from the general salting out and salting in effects, which are thought to be caused mainly by the influence of the dissolved salts on solvent structure,22–24 the diverse functional groups on proteins may give rise to a variety of different specific interactions with co-solvents.
One of the ways to think about the thermodynamics of protein/co-solvent interactions is in terms of preferential hydration and interaction. These concepts assume that a molecule, which is dissolved in an aqueous solution containing a co-solvent, can either have a higher affinity to the co-solvent or to water molecules. A higher affinity to water molecules gives rise to a situation that is called preferential hydration while a higher affinity to the co-solvent is named preferential interaction.25,26
For l-ArgHCl, it is not possible to straightforwardly explain its effect in terms of preferential interaction and hydration alone. Other compounds that show a similar efficacy as suppressors of protein aggregation like, for example, GuHCl, are preferentially bound, and consequently highly denaturing. One possibility to overcome these difficulties would be to postulate an effect of l-ArgHCl on the kinetics of protein aggregation only. Kinetic effects in this case are marked by a selective increase of the free energy of the transition state, that is, the barrier between the unfolded and the aggregated state. A simplified, purely kinetic model for the action of l-ArgHCl, based on a postulated so-called gap effect, was proposed by Baynes et al.27 in 2005. This model predicts a kinetic effect caused by a steric exclusion of co-solvent-molecules from gaps, which are expected to be formed in the transition state of protein-protein association reactions.
At present, it seems impossible to propose any molecular mechanism for the action of l-ArgHCl with a reasonable degree of certainty, as some fundamental information seems to be missing. We are currently not aware on any previous study that has investigated whether the increased apparent solubility of proteins in the presence of l-ArgHCl actually does represent a solution equilibrium or whether it is due to kinetic inhibition of aggregation. The main aim of this article is to decide this question for one model protein.
As solubility measurements in the presence of l-ArgHCl are impractical for well-soluble proteins, we chose a model that shows a relatively low apparent solubility in the absence of co-solvent, namely recombinant plasminogen activator (rPA; BM 06.022), which was first described and characterized by Kohnert et al.28 in 1992. This protein was reported to have a low solubility above pH 4.6 and is mostly insoluble in the pH range of 5 to 8.29 Besides native rPA, we included fully S-carboxymethylated rPA (IAA-rPA) in the solubility studies as a model for non-native, denatured protein states.
Results
Spectroscopical characterization of rPA and IAA-rPA
To establish IAA-rPA as valid model system for the denatured state of rPA, circular dichroism (CD) was used to gather information about differences in the secondary and tertiary structures of both protein forms (Fig. 1). As the solubility of rPA is negligible in the absence of additives, the measurements had to be performed in the presence of 100 mM d,l-arginine hydrochloride (d,l-ArgHCl). Under these conditions, far-UV spectra could only be recorded down to ∼205 nm. Furthermore, IAA-rPA only shows a residual solubility of approx. 0.1 mg mL−1 in 100 mM d,l-ArgHCl (cf. below). Due to these restrictions on the data quality of the recorded far-UV spectra, reliable quantitative estimates of secondary structure content could not be obtained. To reach protein concentrations that were high enough to observe near-UV spectra with an acceptable signal-to-noise ratio, even higher concentrations of ArgHCl up to 1M were necessary.
Figure 1.
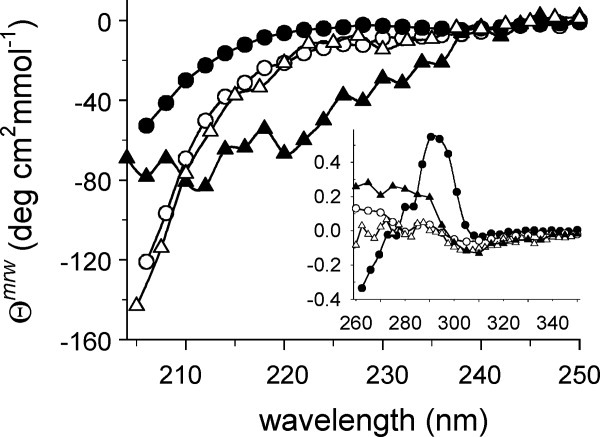
CD spectra of rPA and IAA-rPA. Far-UV CD spectra of rPA (circles) and IAA-rPA (triangles) were recorded under nondenaturing conditions (filled symbols) in 100 mM d,l-ArgHCl and under denaturing conditions (open symbols) in 4M GuHCl. Data are reported as mean molar ellipticity per amino acid residue, θmrw. The inset shows the corresponding near-UV CD spectra. Axis labels and units are the same as for the main panel.
The far-UV spectrum of native rPA showed a shallow minimum at 237 nm, followed by a decrease of the signal down to ∼ −55 deg cm2 mmol−1 at 205 nm. The near-UV spectrum of native rPA (Fig. 1, inset) in the presence of 1M l-ArgHCl showed a maximum at 295 nm and several resolved fine-structure bands in the region between 280 and 260 nm, indicative of a well-ordered tertiary structure.
For the carboxymethylated protein, the far-UV CD spectrum showed a minimum of ∼ −70 deg cm2 mmol−1 at around 210 nm. The near-UV spectrum was significantly different from that of the native protein, and no fine-structure bands were visible. These observations suggest a relatively high content of secondary structure elements in IAA-rPA dissolved in d,l-ArgHCl buffer, along with significant deviations from the tertiary structure of the native protein.
However, the CD spectra of rPA and IAA-rPA in buffer containing 6M GuHCl were practically indistinguishable over the entire monitored wavelength range. The near-UV spectra under these conditions were nearly featureless, whereas the far-UV spectra were in agreement with both protein forms adopting similar sets of random-coil conformations under denaturing conditions.
GuHCl-induced folding/unfolding of rPA and IAA-rPA
As CD had shown significant conformational changes for IAA-rPA induced by GuHCl, it was necessary to investigate if these structural changes were cooperative, that is, whether it was possible to differentiate between different, thermodynamically distinct states of the modified protein. CD had indicated a relatively high content of secondary structure of IAA-rPA in buffer containing 100 mM d,l-ArgHCl. To be able to treat the modified protein as valid model for the denatured state of native rPA, it was necessary to exclude that the conformation, or conformational ensemble, of IAA-rPA under these native-like conditions was separated from the ensemble of random-coil conformations in GuHCl-containing buffer by a cooperative transition.
As a point of reference, the GuHCl-induced folding/unfolding of native rPA was monitored by fluorescence spectroscopy [Fig. 2(A)]. Folding/unfolding of the native protein was found to be fully reversible, and at least two apparently cooperative transitions with midpoints at ∼1.5M and ∼3M GuHCl could be observed, with an additional nonlinear change in the fluorescence signal above 6M GuHCl. Parallel changes were visible in the CD signal at 215 nm [Fig. 2(B)]. The two apparent transitions were ascribed to the separate folding/unfolding of the serine protease and kringle-2 domains of the protein, respectively.
Figure 2.
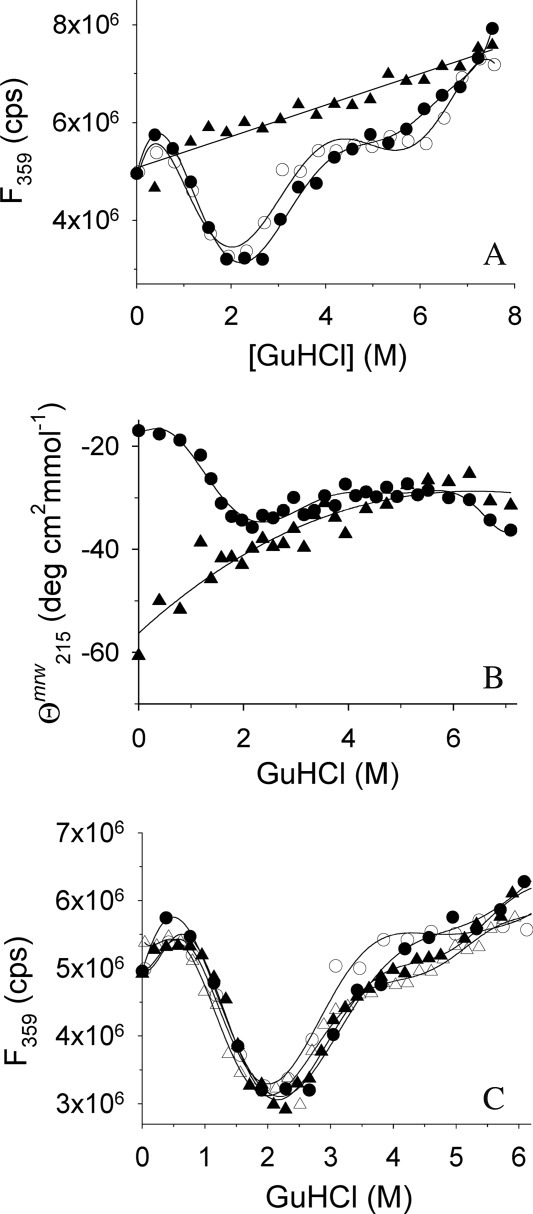
GuHCl-induced unfolding of rPA and IAA-rPA. (A) GuHCl-induced unfolding and refolding of rPA and IAA-rPA, respectively, were monitored by tryptophan fluorescence emission at 359 nm. Excitation wavelength was 280 nm. Data points represent the unfolding of rPA (filled circles), the refolding of rPA (open circles), and the unfolding of IAA-rPA (filled triangles). The protein concentration was 10 μg mL−1. (B) GuHCl-induced unfolding of rPA (filled circles) and IAA-rPA (filled triangles), respectively, were monitored by far-UV CD at 215 nm in the presence of 100 mM d,l-ArgHCl. The protein concentration for rPA was 440 μg mL−1 rPA and 15 μg mL−1–193 μg mL−1 for IAA-rPA. (C) The influence of l-ArgHCl on the GuHCl-induced unfolding and refolding of rPA was monitored by tryptophan fluorescence. In addition to the data from (A), data points are shown for the unfolding (filled triangles) and the refolding of rPA (open triangles) in the presence of 200 mM l-ArgHCl. Lines are meant to guide the eye.
In contrast to native rPA, the carboxymethylated protein did not show any cooperative folding transition over the tested range of GuHCl concentrations [Fig. 2(A,B)]. The observed changes in fluorescence and CD signal, respectively, indicate a loss of secondary structure, when the modified protein is exposed to increasing concentrations of denaturant.
These results are in agreement with the idea that IAA-rPA is present in solution as an ensemble of conformations with average observable properties that gradually relax in response to perturbations of buffer composition, without transitions between defined conformational states separated by significant energy barriers. Using this assumption, the S-carboxymethylated protein was taken as a model for the denatured state of the native protein in the present work.
For the main argument of the present work, it was also necessary to exclude a major influence of l-ArgHCl on the stability of rPA against denaturant-induced unfolding. In all cases described so far, this additive has been found to exert no, or an only marginally destabilizing, effect on the unfolding equilibria of proteins in solution. To investigate, whether this observation also held for rPA, folding/unfolding of the native protein was monitored by fluorescence spectroscopy in the presence of different concentrations of l-ArgHCl [Fig. 2(C)]. Within the error of measurement, no l-ArgHCl-dependent differences between the transition curves could be observed. Folding/unfolding of the protein was fully reversible in all cases. In conclusion, the addition of l-ArgHCl had no apparent effect on the structural stability of native rPA.
Solubility of native rPA
The solubility of native rPA was determined in presence of increasing concentrations of l-ArgHCl by measuring the concentration of dissolved protein in the supernatant [Fig. 3(A)]. As control for ionic strength effects as well as for the influence of the zwitterionic α-amino acid moiety, all measurements were repeated under similar conditions in the presence of NaCl and equimolar amounts of NaCl and glycine, respectively, instead of l-ArgHCl.
Figure 3.
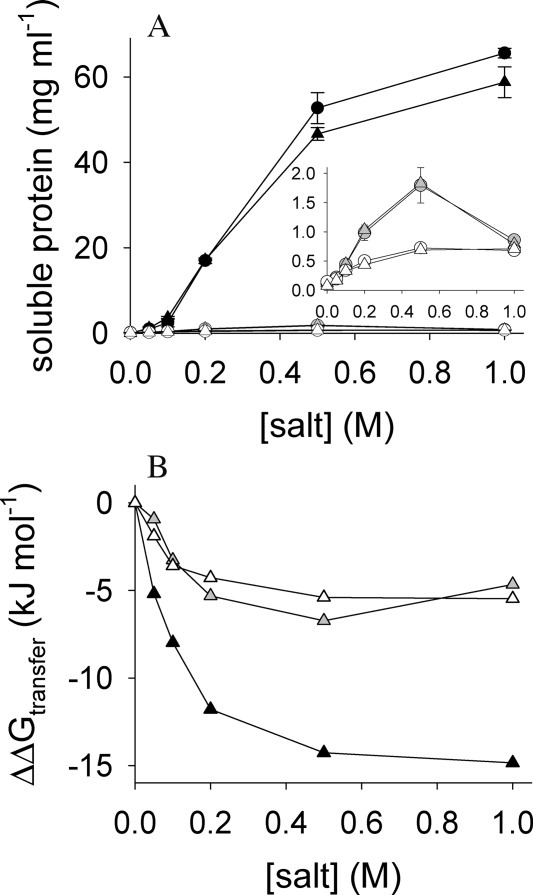
Solubility of rPA. (A) Protein solubility in the presence of increasing concentrations of l-ArgHCl (filled symbols), NaCl (open symbols), and NaCl with an equimolar amount of glycine (gray symbols), respectively, was determined as described in materials and methods. Data points represent solubility values obtained from soluble protein (circles) and from aggregated protein (triangles), respectively, as starting material. Data from two to three independent experiments are represented as mean values ± standard deviation. The inset represents the same data on an expanded protein solubility scale, and axis labels and units are the same as for the main panel. (B) Corresponding transfer free energies calculated from the data in (A).
By definition, the solubility of a substance is defined as the concentration in the solvent phase, which is in equilibrium with a solid phase under a given set of conditions. If such an equilibrium is not established due to additional irreversible processes like, for example, continuous decomposition of the substance, or due to kinetic inhibition, as in the case of supersaturated solutions, solubility in the strict sense cannot be measured. To decide if the observed concentrations of dissolved rPA truly represented equilibrium solubilities, it was necessary to establish that the same values were reached independent of the starting conditions. Thus, the solubility of native rPA was determined on the one hand by dialyzing a highly concentrated protein stock solution in ∼1.2M l-ArgHCl against measurement buffers containing the indicated concentrations of salts. On the other hand, solid precipitate of rPA was prepared by dialyzing the stock solution against additive-free buffer, and the precipitated solid protein was then re-solubilized by buffer exchange to the measurement conditions [Fig. 3(A)].
With increasing concentration of l-ArgHCl, the solubility of native rPA increased up to 66 mg mL−1 in buffer containing 1M l-ArgHCl, while by increasing the NaCl concentration to 1M only 0.66 mg mL−1 of rPA could be kept in solution [Fig. 3(A), inset]. For the buffers containing NaCl/glycine, a maximum solubility of 1.8 mg mL−1 rPA was observed in the presence of 0.5M salt. In the absence of added salts, only 0.07 mg mL−1 protein were detected in the supernatant. Re-solubilization of native rPA from the solid state with increasing concentrations of l-ArgHCl led to a maximum concentration of soluble rPA of 59 mg mL−1 in buffer containing 1M l-ArgHCl, whereas an increase of the NaCl concentration to 1M resulted in only 0.71 mg mL−1 solubilized rPA. In buffer containing 0.5M NaCl/glycine, 1.8 mg mL−1 rPA could be re-solubilized from precipitated protein. In conclusion, l-ArgHCl was found to increase the solubility of native rPA by almost three orders of magnitude, while the combined presence of NaCl and the amino acid glycine led to a 25-fold increase in solubility at best. Concentrations of soluble protein in the supernatant were very similar for all tested buffers, irrespective of the starting material. This clearly indicates that the observed solubility curves represent true equilibrium values. The possibility to re-solubilize rPA, which had been precipitated by removal of l-ArgHCl, indicated that the protein maintained a compact native-like molecular structure in the solid state. This notion was further supported by the observation that the specific enzymatic activity of rPA re-solubilized from the precipitate was identical to that of native protein from the stock solution (data not shown).
This was a precondition for the calculation of the corresponding differences in solid to solute transfer free energies (ΔΔGtransfer) for rPA caused by the presence of l-ArgHCl, NaCl, and NaCl/glycine, respectively, according to
| (1) |
with the universal gas constant R, the temperature T, and S and S0 being the solubilities of rPA in the presence and absence of added salts, respectively [Fig. 3(B)]. Apparently, ΔΔGtransfer was not linearly dependent on the concentrations of either l-ArgHCl or NaCl and glycine, but reached a minimum between 0.5M and 1M of added salt.
Assuming that the presence of the salts had no influence on the structure of the solid state, the standard chemical potential of native rPA in solution was lowered by 15 kJ mol−1 in the presence of 1M l-ArgHCl. The maximal combined effect of ionic strength and the zwitterionic α-amino acid moiety in the presence of 0.5M NaCl/glycine accounted for ∼ −7 kJ mol−1.
Solubility of IAA-rPA
In analogy to the experiments performed with native rPA, solubility measurements of IAA-rPA were carried out in the presence of increasing concentrations of l-ArgHCl, NaCl, and NaCl/glycine, respectively (Fig. 4).
Figure 4.
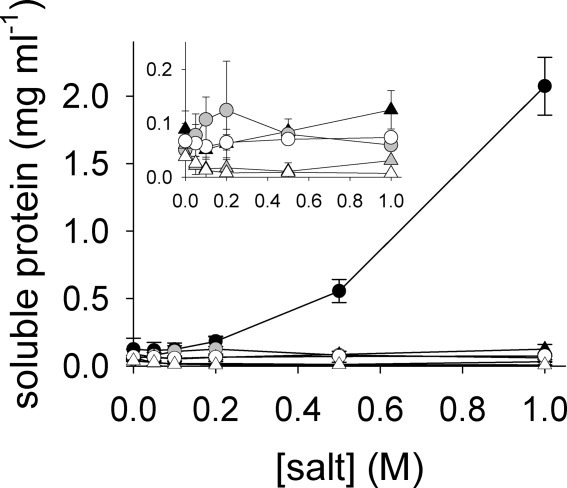
Solubility of IAA-rPA. The solubility of IAA-rPA in the presence of increasing concentrations of l-ArgHCl (filled symbols), NaCl (open symbols), and NaCl with an equimolar amount of glycine (gray symbols), respectively, was determined as described in materials and methods. Data points represent solubility values obtained from soluble protein (circles) and from aggregated protein (triangles), respectively, as starting material. Data from two to three independent experiments are represented as mean values ± standard deviation. The inset represents the same data on an expanded protein solubility scale, and axis labels and units are the same as for the main panel.
With increasing concentrations of l-ArgHCl, the apparent solubility of IAA-rPA also increased up to 2.1 mg mL−1 in buffer containing 1M l-ArgHCl, whereas increasing the NaCl concentration up to 1M resulted in a protein concentration of only 0.074 mg mL−1 in the supernatant. The addition of NaCl had no positive effect on the solubility of IAA-rPA, whereas the addition of NaCl and glycine led to a slightly increased apparent solubility of up to 0.12 mg mL−1 in the presence of 0.2M salt (Fig. 4). These results were obtained using a stock solution of IAA-rPA dissolved in GuHCl as starting material.
Again, to assess if the determined solubilities represented equilibrium values, IAA-rPA was precipitated by removal of GuHCl from the stock solution of denatured protein by dialysis and subsequently dialyzed against buffers containing the indicated concentrations of l-ArgHCl, NaCl, and NaCl/glycine, respectively (Fig. 4). In contrast to native rPA, re-solubilization of IAA-rPA from its solid state could not be effectively achieved. With increasing concentrations of l-ArgHCl, the apparent solubility of IAA-rPA decreased to 0.05 mg mL−1 in 0.1M l-ArgHCl and then increased up to 0.13 mg mL−1. The solubility of IAA-rPA in 1M NaCl decreased to 0.007 mg mL−1, that is, detection of soluble modified protein in the supernatant was hardly possible under these conditions. In the presence of 1M NaCl/glycine, a solubility of rPA of only 0.03 mg mL−1 was observed. Although molar concentrations of l-ArgHCl were able to keep relatively high concentrations of IAA-rPA in solution, the precipitation of the carboxymethylated protein was found to be a practically irreversible process. These findings indicate a high energy barrier between the unfolded state in solution and the solid precipitate of IAA-rPA in the presence of l-ArgHCl. By mass spectrometric analysis, we found that the re-solubilized protein in the supernatant was additionally enriched in over-alkylated forms of IAA-rPA that had been present as impurities in the modified protein preparation. These forms seem to exhibit a slightly increased solubility compared with the correctly S-alkylated protein, possibly due to an increased net charge. Thus, the determined solubility values for the re-solubilization of IAA-rPA from the precipitated denatured material represent upper limits.
It should be noted here that native rPA can be transferred into a similar irreversibly aggregated precipitate by thermal denaturation (not shown).
Concentration dependence of aggregation
In addition to the equilibrium solubility measurements discussed above, we set out to investigate how the observed differences in solubility between the various forms of rPA are reflected in differences in aggregation kinetics.
The aggregation of IAA-rPA, denatured reduced rPA, and of native rPA, respectively, was monitored by light scattering after dilution into renaturation buffer containing different concentrations of l-ArgHCl [Fig. 5(A)]. For the analysis, the initial slopes of the measured light scattering traces were determined. The data was tentatively analyzed using the formalism for a simple nucleation-elongation mechanism.30 In this case, a linear relationship is expected between the logarithms of the initial aggregation rate, as measured by the initial rate of change of the light scattering signal, (dB/dt)0, and of the protein concentration, C.
Figure 5.
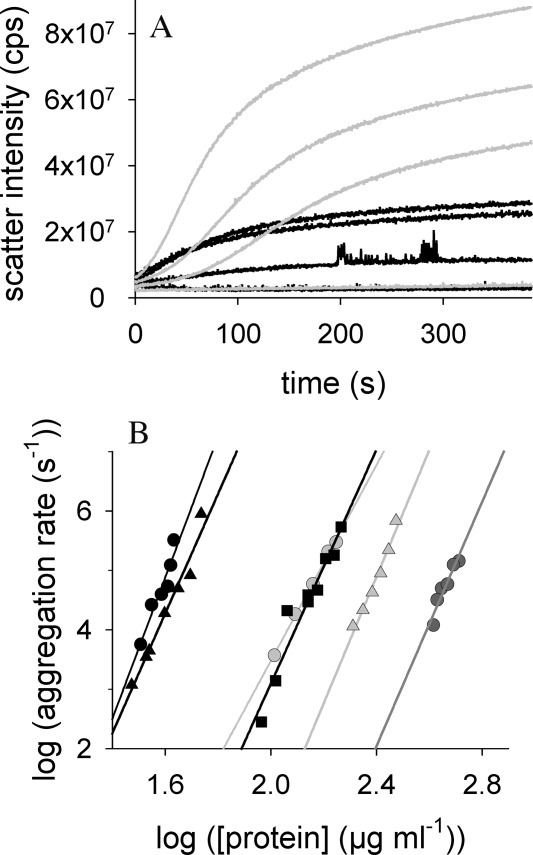
Kinetics of protein aggregation. (A) The time- and protein concentration-dependent formation of rPA- and IAA-rPA aggregates in absence of l-ArgHCl was followed by measuring light scattering intensity at 600 nm. Total protein concentrations were 174, 162, 139, and 103 μg mL−1 for rPA (top to bottom, black lines) and 43, 41, 35, and 32 μg mL−1 for IAA-rPA (top to bottom, gray lines). (B) The initial rates of protein aggregation are plotted against the total protein concentration in the respective experiments. Data points represent aggregation of IAA-rPA in 0 mM (black circles), 200 mM (light gray circles), and 500 mM (dark grey circles) l-ArgHCl, for denatured reduced rPA in 200 mM (black filled triangles) and 500 mM (light gray filled triangles) and for native rPA in absence of l-ArgHCl (black squares). The slopes taken from this double-logarithmic plot (n-values) are summarized in Table I.
| (2) |
Using this relation, it was possible to estimate the apparent order n of the initial aggregation reaction, which may be interpreted as the size of an assumed nucleation seed, from a double logarithmic plot of the initial slope of the aggregation kinetics versus the protein concentration [Fig. 5(B)]. In all cases, a relatively steep dependence of the initial aggregation rates on protein concentration was observed. While the concentration range, in which aggregation could be observed, was strongly dependent on the protein species and on solvent composition, the estimated values of n for the aggregation of IAA-rPA, denatured reduced rPA, and native rPA were found to be in the range between 8 and 12 and are summarized in Table I. The addition of l-ArgHCl did not significantly influence the values of n for IAA-rPA and denatured reduced rPA, indicating similar mechanisms of aggregation for all rPA species under all tested conditions. The presence of l-ArgHCl shifted the curves toward higher protein concentrations without changing the steepness of the concentration dependence [Fig. 5(B)]. Thus, an estimate for the size of the hypothetical nucleation seed for the aggregation of native as well as for denatured forms of rPA of ∼10 was obtained. In all these cases, a series of pre-equilibria leading to a critical transitional stage in the aggregation process must exist, and the observed increase of the apparent solubility of IAA-rPA in the presence of l-ArgHCl might be explained by a shift in these pre-equilibria toward dissociation, and a consequently much reduced concentration of available nucleation seeds.
Table I.
Concentration Dependence of Aggregation Kinetics—n Values
| Protein species | [l-ArgHCl] (M) | n |
|---|---|---|
| IAA-rPA | 0 | 11.9 ± 1.9 |
| 0.2 | 8.2 ± 0.2 | |
| 0.5 | 10.3 ± 1.6 | |
| Denatured reduced rPA | 0.2 | 10.1 ± 0.9 |
| 0.5 | 10.7 ± 0.9 | |
| Native rPA | 0 | 9.8 ± 1.0 |
State of IAA-rPA in solution
The solubility measurements described so far had determined total dissolved protein mass in solution. To determine the molecular mass distribution, the samples were analyzed by asymmetrical flow field flow fractionation/light scattering (AF4/LS) [Fig. 6(A)]. In an AF4 experiment, the injected sample will be concentrated more than 30-fold in a small volume adjacent to the channel wall during the focusing phase, which is necessary to establish an equilibrium distribution of the analyzed molecular species in the flow field, before the actual separation may proceed. As a consequence, a characterization of IAA-rPA in the absence of l-ArgHCl was not possible due to the negligible residual solubility of the modified protein in additive-free buffer, and measurements were performed in buffers containing l-ArgHCl in different concentrations.
Figure 6.
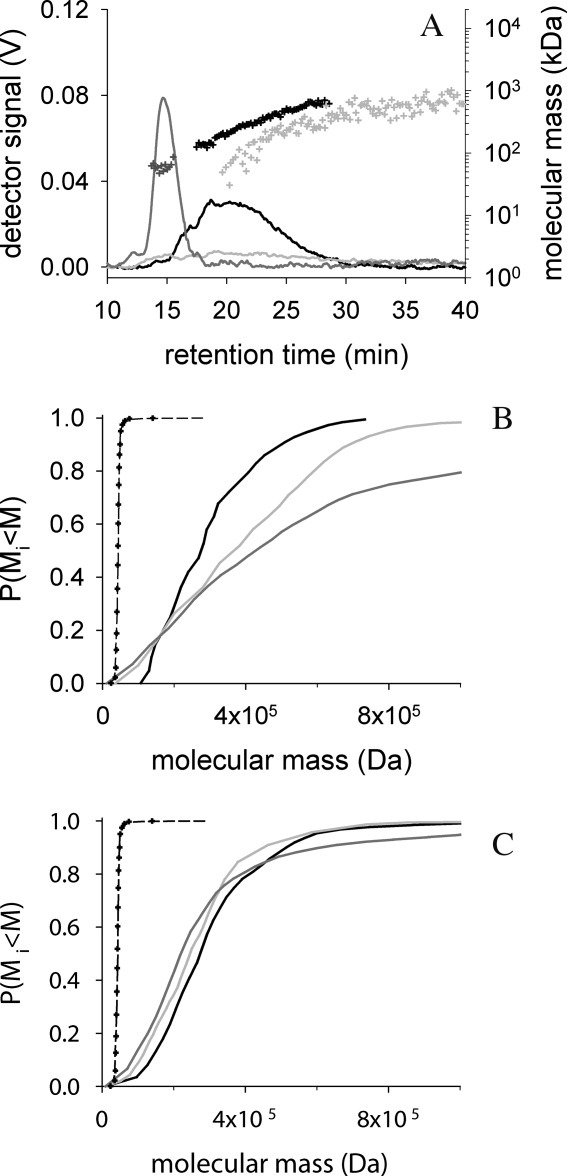
Effect of l-ArgHCl on distribution of rPA species in AF4 experiments. (A) The chromatograms of AF4-measurements in 100 mMl-ArgHCl for 6 μg native rPA (dark gray line) and 7 μg IAA-rPA after focusing for 3 min (black line) and 180 min (gray line), respectively, are shown. The channel height was 490 μm. The channel flow was 1 mL min−1, the focus flow 0.75 mL min−1, and the cross flow 2 mL min−1. The corresponding molecular masses (dots) were calculated from UV-absorption and light scattering signals; for clarity, only every 20th data point is plotted. (B) The cumulative mass distributions of IAA-rPA in 100 mMl-ArgHCl were calculated from the experiments shown in panel (A). Elution was started after 3 min (black line), 60 min (grey line) and 180 min (dark grey line). As reference the cumulative mass distributions of native rPA in 200 mMl-ArgHCl are plotted after 3 min (black dashed line) and after 50 min of focusing (dots). (C) Cumulative mass distributions of IAA-rPA were determined in 500 mMl-ArgHCl. Data are represented as described for panel (B).
Native rPA eluted in a single peak containing a single species of apparently 43 kDa under all tested conditions, in agreement with monomeric protein [Fig. 6(A)]. The ratio of mass-average molecular mass to average molecular mass, Mw/Mn, was 1.0 in all cases, indicating a homogeneous species. IAA-rPA in turn, eluted in a broad peak in a continuous distribution of molecular masses up into the MDa range. In the presence of 100 mM l-ArgHCl, the observed molecular mass distribution for IAA-rPA was dependent on the duration of the focusing phase [Fig. 6(B)]. When the protein was held under focusing conditions from 3 to 180 min, the Mw/Mn ratio increased from 1.2 to 2.9, indicating increasingly populated species of high molecular mass. Along with these changes, the overall percentage of recoverable protein material decreased over focusing time, probably due to the formation of macroscopic aggregates that could no longer be detected by the AF4 method. When the l-ArgHCl concentration was increased to 500 mM, the observable distribution of molecular masses became stable over time [Fig. 6(C)]. The Mw/Mn ratio stayed constant at 1.4 over the entire time range covered by the represented experiment.
Under conditions of a stable molecular mass distribution, analytical ultracentrifugation experiments became possible. The measurements were performed in the presence of 1M l-ArgHCl. Again, native rPA was included for comparison. Apparent sedimentation coefficients sapp were 1.55 S for the native and 2.75 S for the carboxymethylated protein, respectively. The sedimentation equilibrium measurements with rPA and IAA-rPA resulted in apparent molecular masses of 39 kDa for rPA and 103 kDa for IAA-rPA (Fig. 7). Both sets of results were in agreement with monomeric native protein and a mixture of mainly di- and trimeric forms of the modified protein in solution, respectively. To exclude the possibility that IAA-rPA oligomers had been formed by covalent linkage, for example, by formation of intermolecular disulfide bridges on air, the sedimentation equilibrium experiment was repeated with the modified protein dissolved in 4M GuHCl, pH 3 (data not shown). Under these conditions, IAA-rPA appeared as a monomer.
Figure 7.
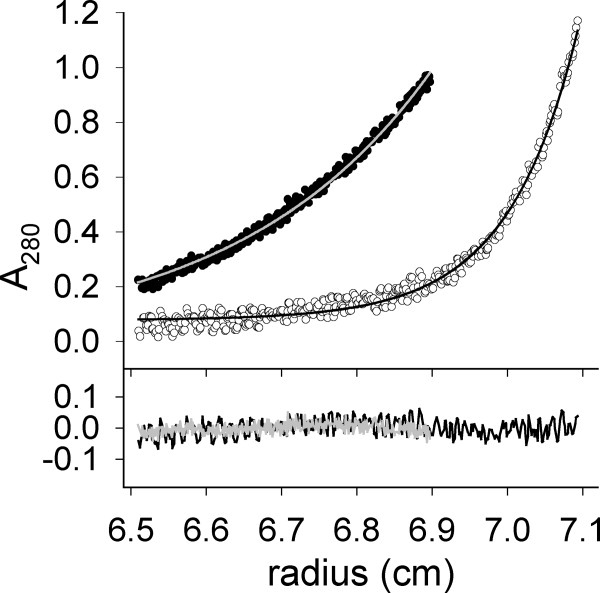
Analytical ultracentrifugation of rPA and IAA-rPA. Sedimentation equilibrium measurements were carried out as described in the text with 380 μg mL−1 rPA (open circles) and IAA-rPA (filled circles), respectively. Solid lines represent single-exponential fits to the data obtained for rPA (black) and IAA-rPA (grey), respectively. The lower panel shows the deviation of the fit from data.
Discussion
In an effort to understand the suppression of protein aggregation by l-ArgHCl, Baynes et al.27 proposed a mechanistic explanation based on the relative size of the arginine molecule, which is assumed to act as a neutral crowder compared with water.31 Such a neutral crowder would interact with protein surfaces with almost the same strength as water molecules. However, if two protein molecules interact, either to form a functional complex or as a step on the pathway to aggregation, gaps between these protein molecules would be transiently formed, before full surface-surface contact is established. In such a case, water molecules would still be able to enter gaps of a certain size, while a neutral crowder would not; this preferential exclusion of crowder molecules from the protein surface would create a thermodynamically unfavorable situation and therefore increase the free energy of the transition state of protein-protein contact formation.
If this postulated gap effect were valid, l-ArgHCl would exert an influence on the kinetics of protein aggregation alone and would not be expected to affect equilibrium solubilities. One of the purposes of the work presented in this article was to determine whether this proposition holds for the tested model protein rPA in its native and in its non-native S-carboxymethylated form, respectively. In the case of native rPA, a straightforward answer was obtained. The native protein equilibrated with a solid precipitate that could be easily re-solubilized into native, enzymatically active protein by simple dilution. Equilibrium solubilities could be determined and were strongly dependent on the concentration of l-ArgHCl in the solvent system. The presence of l-ArgHCl as a co-solvent lowers the chemical potential of the dissolved monomeric protein in solution with respect to its solid state. Any hypothesis that explains the stabilization of protein against aggregation by a mechanism acting on the transition state alone is therefore untenable. The increase in protein equilibrium solubility points into the same direction as recent measurements, in which a significant increase of the solubilities of a series of amino acids as well as of diketopiperazine in the presence of 1M arginine had been demonstrated.32
The only remaining possibility to defend the basic tenets of the neutral crowder hypothesis for arginine would be to hypothesize that the observed effects on equilibrium solubility are caused by a destabilization of the solid state with respect to all soluble species, possibly by molecular mechanisms akin to the gap effect. However, this idea may only be tested by future structural investigations of the solid state.
In the case of IAA-rPA, which was employed as an example for denatured protein states in solution, equilibrium solubilities could not be determined. The precipitated material could only be solubilized by denaturing treatment with GuHCl, but did not measurably equilibrate with the dissolved carboxymethylated protein in the nondenaturing measurement buffers. Apparently, the amorphous aggregate formed from the non-native protein species was thermodynamically much more stable than the solid precipitate formed by native protein. This stabilization might be tentatively ascribed to an increased conformational entropy of the entangled disordered polypeptide molecules in the solid produced from non-native protein, when compared with the well-ordered and structured molecules in the solid obtained by precipitation of native rPA.
Thus, for non-native protein species, the effect of l-ArgHCl on the apparent solubility is determined by its influence on aggregation kinetics. Interestingly, even in the presence of 1M l-ArgHCl, formation of precipitate from the concentrated stock solution of IAA-rPA was observed within minutes, whereas on the other hand, residual solubility was maintained for weeks at an almost constant level. This is in agreement with the very steep concentration dependence of the aggregation kinetics that was observed.
Measurement of the thermodynamic stability of rPA by GuHCl-induced folding/unfolding transition curves revealed no significant shift in the equilibrium between the folded and the unfolded state of native rPA in presence of l-ArgHCl, which is consistent with data published for lysozyme and RNase A.9,33–35 This means that the chemical potential of denatured rPA in solution is lowered parallel to the chemical potential of the folded protein. As it seems likely that the free energy of IAA-rPA is influenced by the same factors as the denatured protein, a similar decrease in the chemical potential of dissolved IAA-rPA by l-ArgHCl with respect to the solid state may be assumed.
The observed decrease in chemical potential of rPA by 14 kJ mol−1 in a solution containing 1M l-ArgHCl would lead to aggregation kinetics that are decelerated by a factor of ∼500, if the chemical potential of the transition state were assumed to be unaffected. However, a much steeper dependency of aggregation kinetics on the concentration of l-ArgHCl was observed for both native and carboxymethylated rPA; at the same protein concentration, aggregation was decelerated by more than 10 orders of magnitude in the presence of 500 mM l-ArgHCl. For all species under all conditions, the apparent size of the nucleation seeds was ∼10, with no significant differences. This indicates the existence of a series of pre-equilibria, which finally result in a critical transitional species in the process of protein aggregation. Thus, the increased apparent solubilities of IAA-rPA and of denatured reduced rPA in presence of l-ArgHCl can be explained by a shift of a series of pre-equilibria towards dissociation, which results in a reduced concentration of nucleation seeds without changing the formal mechanism of protein aggregation.
To summarize the described interpretation of the obtained results, we present an energy scheme for the effects of l-ArgHCl on the aggregation of rPA (Fig. 8). It has been shown that l-ArgHCl increases the solubility of both, the native (N) and the unfolded protein (U), which is equivalent to a reduction of free energy of the protein species in solution. To reflect the fact that l-ArgHCl does not significantly affect the thermodynamic stability of rPA, the relative positions of the native and unfolded state in solution remains unchanged in the presence of additive. The low solubility of rPA under physiological conditions makes the native-like aggregated state (AN) easily accessible. The addition of L-ArgHCl, however results in a relative destabilization of the AN-state versus the native protein in solution. Regrettably, our data do not permit a distinction between a true destabilization of the aggregated state via a gap effect-like mechanism and a stabilization of protein species in solution by interaction with, for example, arginine clusters as proposed by Das et al.36
Figure 8.
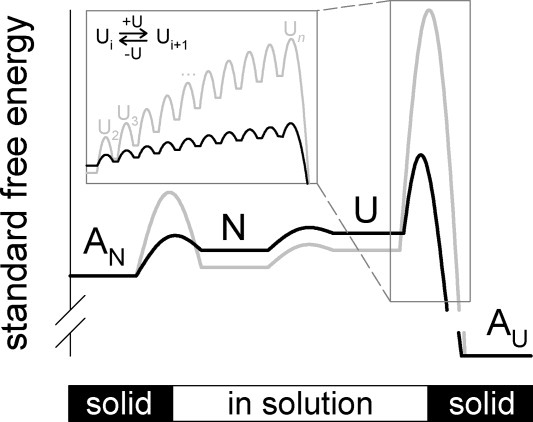
Proposed energy diagram for the effect of l-ArgHCl on protein states in solution. Black lines and letters represent a schematic energy diagram in absence, gray lines and letters symbolize changes in the presence of l-ArgHCl. Abbreviations are N, native rPA in solution; U, unfolded rPA in solution; AN, aggregates of native rPA; and AU, aggregates of unfolded rPA. The inset is meant to illustrate the free energy balance of pre-equilibria leading to formation of the nucleation seed Un via a series of oligomeric species U2, U3, …, etc.
The position of the aggregated state of the unfolded protein (AU) cannot be specified because of the practical irreversibility of aggregation of U under physiological conditions. For the transition state of U to AU a significantly increased free energy in presence of l-ArgHCl was observed, that may be explained by a shift of a series of pre-equilibria toward dissociation.
Over the last two decades, l-ArgHCl has become a widely used additive for in vitro protein refolding. In this article, we present clear evidence that l-ArgHCl acts by a massive increase in the equilibrium solubility of native rPA. Furthermore, we suggest that shifts of the pre-equilibria in the formation of protein aggregates towards dissociation are responsible for the apparently increased solubility of IAA-rPA, our model system for non-native protein states. Any proposed mechanistic model for the action of l-ArgHCl will have to take these findings into account.
Materials and Methods
Materials
A stock solution of pure native rPA (BM 06.022) in a concentration of 4 mg mL−1 was kindly provided by Roche Diagnostics Penzberg. EDTA, Tris base and DTT were purchased from MP Biomedicals LLC. l-Arginine and l-ArgHCl were from Ajinomoto, and bovine serum albumin, d,l-arginine and Tween 80 from Sigma Aldrich Chemie GmbH. GuHCl was kindly provided by Bioselect Nigu GmbH. Chromozyme t-PA was purchased from Roche Diagnostics Penzberg. All other reagents were of analytical grade or higher purity.
Preparation of carboxymethylated rPA (IAA-rPA)
rPA (4 mg mL−1) was denatured and reduced by adding 100 mM DTT to the protein solution, which then was dialyzed for 3 h against 100 volumes of buffer containing 6M GuHCl, 1 mM EDTA, 1 mM DTT, 100 mM Tris/HCl, pH 8.5. DTT was removed by dialyzing the protein solution three times against 50 volumes of 4M GuHCl, pH 3. Afterwards, 2.8 mg mL−1 of denatured reduced rPA were diluted into four volumes of 6M GuHCl, 1 mM EDTA, 100 mM boric acid/NaOH, pH 9, and 12.5 mM iodoacetic acid, incubated for 1 h at 22°C and dialyzed three times against 20 volumes of 4M GuHCl, pH 3.9
Mass spectrometry
For the determination of the molecular mass and the homogeneity of IAA-rPA-preparations, protein samples were desalted via RP-HPLC and analyzed by electron spray ionization-quadrupole-time of flight (Q-TOF)-mass spectrometry using a Q-TOF 2 mass spectrometer (Micromass, Manchester, UK).
Determination of protein concentrations
Protein concentrations were determined photometrically using an extinction coefficient ɛ280 of 1.69 cm2 mg−1 for native rPA28 and an ɛ280 of 1.635 cm2 mg−1 for denatured reduced and IAA-rPA, which was calculated from the amino acid sequence according to Gill and von Hippel.37
Solubility of native rPA and IAA-rPA
For solubility measurements in l-ArgHCl, native rPA stock solution and IAA-rPA in 4M GuHCl, pH 3, respectively, were concentrated using centrifugal ultrafiltration devices (up to 93 mg mL−1 in the case of native rPA), and subsequently dialyzed at 6°C for 48 h against 200 volumes of 1 mM EDTA, 100 mM Tris/HCl, pH 8.5, containing the indicated concentrations of l-ArgHCl. Solubility measurements in NaCl and NaCl/glycine were carried out in an analogous manner. Due to the low solubility of rPA in the NaCl-containing buffers, prior concentration of the protein solutions was not necessary in this case. After dialysis, precipitates were removed by centrifugation for 30 min at 15,000g, 4°C and following ultracentrifugation at 150,000g, 4°C for 20 min. Protein concentrations in the supernatant were determined as described above. For measurement of equilibrium solubilities, rPA was concentrated up to 84 mg mL−1 and dialyzed at 6°C for 24 h against 200 volumes of 1 mM EDTA, 100 mM Tris/HCl, pH 8.5. Afterwards, the aggregated protein suspension was treated as described above. Measurements of IAA-rPA solubility and equilibrium solubility in l-ArgHCl, NaCl, and NaCl/glycine, respectively, were carried out in principle as mentioned above, but the protein was used in concentrations ranging from 0.45 to 4.7 mg mL−1 and the transfer into aggregates was achieved by dialysis of the protein solution for 24 h at 6°C against ∼1000 volumes of 1 mM EDTA, pH 3. The duration of ultracentrifugation at 150,000g and 4°C was prolonged to 1 h.
Measurement of aggregation kinetics
Aggregation was monitored on a Fluoromax 3 spectrofluorimeter (Horiba Jobin Yvon, Edison, NJ) using a 1 cm stirrable fluorescence quartz cell at 20°C. For all measurements, aggregation was monitored via the intensity of scattered light at 600 nm. All buffers were filtered immediately before use. Native rPA was dialyzed for 48 h against 200 volumes of 200 mM l-ArgHCl, 1 mM EDTA, 100 mM Tris/HCl, pH 8.5 and then treated as described above. Aggregation of native rPA was monitored after dilution to an end concentration of l-ArgHCl of 10 mM. IAA-rPA was ultracentrifuged at 150,000g, 4°C for 20 min and diluted into renaturation buffer consisting of 200 mM GuHCl, transferred from the IAA-rPA stock solution, 1 mM EDTA, 100 mM Tris/HCl, pH 8.5 and no, 200 or 500 mM l-ArgHCl.
Denatured reduced rPA was prepared by adding 100 mM DTT to native rPA, which then was dialyzed for 10 h against 100 volumes of buffer containing 6M GuHCl, 1 mM EDTA, 1 mM DTT, 100 mM Tris/HCl, pH 8.5. After addition of 100 mM fresh DTT, denatured reduced rPA was dialyzed against 100 volumes of 4M GuHCl, 5 mM DTT, 1 mM EDTA, 100 mM Tris/HCl, pH 8.5 for 12 h. For the measurement of aggregation, denatured reduced rPA was diluted into renaturation buffer with 200 mM GuHCl, 10 mM DTT and 200 mM or 500 mM l-ArgHCl.
Activity of rPA
Enzymatic activity of rPA was measured by cleavage of 0.4 mM chromozyme t-PA (N-Methylsulfonyl-d-Phe-Gly-Arg-4-nitroanilide acetate) in 90 mM l-ArgHCl, 1 mM EDTA, 0.13% (w/v) Tween 80, 90 mM Tris/HCl, pH 8.5 at 37°C. The increase of the released 4-nitroaniline was monitored photometrically at 405 nm. The slope of the initial 60 s of the reaction was taken and compared with a simultaneously measured reference sample with a known protein concentration. Hydrolytic activities are presented as the percentage of native enzymatic activity.
Measurement of far- and near-UV CD spectra
Far-UV CD of native rPA and IAA-rPA, respectively, had to be recorded in the absence of l-ArgHCl. For that purpose, samples were dialyzed four times for 24 h against 50 volumes of 4M GuHCl, pH 3, followed by two dialysis steps against 10 volumes of buffer containing 100 mM Tris/HCl, pH 8.5, 1 mM EDTA and 1M d,l-ArgHCl. Because of the high optical absorption of d,l-ArgHCl, renatured rPA and IAA-rPA were diluted into nine volumes of 1 mM EDTA, 100 mM Tris/HCl, pH 8.5. Protein concentration were determined photometrically after centrifugation for 1 h at 150,000g. Far-UV CD spectra of denatured IAA-rPA and of denatured reduced rPA were recorded in 4M GuHCl, pH 3. Far-UV CD spectra between 200 and 250 nm wavelength were recorded in 0.1 mm quartz cells using a Jasco J-810 spectropolarimeter (Jasco, Easton, MD). Spectra were accumulated 16 times with a scan rate of 20 nm min−1. The temperature for all measurements was 20°C. Near-UV CD spectra of rPA and IAA-rPA under nondenaturing conditions were recorded after dialyzing the samples two times for 24 h against 50 volumes of 1M l-ArgHCl, 1 mM EDTA, 100 mM Tris/HCl, pH 8.5. Samples were centrifuged for 1 h at 150,000g and 4°C before photometric determination of protein concentrations. Spectra under denaturing conditions were recorded in 4M GuHCl, pH 3. Near-UV CD spectra were measured between 260 and 350 nm in 1 cm quartz cells, and accumulated 50 times with a scan rate of 50 nm min−1.
Measurement of GuHCl-induced folding/unfolding transition curves
For measuring GuHCl-induced unfolding transition-curves by far-UV CD, protein samples were diluted from stock solutions into nine volumes of 100 mM Tris/HCl, pH 8.5, containing 1 mM EDTA, and the indicated concentrations of GuHCl, to final concentrations of 440 μg mL−1 rPA, and 15 μg mL−1 to 193 μg mL−1 IAA-rPA, respectively. The protein stock solutions contained 1M d,l-ArgHCl, 1 mM EDTA, and 100 mM Tris/HCl, pH 8.5. To remove aggregates, IAA-rPA samples were centrifuged for 1 h at 150,000g, 4°C immediately before the experiments. Indicated protein concentrations were determined after this centrifugation step. The measurements were carried out in quartz cells with path lengths of 0.1 or 0.5 mm, and CD signals were monitored at a wavelength of 215 nm. For monitoring GuHCl-induced unfolding by fluorescence emission, protein solutions containing 1 mg mL−1 rPA or IAA-rPA in 1M l-ArgHCl, 1 mM EDTA, 100 mM Tris/HCl, pH 8.5, were diluted into 99 volumes of 100 mM Tris/HCl, pH 8.5, containing 1 mM EDTA and the indicated concentrations of GuHCl and incubated for 24 h at 25°C. For refolding transition curves, the protein was dialyzed two times for 24 h against 200 volumes of 4M GuHCl, pH 3, followed by dilution into 99 volumes of 100 mM Tris/HCl, pH 8.5, containing 1 mM EDTA, and the indicated concentrations of GuHCl and incubation for 24 h at 25°C. Fluorescence emission spectra were recorded using a Fluoromax 3 spectrofluorimeter (Horiba Jobin Yvon, Edison, NJ). The excitation wavelength was 280 nm. Fluorescence emission was recorded between 290 and 440 nm. Spectra were accumulated three times. All measurements were performed at 20°C. Concentrations of GuHCl were determined refractometrically.38
Analytical ultracentrifugation
Before the analytical ultracentrifugation experiments, rPA and IAA-rPA, respectively, were dialyzed for 48 h against 200 volumes of 1M l-ArgHCl, 1 mM EDTA, 100 mM Tris/HCl, pH 8.5, and diluted to a final concentration of 0.38 mg mL−1 in dialysis buffer. Experiments were performed at 20°C in an Optima XL-A (Beckman, Palo Alto, CA) centrifuge using double sector cells and an AnTi50 rotor. Sedimentation velocity and equilibrium runs were carried out at a rotor speed of 40,000 and 12,000 rpm, respectively. Optical scans at 280 nm were recorded every 10 min. Under the employed experimental conditions, protein concentration at the bottom of the cells was ∼3 mg mL−1 during the equilibrium runs.
Asymmetric flow field flow fractionation/light scattering (AF4/LS)
Asymmetric flow field flow fractionation (AF4)39 is a powerful method for the analytical separation of polymers and particles in solution according to size. Separation takes place in a flow channel with one side made up of a semipermeable membrane. While the sample passes along the channel, buffer cross-flow through the semipermeable wall creates a net force perpendicular to the direction of the main channel flow. The molecular species in the sample assume a mass-dependent distribution in the resulting flow field, with larger species being on average closer to the wall than smaller ones. When a laminar flow velocity profile is established along the channel, this situation leads to small species being eluted more quickly than larger ones. By controlling the cross-flow, the separation range of the instrument may be adjusted to cover the entire size range from virus particles and large aggregates to small monomeric proteins (cf., e.g., Refs. 40,41). This analytical separation method may be ideally coupled to an in-line multi-angle laser light scattering (MALS) detector.42 Combined with a concentration detector, for example, a UV absorption or a differential refractive index detector, MALS allows for the direct determination of weight-average molecular masses, MW, of the species in the eluent flow of the AF4. As the concentration detector delivers an independent mass-proportional signal, other molecular mass moments, that is, the average molecular masses, MN, and the z-average molecular masses, MZ, can be calculated as well, and the ratios of these mass moments then serve as indicators of sample homogeneity.
In this work, AF4 measurements were carried out using a Wyatt Eclipse 2 separation system (Wyatt Technology Europe GmbH, Dernbach, Germany) coupled to an Agilent G 1310A isocratic HPLC pump and an Agilent G 1322A degasser (Agilent Technologies, Palo Alto, CA). The height of the separation channel was 490 μm, and the applied membrane consisted of regenerated cellulose with a 10 kDa cutoff. An LKB 2152 UV detector (LKB, Bromma, Sweden) and a Mini Dawn Tristar multi-angle light scattering detector (Wyatt Technology, Santa Barbara, CA) were coupled to the system.
For the experiments, rPA and IAA-rPA were dialyzed against 200 volumes of 1 mM EDTA, 100 mM Tris/HCl, pH 8.5, and 100 mM or 500 mM l-ArgHCl. To remove aggregates, IAA-rPA samples were centrifuged for 30 min at 15,000g and 4°C, followed by an ultracentrifugation step at 150,000g and 4°C for 1 h. Afterwards, protein concentrations were determined photometrically. Samples were injected into the separation channel through a 100 μL loop. The dialysis buffers were used as carrier liquids. Before usage, carrier liquid was filtered with 0.1 μm membranes. For all measurements, inject flow was 0.2 mL min−1, carrier liquid flow was 1 mL min−1, focus flow was 0.75 mL min−1, and the cross flow during the elution phase was 2 mL min−1. AF4 measurements consisted of an equilibration phase of 4 min, and a focusing phase with a duration between 3 and 180 min, followed by an elution phase which was extended to up to 120 min. For the determination of molecular masses from UV absorption and light scattering signals, bovine serum albumin was used as a calibration standard.
Acknowledgments
Rainer Rudolph passed away on December 5, 2009. We lost a great scientist and mentor; his guidance and defining contribution will always be remembered. The authors want to thank Dr. Angelika Schierhorn of the Max-Planck Research Unit Halle for the mass spectrometric analyses and Roche Diagnostics Penzberg for their generous gift of rPA.
References
- 1.Rudolph R, Lilie H. In vitro folding of inclusion body proteins. FASEB J. 1996;10:49–56. [PubMed] [Google Scholar]
- 2.Fahnert B, Lilie H, Neubauer P. Inclusion bodies: formation and utilization. Adv Biochem Eng Biotechnol. 2004;89:93–142. doi: 10.1007/b93995. [DOI] [PubMed] [Google Scholar]
- 3.Lange C, Patil G, Rudolph R. Ionic liquids as refolding additives: N′-alkyl and N′-(ω-hydroxyalkyl) N-methylimidazolium chlorides. Protein Sci. 2005;14:2693–2701. doi: 10.1110/ps.051596605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burgess RR. Refolding solubilized inclusion body proteins. Methods Enzymol. 2009;463:259–282. doi: 10.1016/S0076-6879(09)63017-2. [DOI] [PubMed] [Google Scholar]
- 5.Kiefhaber T, Rudolph R, Kohler H-H, Buchner J. Protein aggregation in vivo: a quantitative model of the kinetic competition between folding and aggregation. Biotechnology (NY) 1991;9:825–829. doi: 10.1038/nbt0991-825. [DOI] [PubMed] [Google Scholar]
- 6.Lee JC, Timasheff SN. The stabilization of proteins by sucrose. J Biol Chem. 1981;256:7193–7201. [PubMed] [Google Scholar]
- 7.Gekko K, Timasheff SN. Mechanism of protein stabilization by glycerol: preferential hydration in glycerol-water mixtures. Biochemistry. 1981;20:4667–4676. doi: 10.1021/bi00519a023. [DOI] [PubMed] [Google Scholar]
- 8.Arakawa T, Tsumoto K, Kita Y, Chang B, Ejima D. Biotechnology applications of amino acids in protein purification and formulations. Amino Acids. 2007;33:587–605. doi: 10.1007/s00726-007-0506-3. [DOI] [PubMed] [Google Scholar]
- 9.Reddy KRC, Lilie H, Rudolph R, Lange C. l-Arginine increases the solubility of unfolded species of hen egg white lysozyme. Protein Sci. 2005;14:929–935. doi: 10.1110/ps.041085005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armstrong N, de Lencastre A, Gouaux E. A new protein folding screen: application to the ligand binding domains of a glutamate and kainate receptor and to lysozyme and carbonic anhydrase. Protein Sci. 1999;8:1475–1483. doi: 10.1110/ps.8.7.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiu FF, Venkatesan N, Wu CR, Chou AH, Chen HW, Lian SP, Liu SJ, Huang CC, Lian WC, Chong P, Leng CH. Immunological study of HA1 domain of hemagglutinin of influenza H5N1 virus. Biochem Biophys Res Commun. 2009;383:27–31. doi: 10.1016/j.bbrc.2009.03.106. [DOI] [PubMed] [Google Scholar]
- 12.Wang XT, Engel PC. An optimised system for refolding of human glucose 6-phosphate dehydrogenase. BMC Biotechnol. 2009;9:19–29. doi: 10.1186/1472-6750-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buchner J, Rudolph R. Renaturation, purification and characterization of recombinant Fab-fragments produced in Escherichia coli. Biotechnology (NY) 1991;9:157–162. doi: 10.1038/nbt0291-157. [DOI] [PubMed] [Google Scholar]
- 14.Rudolph R, Fischer S. 1990. Process for obtaining renatured proteins. US Patent. 4,933,434.
- 15.Lange C, Rudolph R. Suppression of protein aggregation by l-arginine. Curr Pharm Biotechnol. 2009;10:408–414. doi: 10.2174/138920109788488851. [DOI] [PubMed] [Google Scholar]
- 16.Cohn EJ. The relation between the iso-electric point of a globulin and its solubility and acid combining capacity in salt solution. Proc Natl Acad Sci USA. 1920;6:256–263. doi: 10.1073/pnas.6.5.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green AA. Studies in the physical chemistry of the proteins: VIII. The solubility of haemoglobin in concentrated salt solutions. A study of the salting out of proteins. J Biol Chem. 1931;93:495–516. [Google Scholar]
- 18.Green AA. Studies in the physical chemistry of the proteins. IX. The effect of electrolytes on the solubility of haemoglobin in solutions of varying hydrogen ion activity with a note on the comparable behaviour of casein. J Biol Chem. 1931;93:517–542. [Google Scholar]
- 19.Arakawa T, Timasheff SN. Abnormal solubility behavior of beta-lactoglobulsalting-in by glycine and NaCl. Biochemistry. 1987;26:5147–5153. doi: 10.1021/bi00390a038. [DOI] [PubMed] [Google Scholar]
- 20.Ruckenstein E, Shulgin IL. Effect of salts and organic additives on the solubility of proteins in aqueous solutions. Adv Colloid Interface Sci. 2006;123–126:97–103. doi: 10.1016/j.cis.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 21.Hofmeister F. Zur Lehre von der Wirkung der Salze. II. Arch Exp Pathol Pharmakol. 1888;24:247–260. [Google Scholar]
- 22.Nandi PK, Robinson DR. The effects of salts on the free energy of the peptide group. J Am Chem Soc. 1972;94:1299–1308. doi: 10.1021/ja00759a042. [DOI] [PubMed] [Google Scholar]
- 23.Riés-Kautt MM, Ducruix AF. Relative effectiveness of various ions on the solubility and crystal growth of lysozyme. J Biol Chem. 1989;264:745–748. [PubMed] [Google Scholar]
- 24.Zangi R. Can salting-in/salting-out ions be classified as chaotropes/kosmotropes? J Phys Chem B. 2010;114:643–650. doi: 10.1021/jp909034c. [DOI] [PubMed] [Google Scholar]
- 25.Timasheff SN, Inoue H. Preferential binding of solvent components to proteins in mixed water-organic solvent systems. Biochemistry. 1968;7:2501–2513. doi: 10.1021/bi00847a009. [DOI] [PubMed] [Google Scholar]
- 26.Timasheff SN. Protein hydration, thermodynamic binding, and preferential hydration. Biochemistry. 2002;41:13473–13482. doi: 10.1021/bi020316e. [DOI] [PubMed] [Google Scholar]
- 27.Baynes BM, Wang DIC, Trout BL. Role of arginine in the stabilization of proteins against aggregation. Biochemistry. 2005;44:4919–4925. doi: 10.1021/bi047528r. [DOI] [PubMed] [Google Scholar]
- 28.Kohnert U, Rudolph R, Verheijen JH, Weening-Verhoeff EJD, Stern A, Prinz H, Lechner M, Kresse G-B, Buckel P, Fischer S. Biochemical properties of the kringle 2 and protease domains are maintained in the refolded t-PA deletion variant BM 06.022. Protein Eng. 1992;5:93–100. doi: 10.1093/protein/5.1.93. [DOI] [PubMed] [Google Scholar]
- 29.Hu CK, Kohnert U, Stürzebecher J, Fischer S, Llinás M. Complexation of tissue plasminogen activator protease with benzamidine-type inhibitors: interference by the kringle 2 module. Biochemistry. 1996;35:3270–3276. doi: 10.1021/bi9515026. [DOI] [PubMed] [Google Scholar]
- 30.Hofrichter J, Ross PD, Eaton WA. Kinetics and mechanism of deoxyhemoglobin S gelation: a new approach to understanding sickle cell disease. Proc Natl Acad Sci USA. 1974;71:4864–4868. doi: 10.1073/pnas.71.12.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baynes BM, Trout BL. Rational design of solution additives for the prevention of protein aggregation. Biophys J. 2004;87:1631–1639. doi: 10.1529/biophysj.104.042473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arakawa T, Ejima D, Tsumoto K, Obeyama N, Tanaka Y, Kita Y, Timasheff SN. Suppression of protein interactions by arginine: a proposed mechanism of the arginine effects. Biophys Chem. 2007;127:1–8. doi: 10.1016/j.bpc.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 33.Taneja S, Ahmad F. Increased thermal stability of proteins in the presence of amino acids. Biochem J. 1994;303:147–153. doi: 10.1042/bj3030147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arakawa T, Tsumoto K. The effects of arginine on refolding of aggregated proteins: not facilitate refolding, but suppress aggregation. Biochem Biophys Res Commun. 2003;304:148–152. doi: 10.1016/s0006-291x(03)00578-3. [DOI] [PubMed] [Google Scholar]
- 35.Nakakido M, Tanaka Y, Mitsuhori M, Kudou M, Ejima D, Arakawa T, Tsumoto K. Structure-based analysis reveals hydration changes induced by arginine hydrochloride. Biophys Chem. 2008;137:105–109. doi: 10.1016/j.bpc.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 36.Das U, Gururao H, Abdul ES, Pallavi M, Das T, Pasha S, Mann M, Ganguli M, Verma AK, Bhat R, Chandrayan SK, Ahmed S, Sharma S, Kaur P, Singh TP, Srinivasan A. Inhibition of protein aggregation: supramolecular assemblies of arginine hold the key. PLoS One. 2007;11:e1176. doi: 10.1371/journal.pone.0001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 38.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 39.Wahlund KG, Giddings JC. Properties of an asymmetrical flow field-flow fractionation channel having one permeable wall. Anal Chem. 1987;59:1332–1339. doi: 10.1021/ac00136a016. [DOI] [PubMed] [Google Scholar]
- 40.Fraunhofer W, Winter G. The use of asymmetrical flow field-flow fractionation in pharmaceutics and biopharmaceutics. Eur J Pharm Biopharm. 2004;58:369–383. doi: 10.1016/j.ejpb.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 41.Roda B, Zattoni A, Reschiglian P, Moon MH, Mirasoli M, Michelini E, Roda A. Field-flow fractionation in bioanalysis: a review of recent trends. Anal Chim Acta. 2009;635:132–143. doi: 10.1016/j.aca.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 42.Harding SE, Jumel K. Light scattering. Curr Protoc Protein Sci. 1998:7.8.1–7.8.14. doi: 10.1002/0471140864.ps0708s11. [DOI] [PubMed] [Google Scholar]