

Abstract
In the study of rabbit muscle pyruvate kinase (M1-PYK), proline has previously been used as an osmolyte in an attempt to determine a role for preexisting conformational equilibria in allosteric regulation. In this context, osmolytes are small molecules assumed to have no direct interaction with the protein. In contrast to proline's proposed role as an osmolyte, the structure of M1PYK-Mn-pyruvate-proline complex reported herein demonstrates that proline binds specifically to the allosteric site of M1-PYK. Therefore, this amino acid is an allosteric effector rather than a benign osmolyte. Other compounds often used as osmolytes (polyethyleneglycol and glycerol) are also present in the structure, suggesting an interaction with the protein that would, in turn, prevent the usefulness of these compounds in the study of this and most likely other proteins. These findings highlight the need to verify that compounds used as osmolytes to perturb preexisting conformational equilibrium do not directly interact with the protein, a consideration not commonly addressed in the past.
Keywords: allosteric regulation, allostery, pyruvate kinase, M1-PYK, osmolyte, preexisting equilibrium
Statement of Significance for Broader Audience
Osmolyte additions have been used as a means of testing for preexisting equilibrium in allosteric proteins. Before such studies can be useful, the compound used as an osmolyte should not directly interact with the protein of interest. Unfortunately, direct interactions of osmolytes with the protein are seldom evaluated. We show in the case of the pyruvate kinase isozyme found in brain and muscle (M1-PYK) that the compounds previously used as osmolytes directly interact with the protein.
M1-PYK is allosterically inhibited by hydrophobic amino acids.1 Phenylalanine is the inhibitor most often included in allosteric studies. Inhibition results in a decreased affinity of the enzyme for its substrate, phosphoenolpyruvate (PEP). Although the physiological relevance of this inhibition is not well understood, M1-PYK isolated from rabbit muscle has become a model system to study the molecular mechanism of heterotropic K-type allosteric regulation (i.e., the impact that binding of one ligand to a protein has on the protein's affinity for a second, chemically nonidentical ligand2).
One model that has been proposed to describe the allosteric regulation of M1-PYK is based on a preexisting equilibrium between two end-state conformations (see Supporting Information).3 The ability of changes in solution conditions to impact M1-PYKs affinity for both PEP and phenylalanine and to impact the degree homotropic cooperativity in ligand binding has been used as support for this two-state model.4,5 In these studies, osmolytes (inert space-filling solutes) were added to impact the proposed preexisting equilibrium between two conformations. In this context, osmolytes are small molecules that influence the physical properties of water and do not directly interact with the protein. Should these compounds interact directly with the protein, the observed changes in protein properties can result from this direct interaction rather than, or in addition to any potential osmolyte effects. In the previous study, two of the three chemicals chosen to function as osmolytes were amino acids, namely glycine and proline.
In contrast to considering glycine and proline as osmolytes, we have found a series of amino acids to act as allosteric regulators of M1-PYK.1 In our studies, the l-2-aminopropanaldehyde substructure of the amino acid is most important to ligand binding. The hydrophobic bulk of the side chain (not its aromatic nature) is responsible for eliciting the allosteric response. Both glycine and proline were included in our previous studies and evidence that both bind competitively with phenylalanine was provided. Proline, but not glycine, elicits an allosteric response in the protein's affinity for PEP. Structural studies reported herein were initiated to provide further evidence that proline is an allosteric effector (specifically to illustrate that proline binds to the allosteric site of M1-PYK) rather than functioning as a benign noninteracting osmolyte.
Results
A crystal of the M1-PYK-Mn-pyruvate-proline complex was grown as previously described,1 with the exception that proline was substituted for alanine. In contrast to the rapid two-day growth of crystals in the presence of alanine, the crystal grown in the presence of proline required ∼2 weeks. Diffraction data on a crystal maintained at 100 K were collected to a final resolution of 2.4 Å [Fig. 1(A), Table I] using an RU3HR copper rotating anode and RaxisIV++ detector. Like the previously defined structure with alanine bound, the structure with proline bound has two tetramers present in the asymmetric unit. The proline structure is essentially identical to the M1-PYK-alanine structure as indicated by the overall Cα RMSD of 0.3 Å. Similarities are easily appreciated by viewing an overlay of the backbone of subunits from structures with both alanine and proline bound [Fig. 1(B)]. Proline binds to the allosteric site of M1-PYK [Fig. 1(C)] similar to the way alanine binds1 (see Supporting Information); note that this amino acid binding site is not the same as the fructose-1,6-bisphosphate binding site found in other isozymes of pyruvate kinase.
Figure 1.
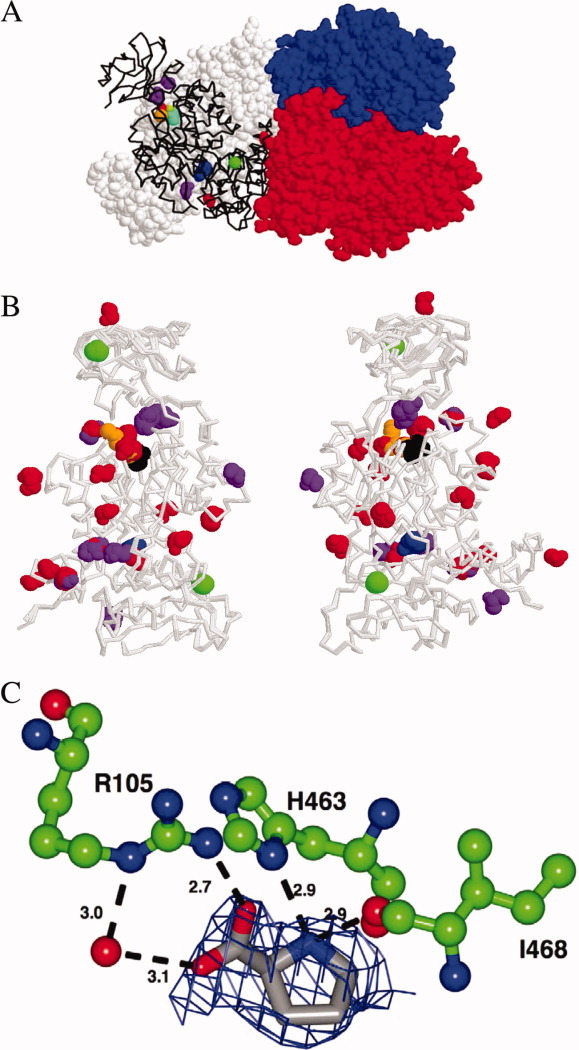
A: Tetramer of M1-PYK with proline bound. Three of the four subunits are in spacefill in red, blue, and white. Ligands (in spacefill) bound to the fourth subunit (black backbone) are glycerol (purple), pyruvate (cyan), K+ (orange), Na+ (green), Mn2+ (lime green), ethylene glycol (red), and proline (blue). B: Two views of an overlay of all 16 subunits of the M1-PYK structures with proline bound and with alanine bound1 included to summarize protein–small molecule interactions represented. Each of the two structures (i.e., one with proline bound and one with alanine bound) has two tetramers in the asymmetric unit, giving rise to the 16 subunits included in the overlay. Backbones of all 16 subunits are gray. To improve contrast, coloring of ligands differs slightly from that in panel A: Na+ (green), glycerol (purple), ethylene glycol (red), pyruvate (black), alanine/proline (blue), and polyethyleneglycol (orange). K+ and Mn2+ are not shown in B. Not all ligands are present in any one subunit. The same site on a subunit can be occupied by glycerol on some subunits but by ethylene glycol in other subunits. Likewise, the site occupied by polyethyleneglycol in one subunit is occupied by ethylene glycol in other subunits. C: Proline bound in allsoteric site of M1-PYK. Those residues directly interacting with proline are illustrated as green ball-and-stick model colored by atom type. A water molecule is represented as an oxygen atom. The potential hydrogen bond distances are given in Angstroms and illustrated by a dashed black line. Proline bound in the allosteric site is rendered as a grey stick model colored by atom type. Fo-Fc density prior to inclusion of proline into the model is rendered at 2.2 s as a blue mesh.
Table I.
Data and Model Statistics for the 2.4 Å Structure of Rabbit Muscle Pyruvate Kinase in Complex With Pyruvate and Proline
| Wavelength | 1.54 Å |
| Space group | P 1 |
| Unit cell | a = 82.4, b = 108.7, c = 144.3 (Å) |
| α = 95.2, β = 93.4, γ = 112.2 (°) | |
| Resolution limits | 36.6–2.4 Å |
| Unique reflections | 139,524 |
| Completenessa (%; all data) | 83 (70) |
| Redundancya | 3.5 (3.0) |
| I/σ(I)a | 8.5 (2.1) |
| Rmergea | 0.09 (0.43) |
| Molecules/ASU | 8 |
| Rwork | 20.6 (30.1)% |
| Rfree | 26.8 (39.1)% |
| Average B factor (Å2) | Protein: 28.4 |
| Pyruvate: 20.5 | |
| Proline: 26.8 | |
| Water: 24.8 | |
| Estimated coordinate error based on maximum likelihood | 0.23 Å |
| Bond length RMSD | 0.02 Å |
| Bond angle RMSD | 1.97° |
Values in parentheses represent statistics for data in the highest resolution shells. The highest resolution shell comprises data in the range of 2.49–2.4 Å.
Subunits show a heterogeneity in many (but not all) of the locations at which Na+, polyethyleneglycol, ethylene glycol, and glycerol interact with the protein; some of these differences may be due to the lower resolution of the M1-PYK-proline structure. Independently, the 2.4 Å resolution of the M1-PYK-Mn-pyruvate-proline complex would not be sufficient to support the assignment of residues and ions, particularly the glycerol and ethylene glycol. However, the structure of this complex crystallizes under the identical conditions and space group as the high-resolution complex with Ala.1 Thus, the assignments of densities that are not consistent with water ions are influenced by the assignments on these same densities in the 1.65 Å structure. At the locations with variable occupancy of polyethyleneglycol, ethylene glycol, or glycerol, the structure of the backbone is equivalent regardless of the polyol occupancy. One glycerol binding site, immediately adjacent to the divalent cation in the active site, is occupied in all subunits; this is an interesting note given that glycerol is the only suitable cryoprotectant.
Discussion
Previous osmolyte studies with M1-PYK have been discussed as support for a model that includes preexisting equilibrium of two conformations.4,5 Unfortunately, the previous conclusions can be challenged as the osmolyte influence on monitored protein properties can be explained by mechanisms other than an allosteric mechanism based on preexisting conformational equilibria. To appreciate this, let us critically evaluate the ability of previous experimental designs to distinguish a role for preexisting equilibrium in the allostery of M1-PYK. In this critical assessment, the primary focus is placed on identifying potential molecular sources for allostery and how these sources relate to monitored protein functions. Three views of allostery (representing two molecular sources for allostery) are contrasted.
A traditional two-state model as represented in most textbooks suggests that the source of allostery originates from a shift of the equilibrium between two enzyme conformations. In this model, the two assumed protein conformations bind substrate, as well as effector, with different affinities. In the absence of ligands, the protein exists in an equilibrium between these two end-point conformations. The free energy difference between the two protein conformational states is constant under all solution conditions and independent of which ligands are or are not bound. Binding of either substrate or effector essentially locks the protein into one of the two conformations. Thus, the only impact changes in solution conditions (i.e., increasing osmolyte concentration) can have is on the ratio of the two conformations when there is no ligand present (i.e., free enzyme). Restated, any change in solution condition that impacts the equilibrium between the two conformations when the protein is unliganded, must alter ligand affinities, the extent of homotropic ligand cooperativity (defined in Ref.2), and the extent of heterotropic allostery (defined in Ref.2). Therefore, these protein properties are all interdependent, as well as being dependent on the distribution between conformations in the absence of ligands. This reasoning is the basis for the original studies that monitor ligand affinities as a function of osmolyte concentration as a means of testing for preexisting equilibrium.
To study the allosteric regulation of M1-PYK, we have previously applied a linked equilibrium analysis of allostery in which the protein's affinity for PEP is linked to the protein's affinity for phenylalanine.1 The source of allostery [i.e., allosteric coupling (Qax)] in a linked equilibrium view of allostery originates from the way a ligand (A) binds to the protein (E) differently when a second ligand (X) is or is not present2,6,7:
| (1) |
Binding of A in the absence of X considers two enzyme complexes (E and AE). Binding of A in the presence of X also considers two enzyme complexes (EX and AEX). Each of the four enzyme complexes in the associated thermodynamic box (free enzyme, enzyme–substrate, enzyme–effector, or substrate–enzyme–effector) can have unique properties (Fig. 2). This analysis makes no assumptions regarding protein conformation; each of the four enzyme complexes can be described as a single conformation, an equilibrium between multiple conformations, or an ensemble of many conformational substates. Therefore, if the impact of osmolyte concentration on the conformation/ensemble of E is different from the impact on the conformation/ensemble of EA, an osmolyte-dependent effect on Kia will be observed. In turn, if the affect of osmolyte concentration on Kia is not the same as the osmolyte influence on Kia/x, an osmolyte-dependent effect on Qax will be observed. The previous molecular crowding experiments did not attempt to monitor this final comparison between Kia and Kia/x.
Figure 2.
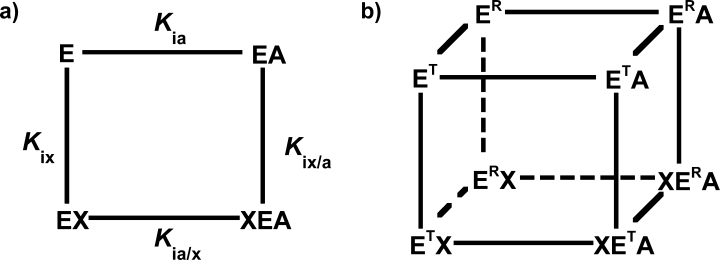
Energy cycles used to explain allosteric effects in rabbit M1-PYK. In both, the enzyme (E) can bind substrate (A) and/or effector (X). The thermodynamic box (a) makes no assumptions regarding the protein and therefore allows each enzyme complex to exist as a single conformational state, an equilibrium of a limited number of states, or an ensemble of many states (i.e., a dynamic structure). In contrast, the thermodynamic cube (b) constrains the protein to two potential conformations (ET or ER); the two conformations are assumed to (1) be equivalent independent of which ligands are or are not bound, (2) have different affinities for substrate, and (3) have different affinities for effector.3 Although the four respective equilibrium constants for the thermodynamic box are included in (a), the associated equilibrium constants are not included for the thermodynamic cube in (b).
Lee3 has recently reviewed data collected for M1-PYK in the context of a proposed expanded two-state model. The resulting thermodynamic cube (Fig. 2) can be viewed as an expansion of the thermodynamic box view of allostery we favor; the expansion being the consideration of each of the four enzyme complexes to a preexisting equilibrium between two protein conformations. Although this thermodynamic cube is an expansion in the number of protein species considered, the explicit consideration limiting the number of conformations to two should be viewed as a restriction compared with the thermodynamic box. The expanded two-state model deviates slightly from the typical textbook presentation of a two-state model; rather than substrate/effector binding locking the protein into exclusively one of the two conformations, ligand binding shifts the equilibrium between the two conformations. Interestingly, the expanded two-state model combines both potential sources of allostery as described for the other two models.
As an aside, much of the previous discussion of the expanded two-state model with regard to the allosteric regulation of M1-PYK focuses on the origin of allostery as a result of a shift in conformational equilibrium.3 The data used to justify this previous discussion have been collected at or near pH 7.0. Phenylalanine binds to M1-PYK with lower affinity at neutral pH than at basic pH.1 It follows that minimal quantity of the ternary complex (Phe-enzyme-PEP) can be formed at pH 7.0. Therefore, this ternary complex is largely unrepresented in the data analyzed in support of a conformational shift as the source of allostery in M1-PYK.3
In all three views of allostery, when the protein does not have ligands bound, any change in solution conditions (pH, temperature, salt or osmolyte concentration, or pressure) might drive a change in the number of protein molecules sampling each accessible conformation/substate (i.e., the unliganded distribution—whether limited to two states or considered as an ensemble—is dependent on solution conditions). In a model based on two conformations in equilibrium, this change in the unliganded conformational equilibrium is the source of allostery. In contrast, in the thermodynamic box view of allostery favored by us, the impact of the shift in the unliganded ensemble may or may not influence the allosteric coupling (Qax). It can now be appreciated that models that incorporate either of the two molecular sources of allostery can accommodate osmolyte-induced changes in the way A or X binds, when the second ligand is absent. Therefore, monitoring these properties as a function of osmolyte concentration cannot be used to confirm preexisting equilibrium in an allosteric system.
Even without these theoretical arguments, the previous studies can be challenged based on the small molecules used as osmolytes, primarily proline. The structural data provided here confirms that proline binds specifically to the previously identified binding site for amino acids effectors.1 Therefore, the structural data is consistent with our previous kinetic data1 and thus the effect proline has on the protein's affinity for PEP must be interpreted as an allosteric effect, and the effect on phenylalanine affinity must be due to competitive binding. Viewing the influence of proline as an allosteric effect is in direct contrast to its previously assumed role as an osmolyte.
Other compounds commonly used to induce molecular crowding are polyethyleneglycol and glycerol. However, the structures with either proline or alanine bound1 contain a polyethyleneglycol molecule (the precipitant used for crystallization), a number of molecules of ethylene glycol (a degradation product of polyethyleneglycol) and glycerol (the cryoprotectant). Even though these compounds may bind to M1-PYK weakly, when used as osmolytes, these molecules are added at high concentrations. This large effective concentration overcomes their weak affinity and results in the molecules populating specific binding sites on the protein. This is not a phenomenon specific to M1-PYK as the observation of ordered polyols and other cryoprotectant molecules is a widely observed phenomenon in high-resolution structures determined by cryo-crystallography. Therefore, it is likely that modification of protein conformation results from direct interactions of these compounds with the protein. On the basis of the observations presented herein, we conclude that these compounds should not be used as osmolytes to test for preexisting equilibrium in M1-PYK. More generally, we suggest that before any compound is used as an osmolyte to test for preequilibrium between conformations in a given protein, potential direct interactions between the protein and the potential osmolyte must be evaluated.
Acknowledgments
The authors appreciate the contributions of Amber Butyn during initial stages of structural refinement. Coordinates and structure factors have been deposited in the RCSB protein databank (http://www.rcsb.org/pdb) under the accession code 3N25.
Glossary
Abbreviations:
- M1-PYK
the pyruvate kinase isozyme found in mammal brain and muscle
- PEP
phospho(enol)pyruvate
- PYK
pyruvate kinase
References
- 1.Williams R, Holyoak T, McDonald G, Gui C, Fenton AW. Differentiating a ligand's chemical requirements for allosteric interactions from those for protein binding. Phenylalanine inhibition of pyruvate kinase(,) Biochemistry. 2006;45:5421–5429. doi: 10.1021/bi0524262. [DOI] [PubMed] [Google Scholar]
- 2.Fenton AW. Allostery: an illustrated definition for the “second secret of life.”. Trends Biochem Sci. 2008;33:420–425. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee JC. Modulation of allostery of pyruvate kinase by shifting of an ensemble of microstates. Acta biochimica et biophysica Sinica. 2008;40:663–669. doi: 10.1111/j.1745-7270.2008.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lonhienne TG, Reilly PE, Winzor DJ. Further evidence for the reliance of catalysis by rabbit muscle pyruvate kinase upon isomerization of the ternary complex between enzyme and products. Biophys Chem. 2003;104:189–198. doi: 10.1016/s0301-4622(02)00366-6. [DOI] [PubMed] [Google Scholar]
- 5.Lonhienne TG, Winzor DJ. Calorimetric demonstration of the potential of molecular crowding to emulate the effect of an allosteric activator on pyruvate kinase kinetics. Biochemistry. 2002;41:6897–6901. doi: 10.1021/bi020064h. [DOI] [PubMed] [Google Scholar]
- 6.Reinhart GD. Quantitative analysis and interpretation of allosteric behavior. Methods Enzymol. 2004;380:187–203. doi: 10.1016/S0076-6879(04)80009-0. [DOI] [PubMed] [Google Scholar]
- 7.Weber G. Ligand binding and internal equilibria in proteins. Biochemistry. 1972;11:864–878. doi: 10.1021/bi00755a028. [DOI] [PubMed] [Google Scholar]