

Abstract
SPP1 is a siphophage infecting the gram-positive bacterium Bacillus subtilis. The SPP1 tail electron microscopy (EM) reconstruction revealed that it is mainly constituted by conserved structural proteins such as the major tail proteins (gp17.1), the tape measure protein (gp18), the Distal tail protein (Dit, gp19.1), and the Tail associated lysin (gp21). A group of five small genes (22–24.1) follows in the genome but it remains to be elucidated whether their protein products belong or not to the tail. Noteworthy, an unassigned EM density accounting for ∼245 kDa is present at the distal end of the SPP1 tail-tip. We report here the gp23.1 crystal structure at 1.6 Å resolution, a protein that lacks sequence identity to any known protein. We found that gp23.1 forms a hexamer both in the crystal lattice and in solution as revealed by light scattering measurements. The gp23.1 hexamer does not fit well in the unassigned SPP1 tail-tip EM density and we hypothesize that this protein might act as a chaperone.
Keywords: phage SPP1, Siphoviridae, Bacillus subtilis, crystal structure
Introduction
Phages represent a highly diverse group of viruses infecting bacteria and are the most populated biological entity on earth.1 The vast majority of them belongs to the Caudovirales order and is composed by a double stranded DNA enclosed in an icosahedral capsid to which is attached a tail. More than 60% of known phages are member of the Siphoviridae family characterized by the presence of a long noncontractile tail, as is the case of SPP1. SPP1 is a virulent Bacillus subtilis phage encapsulating its genome in a 60-nm wide isometric capsid connected to a 160 nm long tail.2,3 Host infection is initiated by the binding of its tail-tip to YueB, a membrane protein with a large ectodomain protruding out of the thick peptidoglycan layer.4,5 This specific and irreversible interaction triggers a cascade of events resulting in DNA ejection into the B. subtilis cytoplasm.
The SPP1 tail and tail-tip have been thoroughly characterized by electron microscopy (EM) and X-ray crystallography resulting in the assignment of most of their components (gp17–gp21) to electron densities.2,6 However, a large volume of electron density remains unassigned at the tail-tip distal end. This observation associated with the identification of five gene products (gp22, gp23, gp23.1, gp24, and gp24.1) coded downstream of gp21, the most distally assigned tail component, led to hypothesize that these gps might be located in the unassigned tail-tip density.2 To bring further structural insights to prove or disprove this hypothesis, we have undertaken the structural study of these orphan gps and we have already reported the gp22 crystal structure.7
We report in this contribution the crystal structure of SPP1 gp23.1 at 1.6 Å resolution. Gp23.1 is a protein of 51 residues with an acidic pI of 3.8. The hexameric stoichiometry observed in solution, using light scattering measurements, is in agreement with the hexameric crystal structure. Based on the lack of bona fide, fitting of our X-ray structure into the tail-tip EM reconstruction and on the fact that we did not identify a catalytic cavity, we propose that gp23.1 might be a chaperone.
Results and Discussion
Gp23.1 crystallization and structure determination
Gp23.1 crystallized either in the orthorombic P212121 or in the hexagonal P6 space groups (Table I). Diffraction was comprised between 2.4 and 1.6 Å. Due to the difficulties encountered for phasing with only one methionine residue per monomer, we decided to introduce two additional methionines by mutating two consecutive leucine residues (Leu 23 and Leu 24). The quality of the Se-SAD data set collected on a crystal of the double mutant allowed us to find the 18 Se sites present in the six molecules of the asymmetric unit and then to phase and build an initial model. Final refinement statistics and models geometry are reported in Table I. A high similarity is observed between all monomers independently of the crystal space group.
Table I.
Data Collection and Refinement Statistics
| SPP1 gp23.1 | Se-peak data set | Native | Native |
|---|---|---|---|
| PDB accession code | 2XF5 | 2XF6 | 2XF7 |
| Data collectiona | |||
| Beamline | X06-SA (SLS) | BM-14 (ESRF) | BM-14 (ESRF) |
| Space group | P212121 | P6 | P212121 |
| Unit cell dimensions (Å) | a = 52.7, b = 66.5, c = 83.0 | a = 52.4, b = 52.4, c = 30.7 | a = 52.4, b = 66.9, c = 82.4 |
| Wavelength (Å) | 0.9795 | 1.7712 | 0.97372 |
| Resolution (Å) | 51.86–2.0 (2.11–2.0) | 45.38–2.12 (2.24–2.12) | 41.27–1.61 (1.70–1.61) |
| Rsymb (%) | 9.4 (46.4) | 5.6 (23.5) | 5.7 (42.1) |
| Mn(I)/σI | 16.0 (4.5) | 33.7 (8.2) | 21.4 (3.3) |
| Completeness (%) | 100 (100) | 99.7 (98.2) | 98.6 (90.6) |
| Redundancya | 10.7 (10.6) | 11.2 (8.3) | 6.3 (4.6) |
| Refinementa | |||
| Resolution (Å) | 44.5–2.0 (2.11–2.0) | 45.38–2.12 (2.37–2.12) | 22.73–1.61 (1.65–1.61) |
| Rwork/Rfree (%) | 16.91/19.74 | 19.1/23.4 | 16.8/20.0 |
| No. of reflections | 20,315 (2792) | 2819 (557) | 37,265 (2071) |
| r.m.s.d. bond lengths (Å)/angle (°) | 0.016/1.4 | 0.014/1.2 | 0.019/1.6 |
Values in parentheses refer to the highest resolution shell.
Rsym = ∑ (|I(h,i) − I(h)|))/∑ (I(h,i).
Gp23.1 monomer structure description
Gp23.1 is a 51-residue long all-α protein that adopts a three-helix bundle fold [Fig. 1(A)]. All residues are visible in the electron density maps (with some variations depending on the monomer considered) except for the first glycine of our construct. Starting from the N-terminus, we observe successively a small stretch of four residues connected to the first α-helix (α1, residues 6–16), a short-loop reaching α2 (residues 21–29) followed by another short-loop reaching α3 (residues 35–43) and finally an extended stretch at the protein C-terminus [Fig. 1(A)]. The overall arrangement of the three α-helices results in the projection of the N- and C-termini on opposite sides of the bundle. The gp23.1 fold is compact with most of the residues observed at the interface between the three α-helices forming a hydrophobic core.
Figure 1.
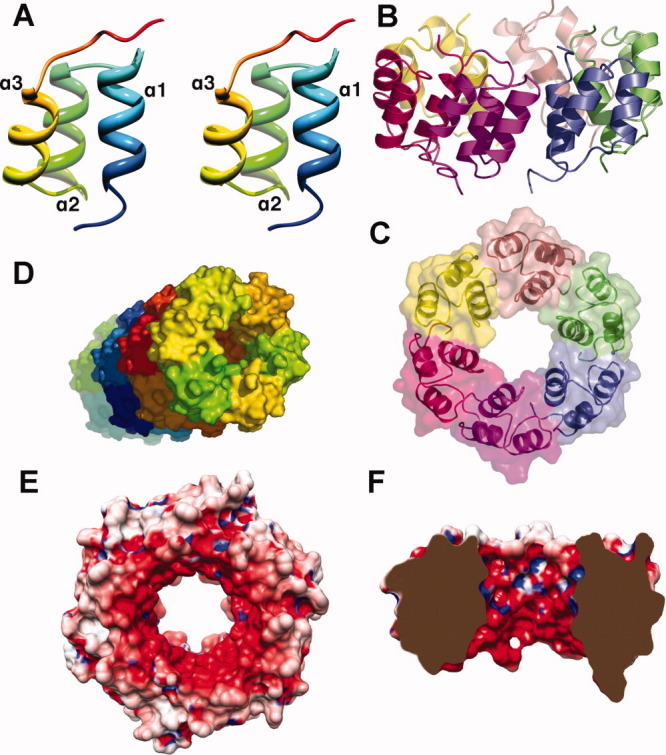
Overall phage SPP1 gp23.1 structure. A: Stereo ribbon representation of the gp23.1 monomer. The protein is colored in rainbow representation from the N-terminus (blue) toward the C-terminus (red). B: Ribbon representation of the gp23.1 hexamer. Each monomer is colored individually. C: Surface representation of the gp23.1 hexamer according to the same colour scheme as in B. D: Crystal packing in the hexagonal space group. A tube is formed through stacking of hexamers with the same orientation. E: Gp23.1 electrostatic potential. The hexamer is rotated 180° relative to C. F: Clipped view of gp23.1 exhibiting the asymmetrical negative charge distribution of the central channel. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Gp23.1 assembles in a hexamer
In the orthorhombic crystals, one gp23.1 hexamer is found in the asymmetric unit whereas only one monomer constitutes the hexagonal crystal asymmetric unit, the hexamer being generated by the crystallographic six-fold axis. The gp23.1 hexamer exhibits a hexagonal-shaped structure that is 58 Å in diameter with an 18 Å wide central channel [Figs. 1(B,C)]. The channel is delineated by the six α1 helices, whereas α2 and α3 helices form the outside surface of the hexamer. The average buried surface area between two neighboring monomers into the hexamer is 475 Å2 per monomer (∼15% of the total monomer accessible surface). Interactions involved at the interface between each two monomers, to ensure hexamer cohesion, include one salt bridge (Lys 40-Glu 32), hydrogen bonds and van der Waals contacts. We used static plus dynamic light scattering measurements to assess gp23.1 mass and size in solution. Our results revealed a mass of 32.9 kDa (theoretical hexamer mass of 33.6 kDa) and a hydrodynamic radius of 2.77 nm demonstrating that the formation of a gp23.1 hexamer is not due to crystal packing interactions but is also observed in solution. Noteworthy, in the gp23.1 hexagonal crystals, the packing results in the formation of tubes made of hexamers stacked with the same orientation [Fig. 1(D)]. This property might be used for biotechnological applications requiring confined environment (nanotubes), as was previously reported for the Pseudomonas aeruginosa Hcp1 building block.8
Gp23.1 biological role
Gp23.1 does not have any significant sequence similarity to any other protein hindering thus the possibility to postulate a function based on sequence conservation. A DALI search using the gp23.1 structure yielded plethora of hits with low significance (2.0 ≤ Z ≤ 4.3) due to the highly common and simple folding motif adopted by this small protein. Nevertheless, none of the hits were convincing to infer a function to gp23.1. The presence of a central channel might be suggestive that gp23.1 is a structural protein allowing DNA transit. Moreover, the observation of the negative electrostatic potential of this conduit [Fig. 1(E)] is reminiscent of the SPP1 gp19.1/Dit protein6 as well as of the SPP1 connector9 and is in agreement with this idea. However, the gp23.1 channel has a diameter two times smaller than in the mentioned proteins and more importantly slightly smaller than that of DNA (∼23 Å) precluding its passage. Fitting our gp23.1 X-ray structure into the unassigned tail-tip distal EM density did not yield satisfactory results because of a mismatch between their six-fold and three-fold respective symmetries. Indeed, the tip end exhibits a three-lobed shape that is not suitable to accommodate the hexameric-shaped gp23.1. We thus exclude the possibility that gp23.1 belongs to the tail-tip. Besides structural proteins, phages express enzymes or proteins involved in a limited number of functions: (i) cell-wall anchoring via the receptor-binding proteins; (ii) cell-wall digestion to allow DNA entry or phage exit (lysozyme and lysin); (iii) virion maturation (proteases); (iv) membrane perforation (holin); (v) phage replication relying on enzymes and DNA-binding proteins; (vi) chaperones involved in the assembly of structural proteins. Since the SPP1 receptor-binding domain has been located in the tail-tip,5 we can rule out the involvement of gp23.1 in the phage adsorption step. Similarly, as gp23.1 does not exhibit any significant sequence or structural resemblance with proteases or glycolytic hydrolases, we believe that it should not harbor such catalytic activity. Gp23.1 is neither a membrane protein nor positively charged excluding a role of holin or DNA/RNA binding protein. We thus propose that gp23.1 might act during phage assembly as a chaperone through interaction with other SPP1 or cellular proteins.
Materials and Methods
Gp23.1 cloning, site-directed mutagenesis, expression and purification
The SPP1 gp23.1 nucleotidic sequence was PCR amplified and cloned by Gateway recombination into the pETG-20A vector.10 The resulting construct encoded a N-terminal fusion with a His6-tagged thioredoxin followed by a tobacco etch virus (TEV) protease cleavage site. Gp23.1 engineering to produce the L23ML24M double mutant was achieved using the Quick Change Multi-Site Directed Mutagenesis kit (Stratagene) and with the following primer 5′-CGGT ACAGCCTCAGAGGAGATGATGCGGGTAGCTGTTA ATGC-3′ according to manufacturer's instructions. The plasmid was transformed in Escherichia coli T7 Express Iq pLysS strain (New England Biolabs) and expression was induced at 25°C overnight using 0.5 mM IPTG in either a Terrific-Broth medium or a minimal medium containing 50 mg/L seleno-methionine for production of seleno-methionine labelled protein.11 After harvesting, cell lysis was done by addition of 0.25 mg/mL lysozyme, a freezing/thawing cycle and sonication. Gp23.1 purification was performed via a first Ni2+-affinity step using 250 mM imidazole for elution. After desalting and TEV protease cleavage (10:1 (w/w) protein:TEV protease ratio, 4°C overnight), a second Ni2+-affinity step was performed followed by a gel-filtration on a preparative Superdex 200 26/60 column. MALDI-TOF mass spectrometry analysis was used to check protein integrity as well as Se-Met incorporation.
Crystallization and structure determination
Initial nano-crystallization screening12 was performed in 96-well Greiner plates with protein concentration varying between 8 and 12 mg/mL. Crystals used for data collection were obtained in: (i) 0.2M Na+SCN-, 2.2M (NH4+)2SO4; (ii) 1.6M Na+-citrate pH 6.5 (cryoprotected with 25% glycerol) and (iii) 0.1M Na+-HEPES pH 7.5, 20% PEG 10,000. Data were collected on BM14 at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) and at the Swiss Light Source (Villigen, Switzerland). Data processing and scaling were done with XDS,13 POINTLESS14 and SCALA.14 Phasing was performed using the SeMet substituted double mutant (L23M-L24M) by the single-wavelength Anomalous Diffraction method using the phenix.autosol wizard.15,16 Resulting phases were the starting point for automatic model building with phenix.autobuild.17,18 Phasing of the two native data set was achieved using MOLREP19 or Phaser.20 Model building and refinement were done with Coot,21 phenix.refine,22 and BUSTER-TNT.23 TLS groups definition was assisted by the TLSMD server.24 Final refinement statistics and quality of the models are summarized in Table I. Structure analysis was helped by the Protein Interfaces, Surfaces and Assemblies server25 and the DALI server.26 Electrostatic potential calculation was performed with pdb2pqr27 and APBS.28 Figures were generated with Chimera.29 The coordinates have been deposited to the PDB with accession codes 2XF5, 2XF6, and 2XF7.
Light scattering measurements
The oligomerization state and size of gp23.1 in solution were studied by MALS/QELS/UV/RI coupled on-line with an analytical SEC column, as described.30–32 MALS, QELS, UV and RI measurements were achieved with a MiniDawn Treos (Wyatt technology), a Dynapro (Wyatt technology), a Photo Diode Array 2996 (Waters), and an Optilab rEX (Wyatt technology), respectively. The SEC column was a 15-mL KW-803 column (Shodex) run at 0.5 mL/min on an Alliance HPLC 2695 system (Waters) in a buffer containing 10 mM Hepes pH 7.5, 150 mM NaCl.
Acknowledgments
We thank Dr. Hassan Belrhali and Prof. Thomas Terwilliger for the help and suggestions they provided.
Glossary
Abbreviations:
- EM
electron microscopy
- MALS/QELS/UV/RI
on-line multi-angle laser light scattering, quasi-elastic light scattering, absorbance and refractive index detectors
- r.m.s.d.
root mean square deviation
- RBP
receptor binding protein
References
- 1.Brussow H, Hendrix RW. Phage genomics: small is beautiful. Cell. 2002;108:13–16. doi: 10.1016/s0092-8674(01)00637-7. [DOI] [PubMed] [Google Scholar]
- 2.Plisson C, White HE, Auzat I, Zafarani A, Sao-Jose C, Lhuillier S, Tavares P, Orlova EV. Structure of bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J. 2007;26:3720–3728. doi: 10.1038/sj.emboj.7601786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tavares P, Santos MA, Lurz R, Morelli G, de Lencastre H, Trautner TA. Identification of a gene in Bacillus subtilis bacteriophage SPP1 determining the amount of packaged DNA. J Mol Biol. 1992;225:81–92. doi: 10.1016/0022-2836(92)91027-m. [DOI] [PubMed] [Google Scholar]
- 4.Sao-Jose C, Baptista C, Santos MA. Bacillus subtilis operon encoding a membrane receptor for bacteriophage SPP1. J Bacteriol. 2004;186:8337–8346. doi: 10.1128/JB.186.24.8337-8346.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sao-Jose C, Lhuillier S, Lurz R, Melki R, Lepault J, Santos MA, Tavares P. The ectodomain of the viral receptor YueB forms a fiber that triggers ejection of bacteriophage SPP1 DNA. J Biol Chem. 2006;281:11464–11470. doi: 10.1074/jbc.M513625200. [DOI] [PubMed] [Google Scholar]
- 6.Veesler D, Gautier R, Lichière J, Auzat I, Tavares P, Bron P, Campanacci C, Cambillau C. Crystal structure of bacteriophage SPP1 Dit (gp 19.1): a paradigm for the adsorption apparatus hub in gram+ infecting phages. J Biol Chem. doi: 10.1074/jbc.M110.157529. (in review). (in review) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veesler D, Blangy S, Spinelli S, Tavares P, Campanacci V, Cambillau C. Crystal structure of the gene 22 product of the Bacillus subtilis SPP1 phage. Prot Sci. 2010;19:1439–1443. doi: 10.1002/pro.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ballister ER, Lai AH, Zuckermann RN, Cheng Y, Mougous JD. In vitro self-assembly of tailorable nanotubes from a simple protein building block. Proc Natl Acad Sci USA. 2008;105:3733–3738. doi: 10.1073/pnas.0712247105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lhuillier S, Gallopin M, Gilquin B, Brasiles S, Lancelot N, Letellier G, Gilles M, Dethan G, Orlova EV, Couprie J, Tavares P, Zinn-Justin S. Structure of bacteriophage SPP1 head-to-tail connection reveals mechanism for viral DNA gating. Proc Natl Acad Sci USA. 2009;106:8507–8512. doi: 10.1073/pnas.0812407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vincentelli R, Bignon C, Gruez A, Canaan S, Sulzenbacher G, Tegoni M, Campanacci V, Cambillau C. Medium-scale structural genomics: strategies for protein expression and crystallization. Acc Chem Res. 2003;36:165–172. doi: 10.1021/ar010130s. [DOI] [PubMed] [Google Scholar]
- 11.Doublie S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- 12.Sulzenbacher G, Gruez A, Roig-Zamboni V, Spinelli S, Valencia C, Pagot F, Vincentelli R, Bignon C, Salomoni A, Grisel S, Maurin D, Huyghe C, Johansson K, Grassick A, Roussel A, Bourne Y, Perrier S, Miallau L, Cantau P, Blanc E, Genevois M, Grossi A, Zenatti A, Campanacci V, Cambillau C. A medium-throughput crystallization approach. Acta Cryst. 2002;D58:2109–2115. doi: 10.1107/s0907444902013938. [DOI] [PubMed] [Google Scholar]
- 13.Kabsch W. Xds. Acta Cryst. 2010;D66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans P. Scaling and assessment of data quality. Acta Cryst. 2006;D62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 15.Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart PH, Hung LW. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Cryst. 2009;D65:582–601. doi: 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD. Automated structure solution with the PHENIX suite. Methods Mol Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 17.Terwilliger T. SOLVE and RESOLVE: automated structure solution, density modification and model building. J Synchrotron Radiat. 2004;11:49–52. doi: 10.1107/s0909049503023938. [DOI] [PubMed] [Google Scholar]
- 18.Terwilliger TC. Automated structure solution, density modification and model building. Acta Cryst. 2002;D58:1937–1940. doi: 10.1107/s0907444902016438. [DOI] [PubMed] [Google Scholar]
- 19.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Cryst. 2010;D66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 20.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Afonine PV, Grosse-Kunstleve RW, Urzhumtsev A, Adams PD. Automatic multiple-zone rigid-body refinement with a large convergence radius. J Appl Cryst. 2009;42:607–615. doi: 10.1107/S0021889809023528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM,, Bricogne G. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Cryst. 2004;D60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- 24.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Cryst. 2006;D62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 25.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB 2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holst M, Saied F. Multigrid solution of the Poisson-Boltzmann equation. J Comput Chem. 1993;14:105–113. [Google Scholar]
- 29.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 30.Campanacci V, et al. Solution and electron microscopy characterization of lactococcal phage baseplates expressed in Escherichia coli. J Struct Biol. doi: 10.1016/j.jsb.2010.02.007. (in press) [DOI] [PubMed] [Google Scholar]
- 31.Veesler D, Blangy S, Siponen M, Vincentelli R, Cambillau C, Sciara G. Production and biophysical characterization of the CorA transporter from Methanosarcina mazei. Anal Biochem. 2009;388:115–121. doi: 10.1016/j.ab.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 32.Veesler D, Dreier B, Blangy S, Lichiere J, Tremblay D, Moineau S, Spinelli S, Tegoni M, Plückthun A, Campanacci V, Cambillau C. Crystal structure and function of a DARPin neutralizing inhibitor of lactococcal phage TP901-1: comparison of DARPin and camelid VHH binding mode. J Biol Chem. 2009;284:30718–30726. doi: 10.1074/jbc.M109.037812. [DOI] [PMC free article] [PubMed] [Google Scholar]