

Abstract
Bacterial degradation of sulfoacetate, a widespread natural product, proceeds via sulfoacetaldehyde and requires a considerable initial energy input. Whereas the fate of sulfoacetaldehyde in Cupriavidus necator (Ralstonia eutropha) H16 is known, the pathway from sulfoacetate to sulfoacetaldehyde is not. The genome sequence of the organism enabled us to hypothesize that the inducible pathway, which initiates sau (sulfoacetate utilization), involved a four-gene cluster (sauRSTU; H16_A2746 to H16_A2749). The sauR gene, divergently orientated to the other three genes, probably encodes the transcriptional regulator of the presumed sauSTU operon, which is subject to inducible transcription. SauU was tentatively identified as a transporter of the major facilitator superfamily, and SauT was deduced to be a sulfoacetate-CoA ligase. SauT was a labile protein, but it could be separated and shown to generate AMP and an unknown, labile CoA-derivative from sulfoacetate, CoA, and ATP. This unknown compound, analyzed by MALDI-TOF-MS, had a relative molecular mass of 889.7, which identified it as protonated sulfoacetyl-CoA (calculated 889.6). SauS was deduced to be sulfoacetaldehyde dehydrogenase (acylating). The enzyme was purified 175-fold to homogeneity and characterized. Peptide mass fingerprinting confirmed the sauS locus (H16_A2747). SauS converted sulfoacetyl-CoA and NADPH to sulfoacetaldehyde, CoA, and NADP+, thus confirming the hypothesis.
Keywords: Anion Transport, Bacterial Metabolism, Coenzyme A, Dehydrogenase, Enzyme Purification, Gene Knockout, Cupriavidus necator, Sulfoacetaldehyde, Sulfoacetate, Sulfoacetyl-CoA
Introduction
Sulfoacetic acid, as the sulfonate ester, was first recognized as a natural product in plant alkaloids (1). Free sulfoacetate was then found to be widespread in plants and algae (2–4). The compound was also detected as an intracellular intermediate in the bacterial degradation of the plant sulfolipid (sulfoquinovosyldiacylglycerol), specifically from its polar head group, sulfoquinovose (5). This sulfolipid is nearly ubiquitous in phototrophic organisms (6) and may represent up to half of the total lipid content in some marine algae (7). The sulfur content of leaves is comprised mainly of sulfolipid and proteins; senescence of deciduous plants thus introduces significant amounts of sulfoquinovose into the soil (8). Sulfoacetate can also be the product of the bacterial assimilation of nitrogen from taurine (9, 10). The latter precursor of sulfoacetate is a major organic solute in marine creatures (11, 12) and in mammals (13), which excrete it in urine (14). Moreover, sulfoacetate is introduced into the environment as sodium lauryl sulfoacetate, a frequent ingredient of cosmetics and personal care products.
Biodegradation of sulfoacetate was first observed by Martelli and Benson (5), and evidence was presented for the hydrolytic cleavage of sulfoacetate to glycolate, but the organism was lost (15, 16). King and Quinn (17) isolated aerobic Gram-positive and Gram-negative bacteria, which degraded sulfoacetate by a different pathway, involving desulfonation via inducible sulfoacetaldehyde acetyltransferase (Xsc)4 (EC 2.3.3.15). Various anaerobic bacteria were also found to utilize sulfoacetate via Xsc (15). The aerobic Ralstonia sp. strain EDS1 dissimilates a range of organosulfonates, including sulfoacetate, via Xsc, and the low molar growth yield with sulfoacetate (60% of the value for all other carbon sources) led to the conclusion that the reduction of sulfoacetate to sulfoacetaldehyde, the organic substrate of Xsc, was metabolically expensive (18). The inducible involvement of Xsc in the degradation of sulfoacetate, taurine, and isethionate was confirmed in Cupriavidus necator (Ralstonia eutropha) H16, where the common pathway also included phosphate acetyltransferase (EC 2.3.1.8), a sulfite exporter TauE (TC 9.A.29.2.1) and sulfite dehydrogenase (SorAB) (EC 1.8.2.1) (Fig. 1) (19, 20).
FIGURE 1.

Sulfoacetaldehyde as the point of convergence of the degradative pathways for sulfoacetate, isethionate, and taurine and the degradative pathway of sulfoacetaldehyde in C. necator H16. Taurine dehydrogenase (TauXY) and isethionate dehydrogenase (IseJ) are inducible, membrane-bound, cytochrome c-coupled enzymes that have not been purified; TauE is a sulfite exporter, located in the cytoplasmic membrane (CM) (19–21, 50). Pta, phosphate acetyltransferase.
It was still unclear which proteins catalyzed the formation of sulfoacetaldehyde from sulfoacetate. However, although the convergence of metabolism of C2 sulfonates at one Xsc (Fig. 1) is widespread (15), some organisms contain paralogues of xsc, e.g. Desulfitobacterium hafniense DCB-2 (Dhaf_0189 and Dhaf_4634). We showed that the presence of more than one xsc gene in a genome can represent complete, individual degradative pathways for different sulfonates (21). Using this idea in D. hafniense DCB-2, where the gene cluster upstream of one xsc was annotated as encoding an acyl-CoA ligase and a NAD(P)-coupled aldehyde dehydrogenase (Dhaf_0190), we hypothesized that this combination of reaction types would convert sulfoacetate to sulfoacetaldehyde via putative sulfoacetyl-CoA. Orthologues of the presumptive sulfoacetaldehyde dehydrogenase (deacylating) (SauS, for sulfoacetate utilization) (Fig. 2A) are widespread, and one sauS gene is found in a four-gene cluster in C. necator H16. We chose to explore the sauRSTU cluster (Fig. 2B) in C. necator H16, with which we had relevant experience (Fig. 1). Strain H16 grows relatively fast with sulfoacetate as a sole source of carbon and energy for growth, and an established protocol to generate in-frame deletions in this organism is available (22, 23). Our hypothesis comprised an IclR-type transcriptional regulator, SauR (H16_A2746), a sulfoacetate-CoA ligase, SauT (H16_A2748), a sulfoacetaldehyde dehydrogenase (acylating), SauS (H16_A2747), and a sulfoacetate transporter belonging to the major facilitator superfamily, SauU (H16_2749) (Fig. 2B).
FIGURE 2.

Initial reactions in the pathway for the dissimilation of sulfoacetate in C. necator H16 (A) and the cluster of genes encoding regulated expression of pathway proteins (B). The locus tags of the sauRSTU cluster are H16_A2746 (sauR) to H16_A2749 (sauU). CM, cytoplasmic membrane.
EXPERIMENTAL PROCEDURES
Materials
Sulfoacetate (99% purity) was purchased from Acros Organics (Geel, Belgium). Other commercial chemicals (∼99% purity) were from Fluka (Seelze, Germany), Merck or Sigma-Aldrich. Sulfoacetyl-CoA is not available commercially, and we failed to synthesize it chemically using protocols to synthesize 3-hydroxybutyryl-CoA (24, 25). Oligonucleotides were synthesized by Microsynth (Balgach, Switzerland). Materials for (RT-)PCR and cloning were purchased from Fermentas GmbH (St. Leon-Rot, Germany) and used as provided by the supplier. Phusion DNA Polymerase from NEB (Ipswich, UK) was used during the construction of deletion mutants. Chromosomal DNA was isolated as described elsewhere (26). Total RNA was isolated using the E.Z.N.A. bacterial kit (Omega Bio-Tek, Doraville, GA). PCR products were purified using QIAquick® spin kit (Qiagen).
Bacteria, Growth Media, and Growth Conditions
C. necator H16 (DSM 428) (27) was isolated in Göttingen, Germany. The organism was grown at 30 °C in freshwater, mineral salt medium (28). Sulfoacetate (10–20 mm) was used routinely as the sole added source of carbon and energy for growth. For enzyme assays with the mutants, which were unable to grow with sulfoacetate, 10 mm acetate served as a growth substrate in the presence of 10 mm sulfoacetate to induce the sulfoacetate degradative enzymes.
Precultures and cultures (5 ml) for the determination of the substrate range were grown in 50-ml screw-cap tubes in a roller. Growth experiments were done on the 50-ml scale in 300-ml Erlenmeyer flasks on a shaker; samples were taken at intervals to measure optical density at 580 nm, to assay protein, and to determine the concentrations of substrate and products.
Strains of Escherichia coli used for site-directed mutagenesis (supplemental Table S1) were grown in LB medium at 37 °C. Appropriate antibiotics were added to medium in the following concentrations: 50 μg ml−1 ampicillin, 20 μg ml−1 tetracycline, and 5 μg ml−1 trimethoprim.
Preparation of Cell-free Extracts
Cell-free extracts were obtained from strain H16 grown on the 1-liter scale in 5-liter Erlenmeyer flasks. The cells were harvested in the midexponential growth phase by centrifugation (30,000 × g, 15 min, 4 °C), washed in 50 mm potassium phosphate buffer, pH 7.2 (containing 5 mm MgCl2), and resuspended in a small volume (2–5 ml) of the same buffer. The cells were disrupted by four passages through a chilled French press set at 138 MPa, and whole cells and cell debris were removed by centrifugation (20,000 × g, 3 min, 4 °C). The membrane and soluble fractions were obtained by ultracentrifugation (200,000 × g, 30 min, 4 °C). DNA was removed by DNase, which was added prior to disruption.
Enzyme Assays
SauT was assayed discontinuously by HPLC as the formation of sulfoacetyl-CoA. The reaction mixture contained (in a final volume of 1 ml): 50 μmol of Tris/HCl buffer, pH 8.0 or 9.0 (containing 5 mm MgCl2), 1 μmol of ATP, 2 μmol of sulfoacetate, 0.5 μmol of CoA, and 0.1–1 mg of protein. SauT activity was estimated as a decrease in concentration of CoA, because no reference material of sulfoacetyl-CoA was available to quantify the novel compound. Sulfoacetaldehyde dehydrogenase (acylating) (SauS) was assayed spectrophotometrically as the sulfoacetaldehyde-dependent reduction of NADP+ at 365 nm, which is the reverse reaction. The reaction mixture contained (in a final volume of 1 ml): 50 μmol of Tris/HCl buffer, pH 9.0 (containing 5 mm MgCl2), 1 μmol of NADP+, 3 μmol of sulfoacetaldehyde, 0.5 μmol of CoA, and 1–100 μg of protein. The reaction was linear for at least 1 min.
Purification of SauS and Identification of the Corresponding Gene
The first step to purify SauS, anion exchange chromatography (MonoQ, HR 10/10; Pharmacia), was performed with the soluble protein fraction at a flow rate of 1 ml min×1. An increasing gradient of sodium sulfate in 50 mm Tris/sulfate buffer, pH 8.7, was applied, and SauS was eluted at 85 mm sodium sulfate. Active fractions were combined, rebuffered on PD10 columns with 50 mm potassium phosphate buffer, pH 6.5, and loaded on to a cation exchange column (MonoS, HR 5/5; Pharmacia). An increasing gradient of sodium sulfate was applied, and SauS was eluted at 95 mm sodium sulfate. N-terminal amino acid sequencing and peptide mass fingerprinting were done by Toplab (Martinsried, Germany) on bands excised from SDS-PAGE gels.
Enzyme Separation of SauT
The first step to separate SauT, anion exchange chromatography with a MonoQ HR 10/10 column, was done with soluble fraction in 50 mm potassium phosphate buffer, pH 6.5. An increasing gradient of sodium sulfate was applied, and SauT eluted at 120 mm sodium sulfate. Fractions were desalted, concentrated, and subjected to hydroxyapatite column chromatography in 10 mm potassium phosphate buffer, pH 6.7. An increasing gradient of potassium phosphate was applied, and SauT eluted at ∼100 mm potassium phosphate.
Analytical Methods
Sulfoacetate was determined by ion chromatography (9). Sulfate was quantified turbidimetrically as an insoluble suspension of BaSO4 (29). Sulfite was quantified as the fuchsin adduct (30). Growth was followed turbidimetrically at 580 nm or assayed as Lowry-type protein in whole cells (31). Protein content of crude cell extracts was determined by protein dye binding (32). Denatured proteins were analyzed on 13% SDS-PAGE gels and stained with Coomassie Brilliant Blue R250 (33). The native molecular weight of separated proteins was calculated after gel filtration by interpolation in a standard curve. The values of Kmapp were derived by hyperbolic curve fitting.
Identification of Sulfoacetyl-CoA
Sulfoacetyl-CoA was visualized by reversed phase HPLC and a diode array detector. The stationary phase was Nucleosil 5-C18 (125 × 3 mm). The mobile phase (0.5 ml min−1) was a 100 mm potassium phosphate solution, pH 5, with a gradient from 0 to 30% methanol. The identity of sulfoacetyl-CoA was confirmed by MALDI-TOF-MS. Samples of reaction mixtures containing putative sulfoacetyl-CoA generated in an assay of SauS were mixed with a matrix of saturated α-cyano-4-hydroxy-cinnamic acid in 50% acetonitrile and 1% trifluoroacetic acid. The dried droplet method was used with 0.8-μl samples on a MALDI steel target, and samples were analyzed in the negative ion mode in an Applied Biosystems 4800 MALDI-TOF/TOF mass spectrometer (34).
Cloning and Sequencing
Standard DNA techniques (35, 36) were used to isolate bacterial plasmid DNA, to transform plasmid DNA into E. coli, and for general DNA handling. Low throughput plasmid DNA was isolated using the QIAprep® miniprep kit, and high throughput isolation of plasmid DNA was done using a boiling method (37). Colony PCR was done as described elsewhere (38). Sequencing was done at GATC (Konstanz, Germany) or Microsynth, and the sequences were analyzed using the Lasergene package from DNASTAR (Madison, WI).
RT-PCR
The primers listed in supplemental Table S2 were used for RT-PCR. PCR was done as described elsewhere (39). The absence of DNA after isolation of RNA was tested by PCR using primers for xsc. Positive controls for the success of RNA isolation were done after reverse transcription using the 16 S rRNA-specific primers 16S-27F and 16S-533R (supplemental Table S2) (40). The PCR products were visualized after separation on 1.5% agarose gels.
Construction of Deletion Mutants in Individual sau Genes
Mutants of C. necator H16 containing in-frame deletions in one of the sauSTU genes were constructed by gene replacement mutagenesis as detailed in the supplemental data. Thereby, a wild type functional gene was replaced by an engineered defective short gene (see below).
Sequence Analyses and Accession Numbers
The sequence of the genome of C. necator H16 (accession numbers NC_008313 and NC_008314) was obtained from the National Center for Biotechnology Information, whose BLAST (41) server was also used. Sequence data were analyzed using different subroutines of the LASERGENE software package (DNASTAR), with SignalP (42), available from the Center for Biological Sequence Analysis, with PROSITE, on the ExPASy Proteomics Server, and with the BLAST server of the Transport Classification Database.
RESULTS
Growth Kinetics
C. necator H16 grew exponentially (μ = 0.14 h−1) with 10–20 mm sulfoacetate as the sole source of carbon and energy (Fig. 3A). Growth was concomitant with substrate utilization and with the stoichiometric recovery of the sulfonate moiety as sulfate (Fig. 3B). The molar growth yield was 5.1 g of protein (mol C)−1, significantly lower than the 6.0–6.6 g of protein (mol C)−1 with taurine, isethionate, or acetate; the latter is a normal value, which indicates mass balance for carbon (43). The specific utilization rate of sulfoacetate was calculated to be 3.8 millikatals (kg of protein)−1.
FIGURE 3.

Semi-log plot (A) of growth of C. necator H16 in 10 mm sulfoacetate-salts medium and the concentrations of substrate and products (B) plotted as function of protein concentration. Filled circles, sulfoacetate; filled triangles, sulfate; open squares, sulfite.
Enzyme Activities and Transcriptional Analyses in C. necator H16
The first scalar enzyme in the postulated pathway (Fig. 2A) was SauT. Extracts of acetate- (Table 1), taurine-, or isethionate-grown cells (not shown) showed no activity of this enzyme. Extracts of sulfoacetate-grown cells catalyzed the CoA- and ATP-dependent conversion of sulfoacetate to sulfoacetyl-CoA (see below) and AMP (including nonspecific activity). This was interpreted as inducible activity of SauT, which was shown by ultracentrifugation of crude extract to be in the soluble fraction. The specific activity of the enzyme was low (Table 1) and difficult to quantify, because we could not stop the reaction without destroying the sulfoacetyl-CoA formed during the reaction.
TABLE 1.
Activities of enzymes and transcription of genes under different growth conditions of C. necator H16
| Specific activitya or transcription |
||
|---|---|---|
| Cells grown with sulfoacetate | Cells grown with acetate | |
| Transcription of sauU (sulfoacetate uptake) | +b | bdlc |
| Sulfoacetate-CoA ligase (SauT) | 0.2 | bdl |
| Transcription of sauT | + | bdl |
| Sulfoacetaldehyde dehydrogenase (SauS) | 10.4 | bdl |
| Transcription of sauS | + | bdl |
a Specific enzyme activity is given in the SI unit millikatal (kg of protein)−1.
b RNA transcript is scored as: −, absent; +, strong. Typical data are shown in supplemental Fig. S1.
c bdl, below detection limit.
The second scalar reaction was SauS (Fig. 2A). Extracts of acetate- (Table 1), taurine-, or isethionate-grown cells (not shown) showed no activity of this enzyme. Extracts of sulfoacetate-grown cells catalyzed the NADP+- and CoA-dependent conversion of sulfoacetaldehyde to sulfoacetyl-CoA (see below). This was interpreted as inducible activity of SauS, which was found to be soluble. The enzyme was highly active (Table 1), which allowed enough sulfoacetyl-CoA to be collected from the HPLC to confirm that SauS was also active in the forward reaction. SauS was identified as the gene product encoded at H16_A2747 (see below). Each of the candidate genes (Fig. 2B) to encode SauU (H16_A2749), SauT (H16_A2748), or SauS (H16_A2747) was found to be transcribed inducibly (Table 1), which corresponded to the inducible nature of the degradative pathway (Table 1).
Growth of Deletion Mutants
We were able to generate three deletion mutants, each with an in-frame mutation in sauU (H16ΔsauU), sauT (H16ΔsauT), or sauS (H16ΔsauS), respectively (Fig. 4). None of these mutants grew with sulfoacetate, but each grew with acetate, taurine, isethionate, or sulfoacetaldehyde, as the wild type did. The mutations were specific for sulfoacetate metabolism (Fig. 1).
FIGURE 4.
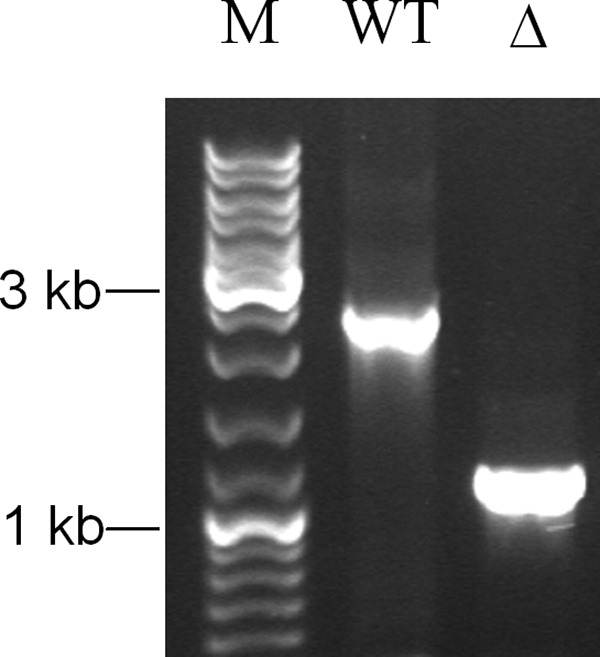
Typical confirmation by PCR that a deletion mutant had been generated. Amplicons of chromosomal DNA from strains H16 (WT) and H16ΔsauU (Δ) were obtained using primers h16_sauUproof_ f2 and h16_sauUproof_ r1 (supplemental Table S3). M, marker (GeneRuler DNA ladder mix).
Mutant H16ΔsauU synthesized neither SauS nor SauT (Table 2), so presumably no sulfoacetate entered the cell to enable induction to occur. In contrast, mutant H16ΔsauT expressed SauS (Table 2) and mutant H16ΔsauS expressed SauT (Table 2), so regulation of the gene cluster, presumably by SauR (Fig. 2B), was unaffected. No mutation in sauR was obtained. There appears to be irrefutable evidence for the functions of SauSTU.
TABLE 2.
Phenotypes and enzyme activities of different mutants of C. necator H16
| H16ΔsauS | H16ΔsauT | H16ΔsauU | |
|---|---|---|---|
| Growth with sulfoacetate | |||
| Activity of sulfoacetaldehyde dehydrogenase (SauS) | bdla | 22.3b | bdl |
| Activity of sulfoacetate-CoA ligase (SauT) | 0.03 | bdl | bdl |
a bdl, below detection limit.
b Specific enzyme activity is given in the SI unit millikatal (kg of protein)−1.
Identification of Sulfoacetyl-CoA as the Intermediate in Sulfoacetate Degradation
Putative sulfoacetyl-CoA was detected by HPLC as a novel peak generated during the activation of sulfoacetate by SauT and during the oxidation of sulfoacetaldehyde by SauS. The UV spectrum of the unknown involved maxima at 212 and 257 nm and a minimum at 225 nm, similar to HSCoA but with a slightly shorter (0.7 min) retention time. The compound had a half-life of ∼2 h under these conditions (pH 9.0), with shorter half-lives at higher and lower pH values. Samples taken during the reaction of purified SauS with sulfoacetaldehyde, NADP+, and HSCoA were analyzed by MALDI-TOF-MS in the negative ion mode. The formation of a compound (m/z = 888.7 = [M − 1]−) was detected. The value of M (889.7) corresponds to that calculated (889.6) for protonated sulfoacetyl-CoA. This was taken as confirmation of the identity of sulfoacetyl-CoA.
Separation of SauT
Two separative steps with SauT were possible before activity was lost. This preparation showed sulfoacetate-dependent formation of sulfoacetyl-CoA and of AMP, but SauT was insufficiently concentrated to be visible on SDS-PAGE gels.
Purification and Characterization of SauS
SauS was purified 175-fold to apparent homogeneity (Fig. 5) in two steps (Table 3). The sequence of six N-terminal amino acids was determined to be SVQILH. This corresponded to the derived sequence of only one ORF (H16_A2747) in the genome of C. necator H16, which confirmed our hypothetical locus for sauS (Fig. 2B). Peptide mass fingerprinting of SauS confirmed this conclusion. The derived molecular mass of mature SauS, 51.5 kDa, corresponds to the value (∼52 kDa) for the denatured protein (Fig. 5) interpolated into the standard curve. Separation of native SauS on a calibrated gel filtration column allowed a molecular mass of 115 kDa to be calculated. Given the errors in the method (44), we tentatively postulate that native SauS is a dimer.
FIGURE 5.
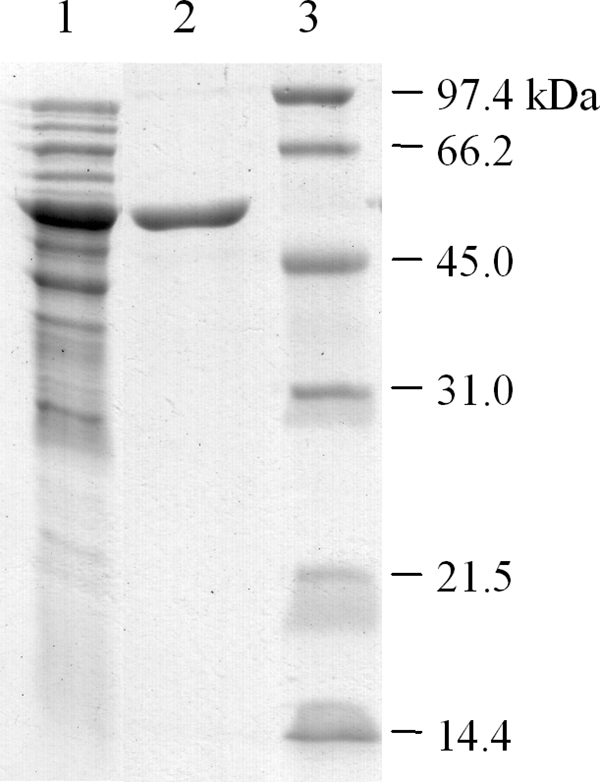
Electropherogram of SauS at different stages of purification. Lane 1, SauS after anion exchange chromatography; lane 2, SauS after cation exchange chromatography; lane 3, molecular mass markers.
TABLE 3.
Purification of sulfoacetaldehyde dehydrogenase (SauS) from C. necator H16
| Purification step | Specific activity | Volume | Total protein | Recovery | Purification |
|---|---|---|---|---|---|
| millikatals kg−1 | ml | mg | % | fold | |
| Crude cell extract | 7 | 2.1 | 53 | 100 | 1 |
| Soluble fraction | 9 | 2.0 | 37 | 94 | 1 |
| Anion exchanger (pool) | 35 | 3.5 | 4 | 37 | 5 |
| Cation exchanger | 1191 | 0.9 | 0.002 | 1 | 175 |
SauS was specific for NADP+ as a cofactor that could not be replaced by NAD+. The Kmapp values for NADP+, CoA, and sulfoacetaldehyde were 64, 102, and 330 μm, respectively. The enzyme was specific for sulfoacetaldehyde; none of the 10 tested compounds was a substrate: acetaldehyde, phosphonoacetaldehyde, glycolaldehyde, formaldehyde, propionaldehyde, succinic semialdehyde, glyoxylate, betaine aldehyde, glyceraldehyde, and 2-oxobutanoic acid. None of these compounds inhibited the enzyme reaction. The optimal activity of the enzyme was obtained in 50 mm Tris/HCl buffer, pH 9.0, containing 5 mm magnesium chloride. The enzyme could be stored at 4 or −18 °C but lost 30% of its activity after 1 week.
DISCUSSION
The hypothetical pathway contains three steps, one vectorial and two scalar, and a novel metabolic intermediate, sulfoacetyl-CoA (Fig. 2A). Evidence for sulfoacetyl-CoA was obtained by HPLC and confirmed by MALDI-TOF-MS. Our earlier hypothesis, a phosphorylated intermediate analogous to steps in the biosynthesis of, for example, lysine or proline (18), is thus invalid. Sulfoacetyl-CoA must be almost as ubiquitous as sulfoacetate, but it will be almost undetectable in the environment, partly because it is intracellular and partly because it is so labile. We suggested that sulfoacetate is a ubiquitous natural product (see the Introduction), although there are no direct determinations of this. The biodiversity of sulfoacetate degradation5 may be considered to support this hypothesis, because organisms have evolved and sustained so many different variants of the degradative pathway. The reactions we have established (Fig. 2) seem to be novel in degradative pathways.
The weakest portion of our hypothesis is the identification of the transcriptional regulator, SauR, attributed to locus tag H16_A2746. The gene cluster, sauSTU, is inducibly transcribed, and the enzymes are expressed inducibly (Table 1), so a regulator is involved. The regulator encoded adjacent to sauSTU was suggested as the simplest hypothesis, given the conserved sauRSTU cluster in all relevant betaproteobacteria.5
The first step in the pathway is transport, which involves SauU in C. necator H16 (Fig. 2). The cytoplasmic membrane is apparently impermeable for sulfoacetate unless a transporter is present, because deletion mutant H16ΔsauU is obviously unable to induce expression of the sau cluster (Table 2). This emphasizes the impermeability of membranes to sulfonates pointed out elsewhere (45, 46) and supports the idea that nature uses the sulfonate substituent to prevent a molecule crossing a membrane. The sequence of SauU indicates that it belongs to the major facilitator superfamily (TC 2.A.1.-.-) of transporters. The closest orthologues (TC 2.A.1.14.-) have ∼30% sequence identity and share the predicted structure of 12 transmembrane helices.
Sulfoacetate-CoA ligase, SauT, was separated sufficiently to allow us to confirm a systematic name, sulfoacetate:CoA ligase (AMP-forming) (EC 6.2.1.-). This enzyme represents one major energy drain in the pathway (with SauS as another) compared with, for example, taurine or isethionate (Fig. 2A), given that each named growth substrate yields only one acetyl-CoA for energy conservation and anabolism (Fig. 1). This degradative pathway (Fig. 2A) makes it easy to understand how 20–40% (this paper and Ref. 18) reductions in molar growth yield occur.
Sulfoacetaldehyde dehydrogenase (acylating) (SauS), apparently the marker enzyme for the pathway,5 has been purified in this study. It shows the highest similarity to aldehyde dehydrogenases (acylating) (EC 1.2.1.10), a reaction it does not catalyze. (Acet)aldehyde dehydrogenases (EC 1.2.1.-) (47) are thought to be NAD+-dependent enzymes (also reacting with NADP+ but at a lower rate), existing as dimers (48), tetramers, or polymers. Purified sulfoacetaldehyde dehydrogenase (acylating) from C. necator H16 is presumed to form a dimer in solution but acts solely with NADP+ as a cofactor, and sulfoacetaldehyde and CoA act as substrates. SauS must be distinguished both from sulfoacetaldehyde dehydrogenase, SafD (EC 1.2.1.73), which is specific for NAD+ and generates sulfoacetate from sulfoacetaldehyde (10), and from sulfoacetaldehyde reductase, IsfD (EC 1.1.1.-), which generates isethionate from sulfoacetaldehyde and NADP+ (49). We propose that SauS, nonidentical with any reported enzyme, should have the accepted name “sulfoacetaldehyde dehydrogenase (acylating)” and the systematic name “2-sulfoacetaldehyde:NADP+ oxidoreductase (CoA-acylating)” (EC 1.2.1.-).
Supplementary Material
Acknowledgments
We are grateful to Gertrud Stahlhut for excellent technical advice on mutagenesis, to Valentina Vongrad for data generated during a practical course for advanced students, and to Karin Denger for discussions and for critically reading the manuscript. We also thank Tobias Erb (Freiburg, Germany) for sharing his experience on synthesizing CoA-esters and the late Hal Dixon (Cambridge, UK) for phosphonoacetaldehyde.

The on-line version of this article (available at http://www.jbc.org) contains supplemental text, Tables S1–S3, and Fig. S1.
S. Weinitschke, M. Buhmann, T. H. M. Smits, and A. M. Cook, manuscript in preparation.
- Xsc
- sulfoacetaldehyde acetyltransferase.
REFERENCES
- 1.Folkers K., Koniuszy F., Shavel J. (1944) J. Am. Chem. Soc. 66, 1083–1087 [Google Scholar]
- 2.Gupta S. D., Sastry P. S. (1988) Arch. Biochem. Biophys. 260, 125–133 [DOI] [PubMed] [Google Scholar]
- 3.Lee R. F., Benson A. A. (1972) Biochim. Biophys. Acta. 261, 35–37 [DOI] [PubMed] [Google Scholar]
- 4.Shibuya I., Yagi T., Benson A. A. (1963) in Studies on Microalgae and Photosynthetic Bacteria, pp. 627–636, The University of Tokyo Press, Tokyo [Google Scholar]
- 5.Martelli H. L., Benson A. A. (1964) Biochim. Biophys. Acta. 93, 169–171 [DOI] [PubMed] [Google Scholar]
- 6.Harwood J. L. (1980) in The Biochemistry of Plants (Stumpf P. K. ed) pp. 301–320, Academic Press, New York [Google Scholar]
- 7.Dembitsky V. M., Rozentsvet O. A., Pechenkina E. E. (1990) Phytochemistry 29, 3417–3421 [Google Scholar]
- 8.Harwood J. L., Nicholls R. G. (1979) Biochem. Soc. Trans. 7, 440–447 [DOI] [PubMed] [Google Scholar]
- 9.Denger K., Weinitschke S., Hollemeyer K., Cook A. M. (2004) Arch. Microbiol. 182, 254–258 [DOI] [PubMed] [Google Scholar]
- 10.Krejcík Z., Denger K., Weinitschke S., Hollemeyer K., Paces V., Cook A. M., Smits T. H. (2008) Arch. Microbiol. 190, 159–168 [DOI] [PubMed] [Google Scholar]
- 11.Allen J. A., Garrett M. R. (1971) Adv. Mar. Biol. 9, 205–253 [Google Scholar]
- 12.Yin M., Palmer H. R., Fyfe-Johnson A. L., Bedford J. J., Smith R. A., Yancey P. H. (2000) Physiol Biochem. Zool. 73, 629–637 [DOI] [PubMed] [Google Scholar]
- 13.Huxtable R. J. (1992) Physiol. Rev. 72, 101–163 [DOI] [PubMed] [Google Scholar]
- 14.Stipanuk M. H. (2004) Annu. Rev. Nutr. 24, 539–577 [DOI] [PubMed] [Google Scholar]
- 15.Cook A. M., Denger K. (2002) Arch. Microbiol. 179, 1–6 [DOI] [PubMed] [Google Scholar]
- 16.Martelli H. L., Souza S. M. (1970) Biochim. Biophys. Acta. 208, 110–115 [PubMed] [Google Scholar]
- 17.King J. E., Quinn J. P. (1997) Microbiology 143, 3907–3912 [DOI] [PubMed] [Google Scholar]
- 18.Denger K., Cook A. M. (2001) Arch. Microbiol. 176, 89–95 [DOI] [PubMed] [Google Scholar]
- 19.Denger K., Weinitschke S., Smits T. H., Schleheck D., Cook A. M. (2008) Microbiology 154, 256–263 [DOI] [PubMed] [Google Scholar]
- 20.Weinitschke S., Denger K., Cook A. M., Smits T. H. (2007) Microbiology 153, 3055–3060 [DOI] [PubMed] [Google Scholar]
- 21.Weinitschke S., Sharma P. I., Stingl U., Cook A. M., Smits T. H. (2010) Appl. Environ. Microbiol. 76, 618–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lenz O., Schwartz E., Dernedde J., Eitinger M., Friedrich B. (1994) J. Bacteriol. 176, 4385–4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeffke T., Gropp N. H., Kaiser C., Grzeszik C., Kusian B., Bowien B. (1999) J. Bacteriol. 181, 4374–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chohan S. N., Copeland L. (1998) Appl. Environ. Microbiol. 64, 2859–2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stadtman T. C., Elliott P. (1957) J. Biol. Chem. 228, 983–997 [PubMed] [Google Scholar]
- 26.Desomer J., Crespi M., Van Montagu M. (1991) Mol. Microbiol. 5, 2115–2124 [DOI] [PubMed] [Google Scholar]
- 27.Pohlmann A., Fricke W. F., Reinecke F., Kusian B., Liesegang H., Cramm R., Eitinger T., Ewering C., Pötter M., Schwartz E., Strittmatter A., Voss I., Gottschalk G., Steinbüchel A., Friedrich B., Bowien B. (2006) Nat. Biotechnol. 24, 1257–1262 [DOI] [PubMed] [Google Scholar]
- 28.Thurnheer T., Köhler T., Cook A. M., Leisinger T. (1986) J. Gen. Microbiol. 132, 1215–1220 [Google Scholar]
- 29.Sörbo B. (1987) Methods Enzymol. 143, 3–6 [DOI] [PubMed] [Google Scholar]
- 30.Ruff J., Denger K., Cook A. M. (2003) Biochem. J. 369, 275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennedy S. I., Fewson C. A. (1968) Biochem. J. 107, 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 33.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 34.Hollemeyer K., Velagapudi V. R., Wittmann C., Heinzle E. (2007) Rapid Commun. Mass. Spectrom. 21, 336–342 [DOI] [PubMed] [Google Scholar]
- 35.Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K. (1987) Current Protocols in Molecular Biology, pp. 2.0.1–2.1.10, John Wiley & Sons, New York [Google Scholar]
- 36.Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., pp. 1.21–1.32, 1.74–1.84, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 37.Holmes D. S., Quigley M. (1981) Anal. Biochem. 114, 193–197 [DOI] [PubMed] [Google Scholar]
- 38.Coenye T., Goris J., Spilker T., Vandamme P., LiPuma J. J. (2002) J. Clin. Microbiol. 40, 2062–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Innis M. A., Gelfand D. H., Sninsky J. J., White T. J. (1990) PCR Protocols: A Guide to Methods and Applications, pp. 3–12, Academic Press, Inc., San Diego [Google Scholar]
- 40.Weisburg W. G., Barns S. M., Pelletier D. A., Lane D. J. (1991) J. Bacteriol. 173, 697–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bendtsen J. D., Nielsen H., von Heijne G., Brunak S. (2004) J. Mol. Biol. 340, 783–795 [DOI] [PubMed] [Google Scholar]
- 43.Cook A. M. (1987) FEMS Microbiol. Rev. 46, 93–116 [Google Scholar]
- 44.le Maire M., Ghasi A., Moller J. V. (1996) ACS Symp. Ser. 635, 36–51 [Google Scholar]
- 45.Graham D. E., Xu H., White R. H. (2002) J. Biol. Chem. 277, 13421–13429 [DOI] [PubMed] [Google Scholar]
- 46.Mampel J., Maier E., Tralau T., Ruff J., Benz R., Cook A. M. (2004) Biochem. J. 383, 91–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Powlowski J., Sahlman L., Shingler V. (1993) J. Bacteriol. 175, 377–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Söhling B., Gottschalk G. (1993) Eur. J. Biochem. 212, 121–127 [DOI] [PubMed] [Google Scholar]
- 49.Krejcík Z., Hollemeyer K., Smits T. H., Cook A. M. (2010) Microbiology 156, 1547–1555 [DOI] [PubMed] [Google Scholar]
- 50.Brüggemann C., Denger K., Cook A. M., Ruff J. (2004) Microbiology 150, 805–816 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.