

Abstract
It is conceivable that stimulating proteasome activity for rapid removal of misfolded and oxidized proteins is a promising strategy to prevent and alleviate aging-related diseases. Sulforaphane (SFN), an effective cancer preventive agent derived from cruciferous vegetables, has been shown to enhance proteasome activities in mammalian cells and to reduce the level of oxidized proteins and amyloid β-induced cytotoxicity. Here, we report that SFN activates heat shock transcription factor 1-mediated heat shock response. Specifically, SFN-induced expression of heat shock protein 27 (Hsp27) underlies SFN-stimulated proteasome activity. SFN-induced proteasome activity was significantly enhanced in Hsp27-overexpressing cells but absent in Hsp27-silenced cells. The role of Hsp27 in regulating proteasome activity was further confirmed in isogenic REG cells, in which SFN-induced proteasome activation was only observed in cells stably overexpressing Hsp27, but not in the Hsp27-free parental cells. Finally, we demonstrated that phosphorylation of Hsp27 is irrelevant to SFN-induced proteasome activation. This study provides a novel mechanism underlying SFN-induced proteasome activity. This is the first report to show that heat shock response by SFN, in addition to the antioxidant response mediated by the Keap1-Nrf2 pathway, may contribute to cytoprotection.
Keywords: Aging, Gene Regulation, Heat Shock Protein, Neurodegeneration, Proteasome, HSF1, Hsp27, Sulforaphane
Introduction
The ubiquitin-proteasome system (UPS)2 is a nonlysosomal protein degradation mechanism that plays a primary role in the control of protein turnover in mammalian cells as well as in the removal of abnormal proteins (1, 2). This tightly regulated proteasome complex is thus involved in the cell cycle, cellular signal transduction, transcriptional regulation, stress responses, cell differentiation, and metabolic adaptation (3, 4). Thus, UPS dysfunction has been implicated in the development of many diseases. Particularly, aging-related reduction of proteasome activity, which results in an accumulation of oxidized and ubiquitinated proteins and protein aggregation, has been attributed to diseases such as cataract formation, cardiac dysfunctions (transient ischemia or reperfusion, pressure overload, and inclusion body myositis) and neurodegenerative diseases (Alzheimer, Parkinson, and Hungtington) (3–6). Also, decreased proteasome activity leads to cellular senescence in mammals (7). These findings suggest that UPS is a potential therapeutic target for these diseases.
Sulforaphane (SFN), an isothiocyanate (ITC), is originally isolated from broccoli (8). It has been shown to be an effective cancer preventive agent in a variety of cancer cells and animal models, and the intake of dietary ITCs is associated with reduced human cancer risk (9–12). The major mechanism by which SFN, an electrophile, protects cells is believed to be through induction of direct and indirect antioxidant response, including up-regulation of glutathione and activation of NF-E2-related factor 2 (Nrf2)-mediated phase II enzymes and antioxidant enzymes that relieve cells from oxidant stress and carcinogenic attacks (11). However, its chemopreventive activity also includes other mechanisms, such as induction of cell cycle arrest and apoptotic pathways, inhibition of angiogenesis, and anti-inflammatory activity (9). Recently, SFN has been shown to enhance mammalian proteasome activity through induction of 26 S proteasome subunit PSMB5 (13). Both induction of PSMB5 and proteasome activation have been associated with antioxidant response mediated through the Keap1-Nrf2 pathway (13–16).
Despite extensive studies centering on the Keap1-Nrf2 signaling pathway by SFN, its effects on the heat shock response, a highly conserved and fundamental cytoprotective mechanism to heat shock or chemical stress, remain unexplored. Heat shock response features induced expression of heat shock proteins (HSPs) for repair of misfolded proteins caused by stressed conditions (17). Heat shock transcription factor 1 (Hsf1) plays an essential role in regulating expression of HSPs. When cells are under unstressed conditions, the majority of Hsf1 is sequestered by chaperones, including Hsp90 and Hsp70, and exists as a lament monomer in the cytoplasm. Upon stress, Hsf1 undergoes multistep activation, including release from HSP complexes, hyperphosphorylation, homotrimerization, nuclear translocation, and activation of genes with heat shock element (HSE) in their promoter regions (17, 18).
In particular, expression of heat shock protein 27 (Hsp27) (19), a small HSP that can be induced by a wide variety of stresses including heat shock, oxidative stress, anticancer drugs, and radiation (20), has been associated with elevated proteasome activity and rapid degradation of UPS substrates IκBα and p27kip1 (21, 22). Additionally, functions of Hsp27 include redox homeostasis, chaperone activity, thermo-tolerance, antiapoptosis, and regulation of cytoskeleton and cell development (23–25). Here, we report that SFN induces a rapid and significant heat shock response mediated by Hsf1. Also, our results indicate that Hsp27 expression, rather than its phosphorylation, underlies proteasome activity enhancement induced by SFN. This study shows for the first time that the SFN-induced heat shock response, like the antioxidant response, may mediate cytoprotection. This novel discovery may improve our knowledge of the chemopreventive activity by SFN and point to a new direction for its future research, which might provide novel leads for the discovery of compounds with more potent induction of both chaperone and proteasome activities.
MATERIALS AND METHODS
Cell Lines and Reagents
HeLa, COS-1 cell lines were grown in DMEM supplemented with 10% (v/v) fetal bovine serum, 100 unit/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) at 37 °C, 5% CO2. Rat colon carcinoma REG cells (no detectable Hsp27), REG-Hsp27 cells (REG cells stably transfected with wild-type Hsp27) were grown in Ham's F-10 medium supplemented with 10% (v/v) fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). SFN was generously provided by Dr. Stephen Hecht (University of Minnesota). Proteasome substrates Suc-LLVY-AMC, Boc-LRR-AMC, and Z-LLE-AMC, proteasome inhibitors MG132 and Epoxomicin, and p38 phosphorylation inhibitor SB203580 were purchased from Calbiochem. Actinomycin D and cycloheximide were purchased from Sigma-Aldrich. OGX-427, a second generation of antisense oligonucleotides against the Hsp27 gene was a gift from OncoGeneX (British Columbia, Canada). The control oligonucleotides for gene silencing experiment were provided by Dr. Jacques Landry (Universitaire de Québec, Canada).
Cell Lysate Preparation
For immunoblots, cytoplasmic proteins were extracted as supernatant after cells were lysed in buffer A containing 50 mm Tris-HCl, 10 mm KCl, 5 mm MgCl2, 0.5% Nonidet P-40, 1 mm DTT, and a protease inhibitor tablet, and centrifuged at 1,000 × g for 5 min. The pellet containing nuclear proteins was washed twice in buffer A and dissolved in buffer B containing 65 mm Tris-HCl, pH 8.0, 2% SDS, 150 mm NaCl, and 50 mm DTT.
Immunoblotting and Antibodies
Cell lysate with 20 μg of proteins was loaded on a SDS-polyacrylamide gel and separated by electrophoresis and then electroblotted onto polyvinylidene difluoride membranes (PVDF; Millipore). Immunoblot analysis was performed with specific antibodies and enhanced chemiluminescence-based detection (GE Healthcare). Antibodies against p38-MAPK and phosphorylated p38 were purchased from Cell Signaling (Danvers, MA). Antibodies against Hsp27, phosphorylated Hsp27, Hsf1, heat shock cognate protein 70, Hsp70, and Hsp90 were obtained from Assay Designs (Ann Arbor, MI). Antibodies against β-tubulin and ubiquitin were purchased from Sigma-Aldrich. Antibodies against p53 and Nrf2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Immunoprecipitation
Cells were treated with either 10 μm SFN for 15 and 30 min or heat shock at 42 °C for 30 min before being lysed in the radioimmune precipitation assay buffer containing 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm PMSF, 1 mm EDTA, 5 μg/ml aprotinin. Cell lysate was centrifuged at 10,000 × g for 10 min. The supernatant was incubated individually with anti-Hsp70 or anti-Hsp90 (1 μg) at 4 °C overnight followed by incubation with protein G-agarose beads (Santa Cruz Biotechnology) at room temperature for 1 h. The beads were washed four times with PBS and 0.1% Triton X-100 before being boiled in 2% SDS buffer and loaded for SDS-PAGE.
Immunofluorescence Staining
The assay was performed according to a published method (26). Briefly, 104 cells were cultured on microscope coverglass (12-mm diameter; FisherBrand) placed in individual wells of a 24-well tissue culture plate (BD Bioscience) for 24 h before being treated with either 5 μm SFN for heat shock for up to 1 h. The cells were then fixed with 3% paraformaldehyde in PBS (20 min), quenched with 50 mm NH4Cl in PBS, and permeabilized for 30 min with 1% BSA and 0.1% Triton X-100 in PBS. Cells were incubated overnight at 4 °C with mouse monoclonal anti-Hsf1 (1:400, clone E-4; Santa Cruz Biotechnology) and rabbit polyclonal anti-Nrf2 (1:400, clone C-20; Santa Cruz Biotechnology), rinsed three times with 0.1% BSA and 0.075% saponin in PBS, then incubated with Alexa Fluor 546-conjugated goat anti-mouse IgG and Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:800 for both; Invitrogen) for 1 h at room temperature. Once the labeling was complete, the coverglasses were rinsed twice with PBS and mounted onto microscope slides with a small drop of ProLong AntiFade reagent containing DAPI (Invitrogen). The samples were viewed, and images were taken with a Fluoview-FV300 Laser Scanning Confocal system (Olympus, Tokyo, Japan).
Reverse Transcription-PCR (RT-PCR)
Total RNA was extracted from cell lines using the RNeasy kit (Qiagen, Valencia, CA) with an on-column DNase digestion. A total of 1 μg of RNA was used as a template using the SuperScript One-step reverse transcription-PCR system (Invitrogen) The sequences of primers used in PCR were as follows: Hsp27: forward primer, 5′-CACGAGGAGCGGCAGGACGAG and reverse primer, 5′-CAGTGGCGGCAG CAGGGGTGG; β-actin: forward primer, 5′-TGACGGGGTCTACCCACACTGTG and reverse primer, 5′-CATTGCGCTGGACGATGGAGGG. RT-PCR products were separated on a 2% agarose gel containing ethidium bromide and visualized by ultraviolet light.
Ectopic Expression of Hsp27
A plasmid that contained Hsp27 (a gift from Dr. Tsuruo at the Japanese Foundation for Cancer Research, Tokyo, Japan) (25) was transfected to COS-1 cells using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. SFN treatment started after 48 h of transfection.
Hsp27 Knockdown
HeLa cells were treated at 50–60% confluence with 50 nm OGX-427 (27) or control oligonucleotides in serum-free DMEM without antibiotics using OligofectamineTM transfection reagent (Invitrogen). After 4 h of incubation at 37 °C, DMEM with 3× the normal concentration of serum was added, resulting in a final concentration of FBS of 10%. SFN treatment started after 48 h of transfection.
Hsf1 Knockdown
Hsf1 knockdown was achieved by lentiviral-based Hsf1 shRNA (5′-GCAGGTTGTTCATAGTCAGAA-3′). A GFP-targeted shRNA (5′-GCAAGCTGACCCTGAAGTTCA-3′) was used as a control. The virus particles were generated in HEK-293T cell by transfection of shRNA-carrying plasmids and pCMV-dR8.2 dvpr and pCMV-VSVG. Viral transduction was achieved by incubating target cells with virus-containing medium with 5 μg/ml Polybrene (Sigma). SFN treatment started after 24 h of transduction.
Assay for Proteasome Activity
Proteasome activity was analyzed based on a previously published method (15). After treatment, cells were harvested and lysed with proteasome lysis buffer (50 mm Tris-HCl, pH 7.8, 20 mm KCl, 5 mm MgCl2, 1 mm DTT, 1 mm ATP, 10% glycerol, and 0.04% Nonidet P-40) by repeated pipetting, followed by incubation on ice for 20 min. Cell lysate with 30 μg of proteins was incubated with 20 μm fluorogenic peptide substrates in 100-μl volume for 60 min at 37 °C in the dark. Cold PBS was used to dilute samples to 200 μl before fluorescence was measured by a Bio-Tek 96-well plate reader (Winooski, VT) with an excitation filter of 365 ± 20 nm and an emission filter of 485 ± 25 nm.
Luciferase Assay
HeLa cells were seeded in 6-well plates to 70–80% confluence and transfected with HSE-luc, a plasmid containing an inducible human Hsp70B promoter-driven luciferase reporter gene, by Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. After 24 h of growth, cells were treated with either 5 or 10 μm SFN for up to 4 h. As a positive control, cells were heat-shocked at 42 °C for 30 min and incubated at 37 °C for 3 h and 30 min. Luciferase assays were performed by using Dual-Luciferase Reporter Assay system (Promega, Madison, WI) according to the manufacturer's instructions. Firefly luciferase activity value was normalized to Renilla activity value. Promoter activity was presented as the fold of change compared with the vehicle-treated control.
Data Analysis
At least three independent experiments were conducted for all analyses. Values are expressed as means ± S.D. A Student's t test was used to compare averaged values, and p values of <0.05 were considered statistically significant.
RESULTS
SFN Induces Proteasome Activities in Both HeLa and COS-1 cells
To study the effects of SFN on proteasome activity, both HeLa and COS-1 cells were treated with 10 and 7.5 μm SFN, respectively, for up to 8 h. Proteasome activities were measured using three fluorescent-labeled peptide proteasome substrates corresponding to three distinctive proteasome activities. Results (Fig. 1) show that SFN treatment induced time-dependent chymotrypsin-like and caspase-like, but not trypsin-like, proteasome activity in both cell lines, which is consistent with a previous report showing the same effects in murine neuroblastoma Neuro2A cells (13). The results suggest that the induction of proteasome activity by SFN may be cell line-nonspecific. To confirm that the rise of caspase-like activity is proteasome-specific, we measured the population of apoptotic cells following 24 h of SFN treatment using Annexin V-propidium iodide staining. The results indicate that SFN at 7.5 or 10 μm did not induce noticeable apoptosis in cells (data not shown), ruling out contributions of apoptosis-related caspase activation. The notion that the activity is specifically associated with the proteasome is also supported by the observation that induction was completely attenuated by treatment with Epoxomicin, a specific proteasome inhibitor, at 10 μm for 1 h (Fig. 1A).
FIGURE 1.

SFN enhances proteasome activities in HeLa and COS-1 cells. Kinetic analysis of chymotrypsin-like, caspase-like, and trypsin-like proteasome activities measured by using Suc-LLVY-AMC, Z-LLE-AMC, and Boc-LRR-AMC substrates, respectively, in HeLa (A) and COS-1 (B) cells treated with 10 and 7.5 μm SFN, respectively. Treatment with 10 μm Epoxomicin (Epox), a known proteasome inhibitor, for 1 h was used to indicate that SFN-induced activities were proteasome-dependent. White bars, chymotrypsin-like activity; striped bars, trypsin-like activity; black bars, caspase-like activity. *, p < 0.05; **, p < 0.01. Error bars, S.D.
SFN-induced Proteasome Activation Requires de Novo Protein Synthesis
The time course of the proteasome induction suggests that SFN-induced proteasome activation may relate to de novo protein synthesis. To test this, we pretreated HeLa and COS-1 cells with the transcription inhibitor actinomycin D or the protein synthesis inhibitor cycloheximide for 1 h and then treated them with SFN for another 2 h. Results (Fig. 2A and B) show that both actinomycin D and cycloheximide completely blocked SFN-induced proteasome activation, but neither compound alone had any effect on proteasome activity, indicating that SFN-induced proteasome activation is likely mediated through de novo protein synthesis.
FIGURE 2.

SFN-induced proteasome activation dependents on de novo protein synthesis and SFN induces Hsp27 expression. HeLa (A) and COS-1 (B) cells were pretreated with actinomycin D (Act.D) or cycloheximide (CHX) for 1 h and then treated with SFN (10 μm for HeLa and 7.5 μm for COS-1). Chymotrypsin-like activity was measured using substrate Suc-LLVY-AMC. Hsp27 protein (C) and mRNA (D) levels were increased in SFN-treated cells. Cells were treated with SFN for up to 8 h. Hsp27 protein and mRNA levels were examined by immunoblotting and RT-PCR, respectively. β-Actin was used as loading control for both assays. Band intensity was estimated using ImageJ software and marked on top of each band. *, p < 0.05; **, p < 0.01. Error bars, S.D.
SFN Induces Rapid Hsp27 Up-regulation
To study whether SFN induces Hsp27, a protein known to be associated with proteasome activity regulation (21, 22), we treated both HeLa and COS-1 cells with 10 and 7.5 μm SFN, respectively, for up to 8 h. Hsp27 transcript and protein levels were examined by RT-PCR and immunoblotting, respectively. Results (Fig. 2, C and D) show that expression of Hsp27 was substantially induced by SFN as early as 2 h after treatment. Hsp27 mRNA and protein level increases were time-dependent in both cell lines.
SFN Activates Hsf1
Hsp27, like other HSPs, is regulated by Hsf1 (17, 18). To determine whether SFN activates Hsf1, we treated HeLa cells with 10 μm SFN, extracted nuclear proteins, and performed immunoblots. The results (Fig. 3A) show that Hsf1 was significantly and rapidly enriched in the nucleus in a time-dependent and concentration-dependent manner by SFN treatment, suggesting that Hsf1 was translocated into the nucleus, a sign of activation, by SFN. The Hsf1 activation is also supported by a progressive mass increase of Hsf1, presumably due to Hsf1 hyperphosphorylation, another signature event within Hsf1 activation (17). To confirm nuclear translocation of Hsf1, we immunostained Hsf1 in cells before and after SFN treatment and obtained fluorescent images using laser confocal microscopy. The results (Fig. 3B) show that Hsf1, stained in red, is localized to both the cytoplasm and the nucleus before SFN treatment. However, nuclear translocation of Hsf1 was rapid after either SFN or heat shock treatments. Nrf2, a signaling protein known to be activated by SFN through nuclear translocation (11), was used as an assay control. It is noted that heat shock did not induce substantial enrichment of Nrf2 in the nucleus.
FIGURE 3.

SFN activates Hsf1. A and B, SFN induces Hsf1 nuclear translocation and mass increase. A, HeLa cells were treated with 10 μm SFN for up to 4 h (left panel) and SFN at different concentrations for 4 h (right panel). HS, heat shock at 42 °C for 1 h and recovery at 37 °C for 3 h. p53 was used as a loading control for the nuclear proteins. B, HeLa cells were treated with either vehicle dimethyl sulfoxide (DMSO), 5 μm SFN, or heat shock for 1 h before immunofluorescent staining. Hsf1 was stained in red, Nrf2 in green, and the nucleus in blue by DAPI. C, SFN induces dissociation of Hsf1 from Hsp90 and Hsp70. HeLa cells were treated with 10 μm SFN or heat shock for up to 30 min before cell lysate was used to immunoprecipitate (IP) Hsp90 and Hsp70 individually.
Literature indicates that both Hsp90 and Hsp70 sequester Hsf1 in HSP complexes (28, 29). To demonstrate further that SFN activates Hsf1, we performed immunoprecipitation of Hsp90 and Hsp70 before and after SFN treatment. The results (Fig. 3C) show that both SFN and heat shock caused the dissociation of Hsf1 from Hsp90 and Hsp70, confirming that SFN activates Hsf1.
SFN Induces Heat Shock Response
To demonstrate SFN-induced and Hsf1-mediated heat shock response, we transiently transfected a luciferase reporter plasmid carrying Hsp70-based HSE and treated cells with SFN. The results (Fig. 4A) show that SFN activated Hsf1-mediated transcription. The activation is also time- and concentration-dependent. To confirm the activation, we checked protein levels of Hsf1 and some of its downstream gene products. Fig. 4B shows that SFN induced substantial expression of Hsf1 and Hsp70. However, the level of Hsp90 and Hsc70 remained the same. Taken together, these results indicate that SFN induces heat shock response, including overexpression of Hsp27, in cells.
FIGURE 4.
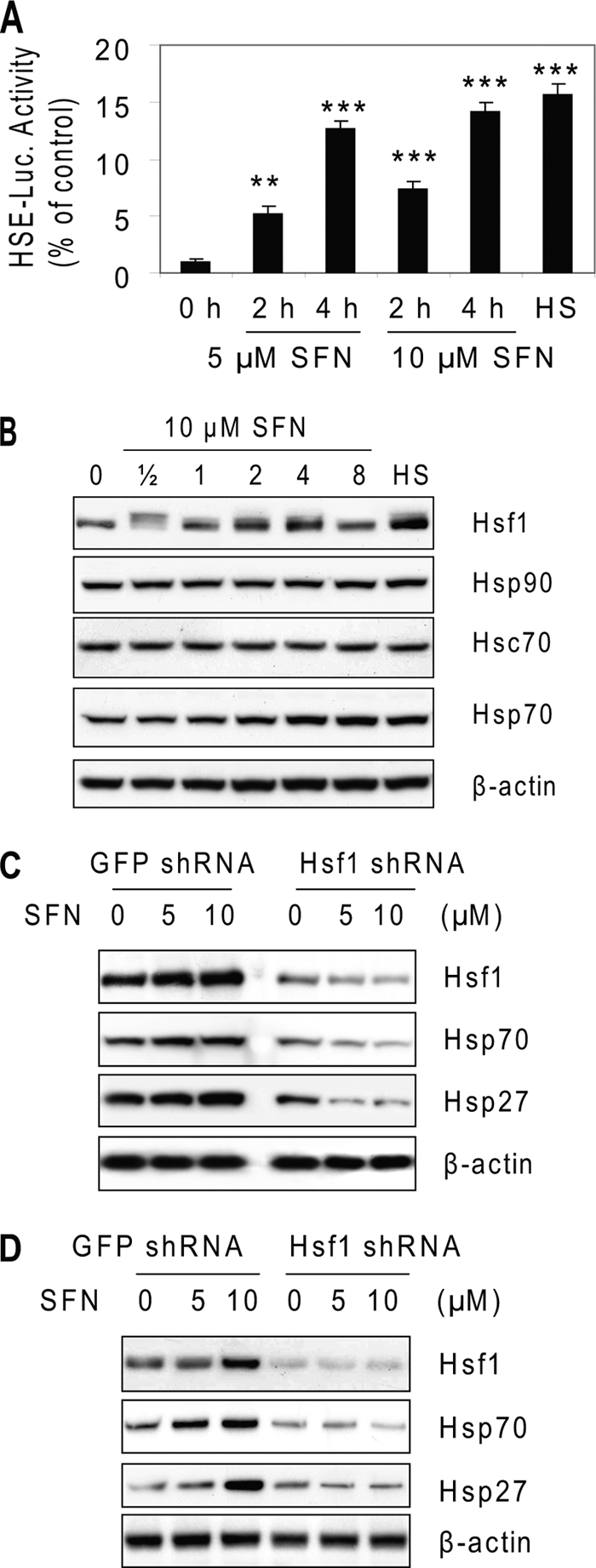
SFN induces Hsf1-mediated heat shock response. A, SFN activates Hsp70-based HSE-binding luciferase activity. HeLa cells were transiently transfected with Hsp70-Luc for 24 h and treated with 5 or 10 μm SFN for up to 4 h. **, p < 0.01; ***, p < 0.001. Error bars, S.D. B, SFN induces substantial expression of Hsf1 and Hsp70, but not Hsp90 and Hsc70. HeLa cells were treated with 10 μm SFN for up to 8 h before cell lysate was used for immunoblots. C, Hsf1 regulates overexpression of Hsp70 and Hsp27 by SFN in HeLa cells. HeLa cells were transduced by lentiviral-based Hsf1 shRNA for 24 h before being treated with 5 and 10 μm SFN for 8 h. GFP-targeted shRNA was used as a control. D, SFN activates Hsf1-mediated heat shock response in COS-1 cells. COS-1 cells were transduced by lentiviral-based Hsf1 shRNA for 24 h before being treated with 5 and 10 μm SFN for 8 h.
To ascertain the role of Hsf1 in regulating HSPs, especially Hsp27, we knocked down Hsf1 with lentiviral-based shRNA before SFN treatment. The results (Fig. 4C) show that Hsf1 was effectively knocked down by shRNA, and SFN-induced expression of Hsf1, Hsp70, and Hsp27 were abolished after Hsf1 knockdown, indicating that Hsf1 regulates overexpression of Hsp27 by SFN. Also, SFN-activated the Hsf1-mediated heat shock response was observed in COS-1 cells (Fig. 4D), indicating that the activation is unlikely cell type-specific.
Overexpression of Hsp27 Reinforces SFN-induced Proteasome Activation
We next questioned whether SFN-induced Hsp27 expression is functionally associated with proteasome activity. To answer this, we transiently transfected COS-1 cells with a plasmid carrying Hsp27 or an empty plasmid. COS-1 cells were chosen because the basal Hsp27 level is relatively low compared with HeLa cells. Results (Fig. 5A) show that SFN induced chymotrypsin-like activity >3-fold in cells with ectopically expressed Hsp27, compared with only 1.5-fold in control cells. The Fig. 5A inset shows that Hsp27 was selectively overexpressed with no substantial change in Hsc70. The results suggest that selective overexpression of Hsp27 may be related to SFN-induced proteasome activation. Also, proteasome activity in untreated cell lines was similar, suggesting that proteasome activity induction is triggered primarily by SFN treatment.
FIGURE 5.

Hsp27 expression regulates proteasome activities. A, ectopic expression of Hsp27 reinforces SFN-induced proteasome activation. COS-1 cells were transiently transfected with either a control plasmid (white bars) or a Hsp27-carrying plasmid (black bars) for 48 h before being treated with 7.5 μm SFN. Inset shows Hsp27 and Hsc70 levels by immunoblotting. B, silencing Hsp27 abolishes SFN-induced proteasome activation. HeLa cells were transiently transfected with either a control oligonucleotides (white bars) or OGX-427 for 48 h before being treated with 10 μm SFN. C, Hsp27 is required in SFN-induced proteasome activation. Parental REG cells (no detectable Hsp27) and REG stably transfected with wild-type Hsp27 were treated with 5 μm SFN. Inset shows Hsp27 and Hsc70 immunoblot images. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.D.
Silencing of Hsp27 Inhibits SFN-induced Proteasome Activation
To confirm the role of Hsp27 in SFN-induced proteasome activation, we pretreated HeLa cells with 50 nm OGX-427, a second generation antisense oligonucleotides against Hsp27 (27), prior to SFN treatment. HeLa cells were chosen because the basal Hsp27 level is relatively high. Results (Fig. 5B) show that OGX-427 significantly reduced Hsp27 protein expression (see inset) and inhibited SFN-induced proteasome activation, suggesting that Hsp27 expression is required for SFN-induced proteasome activation.
Hsp27 Expression in REG Cells Confers SFN-induced Proteasome Activation
To confirm the relationship between the Hsp27 expression level and proteasome activation further, we treated a pair of isogenic cells, parental rat colon cancer REG cells (no detectable Hsp27) and REG-Hsp27 cells (stably transfected with wild-type Hsp27), with 5 μm SFN before measuring proteasome activity. Results (Fig. 5C) show that the SFN-stimulated chymotrypsin-like activity was observed only in REG-Hsp27 cells, but not in parental REG cells, confirming that Hsp27 expression is essential for proteasome activation by SFN. Hsp27 and Hsc70 levels are shown in the inset.
Phosphorylation of Hsp27 Is Unrelated to SFN-mediated Proteasome Activation
Hsp27 can be phosphorylated at serines 15, 78, and 82 by p38-regulated/activated protein kinase and mitogen-activated protein kinases (MAPK)-associated protein kinases (MAPKAP kinases 2/3), which are activated through phosphorylation by p38 kinase (23, 30). Hsp27 phosphorylation has been associated with its reduced oligomerization and chaperone activity (23). A previous report showed that SFN activates the p38 MAPK pathway (31). To investigate whether Hsp27 phosphorylation plays a role in SFN-induced proteasome activation, we pretreated HeLa cells with 10 μm SB203580, a specific p38 MAPK inhibitor, for 1 h followed by treatment with SFN for 4 h. Results (Fig. 6A) show that SFN moderately induced phosphorylation of both p38 and Hsp27, and SB203580 effectively blocked their phosphorylation. However, inhibition of Hsp27 phosphorylation did not have substantial effects on SFN-induced proteasome activation (Fig. 6B), suggesting that Hsp27 phosphorylation may not be related to SFN-induced proteasome activation.
FIGURE 6.

Phosphorylation of Hsp27 is unrelated to SFN-induced proteasome activation. A, phosphorylation and protein levels of both p38 and Hsp27 in HeLa cells treated with 10 μm SFN in the presence or absence of 10 μm SB203580, a specific p38 MAPK inhibitor are shown. Cell lysates were collected after 4-h treatment. B, inhibition of Hsp27 phosphorylation did not block SFN-induced proteasome activation. C, SFN promoted similar levels of proteasome activity in cells transfected with three Hsp27 variants. Parental REG cells were transiently transfected with Hsp27-wt, Hsp27–3A, Hsp27–3D, and control vector pcDNA3 for 48 h before treated with 5 μm SFN. Inset shows Hsp27 and Hsc70 immunoblot images. *, p < 0.05; **, p < 0.01. Error bars, S.D.
To ascertain the role of Hsp27 phosphorylation, we transiently transfected REG cells with plasmids carrying Hsp27-wt, Hsp27–3A, and Hsp27–3D, for 48 h prior to SFN treatment. Hsp27–3A mimics dephosphorylated Hsp27 with three phosphorylatable serines replace with alanine residues, whereas three serines in Hsp27–3D were replaced with aspartates, mimicking phosphorylated Hsp27. Results (Fig. 6C) show that SFN-induced proteasome activation was observed in cells transfected with Hsp27 variants, but not those with an empty vector. No substantial differences were shown in cells with three Hsp27 variants, confirming that Hsp27 expression, but not phosphorylation, may be relevant to proteasome activation by SFN.
SFN Treatment Alleviates Cells from Proteasome Inhibition
To study the biological significance of the heat shock response induced by SFN, we transiently transfected HeLa cells with either pcDNA3 vector, a plasmid carrying Hsp27, or a plasmid carrying Hsf1 for 24 h. Cells were treated with 10 μm SFN for 2 h and recovered in fresh medium overnight followed by 500 nm MG132, a specific proteasome inhibitor, for 30 min. Results (Fig. 7) show that MG132, as expected, caused accumulation of ubiquitinated proteins (second lane); SFN-treated cell lysates had reduced amounts of ubiquitinated proteins (third through fifth lanes compared with second lane), suggesting that SFN alleviated MG132-induced proteasome inhibition. Cells transfected with either Hsp27 (fourth lane) or Hsf1 (fifth lane) had a greater reduction in the buildup of ubiquitinated proteins, confirming the role of Hsf1-mediated heat shock response and more specifically, Hsp27, in proteasome regulation. It is also noted that heat shock of cells also reduced the amount of ubiquitinated proteins, further supporting the role of heat shock response.
FIGURE 7.

Both SFN and heat shock treatments alleviate accumulation of ubiquitinated proteins by proteasome inhibitor MG132. HeLa cells were transiently transfected with control vector pcDNA3, a Hsp27-carrying plasmid, or a Hsf1-carrying plasmid for 24 h. Cells were pretreated with 10 μm SFN for 2 h, the culture medium was replenished, cells were incubated in fresh medium overnight, and then cells were treated with 500 nm MG132 for 30 min. Cell lysate was used to blot for ubiquitinated proteins. Heat shock was carried out at 42 °C for 1 h, followed by 37 °C overnight.
DISCUSSION
SFN is a promising cancer preventive agent. Previous studies indicate that its major cell protective mechanism is the induction of the Keap1-Nrf2 pathway (8–16). SFN-enhanced proteasome activity in murine neuroblastoma cells and induction of proteasome subunit PSMB5 have also been attributed to Nrf2-mediated antioxidant response (13). However, recent studies suggest that Hsf1, instead of Nrf2, is related to the stress-induced proteasome subunit expressions. A report revealed that a variety of HSPs and proteasome subunits, including Hsp27 and PSMB5, are significantly induced by WY-14643, a peroxisome proliferator-activated receptor α inducer, in nrf2-null mice (32). More recently, the interaction between Hsf1 and peroxisome proliferator-activated receptor α on a genetic level is considered a deciding factor in the regulation of some HSPs and proteasome subunit genes (33). Additionally, a previous study indicated that proteasome inhibition triggers Hsf1 activation, and a Hsf1-mediated rapid heat shock response has been implicated in modulating the UPS pathway (34). In this study, we discovered that SFN induces a rapid and significant Hsf1-mediated heat shock response and that SFN-induced Hsp27 expression, not its phosphorylation, underlies SFN-induced proteasome activity enhancement. Our results not only confirm the link between the heat shock response and proteasome activity but also specify the role of Hsp27 in stress-induced proteasome activation.
Like Nrf2-mediated antioxidant response, Hsf1-mediated heat shock response is also sensitive to redox conditions (34–36). It can be induced by a variety of thiol-reactive and electrophilic compounds, such as hydrogen peroxide, menadione, cadmium, arsenic trioxide, 15-deoxy-PGJ2, 4-hydroxynonenal, and iodoacetamide (35–42). However, the detailed mechanisms by which these compounds activate Hsf1 are unclear. It is possible that Hsf1 is activated indirectly through sensing accumulation of oxidized and misfolded proteins. It is also likely that the activation is due to direct impact on Hsf1 through thiol oxidation, S-glutathiolation, or disulfide formation (34–36). It has been proposed that SFN may covalently modify certain key cysteines on Keap1 for the activation of Nrf2 (11). Therefore, the possibility that SFN activates Hsf1 through covalent binding exists. The upstream mechanisms by which SFN activates Hsf1 require further investigation.
Both Hsf1-mediated heat shock response and Nrf2-mediated antioxidant response impart strong protection to stressed cells, including redox homeostasis (11, 23). Recently, 4-hydroxynonenal has been shown to activate both Nrf2 and Hsf1 as antiapoptotic responses (43). Interestingly, Hsf1-mediated cytoprotection is even stronger than that mediated by Nrf2. In addition to their overlapping functions, two seemingly parallel pathways may have cross-talk. Heme oxygenase 1, a known phase II enzyme with antioxidant activity, is a bona fide HSP (Hsp32) (43). Expression of NAD(P)H:quinone oxidoreductase, the most studied phase II enzyme, can be up-regulated by heat shock treatment of cells (44). Also, Hsp27 expression levels have been associated with activity of some detoxifying enzymes, including glucose-6-phosphate dehydrogenase, phase II enzymes glutathione S-transferase, and glutathione reductase (45). Understanding their relationships will improve our knowledge of both pathways and give insight to the biological activity of SFN.
The mechanism by which Hsp27 activates the proteasome and expedites removal of ubiquitinated proteins is still unclear. It is conceivable that Hsp27 stimulates the UPS system through protein interaction. αB-crystallin and αB-crystallin domain-containing Hsp27 have been shown to interact directly with ubiquitin and favorably bind to polyubiquitin chains (46, 47). Also, they are able to bind to the 26 S proteasome subunits (21, 22). Additionally, Hsp27 has been found to induce p27kip1 ubiquitination and degradation in a phosphorylation-independent manner. This is consistent with our results that Hsp27 phosphorylation is not related to proteasome activation by SFN.
Unlike SFN, benzyl ITC and phenethyl ITC, two other often-studied ITCs, induced neither Hsp27 protein accumulation nor proteasome activation at the same concentration range. In fact, both hydrophobic ITCs at concentrations above 5 μm rapidly and significantly inhibit activities in both 20 S and 26 S proteasome complexes, presumably through direct binding.3 Proteasome inhibition is also evident when cells are treated with more than 40 μm SFN.3 Our previous studies also revealed a contrasting structure-activity relationship among these three ITCs. Benzyl ITC and phenethyl ITC, which have much higher binding affinities toward tubulin than SFN, cause significant microtubule disruption, tubulin precipitation, tubulin degradation, and ultimately cell cycle arrest at the G2/M phase and apoptosis. SFN, on the contrary, is relatively inert in tubulin-related biological activities (26, 48–50).
Both HSPs and UPS are two key aspects of the protein quality control mechanism, which guarantees normal cellular functions and viability. Under stressed conditions, HSPs are dispatched to correct conformation and repair misfolded proteins whereas UPS is activated to remove proteins that are beyond repair. In this study, we showed that small molecular SFN can induce expression of HSPs and stimulate proteasome activity. These dual effects suggest SFN as a promising agent for the prevention and relief of cancer and various aging-related diseases, which commonly feature declined heat shock response and decreased proteasome activity.
Acknowledgments
We thank Dr. J. Christopher Corton (National Health and Environmental Effects Research Laboratory, U.S. Environmental, US EPA) and Dr. Jürgen Dohmen (University of Cologne, Germany) for fruitful discussion; Dr. Stephen Hecht (University of Minnesota) for providing the SFN; OncoGeneX (British Columbia, Canada) for providing OGX-427; Dr. Jacques Landry (Universitaire de Québec, Canada) for providing control oligonucleotides for Hsp27 gene silencing; Dr. Takashi Tsuruo (Japanese Foundation for Cancer Research, Japan) and Dr. Bianca Brundel (University Medical Center Groningen, Netherlands) for providing Hsp27-carrying plasmids; Dr. Carmen Garrido (INSERM UMR-866, France) for providing REG cells and Hsp27-carrying plasmids; Dr. Richard Morimoto (Northwestern University) for providing HSE-Luc and Hsf1-myc plasmids; and Lena Jia for the editorial assistance.
This work is supported by National Institutes of Health Grant CA100853 through the National Cancer Institute (to F.-L. C.).
L. Mi, N. Gan, and F.-L. Chung, unpublished data.
- UPS
- ubiquitin-proteasome system
- HSE
- heat shock element
- Hsf1
- heat shock transcription factor 1
- HSP
- heat shock protein
- ITC
- isothiocyanate
- Nrf2
- NF-E2-related factor 2
- SFN
- sulforaphane.
REFERENCES
- 1.Goldberg A. L. (2003) Nature 426, 895–899 [DOI] [PubMed] [Google Scholar]
- 2.Hoffman L., Rechsteiner M. (1996) Curr. Top. Cell Regul. 34, 1–32 [DOI] [PubMed] [Google Scholar]
- 3.Gottesman S., Wickner S., Maurizi M. R. (1997) Genes Dev. 11, 815–823 [DOI] [PubMed] [Google Scholar]
- 4.Groll M., Clausen T. (2003) Curr. Opin. Struct. Biol. 13, 665–673 [DOI] [PubMed] [Google Scholar]
- 5.Dahlmann B. (2007) BMC Biochem. 8, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu X., Patterson E., Kem D. C. (2009) Curr. Opin. Pharmacol. 9, 167–172 [DOI] [PubMed] [Google Scholar]
- 7.Stadtman E. R. (1992) Science 257, 1220–1224 [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y., Talalay P., Cho C. G., Posner G. H. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 2399–2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.World Health Organization (2004) IARC Handbook on Cancer Prevention, Vol. 9: Cruciferous Vegetables, Isothiocyanates and Indoles, IARC, Lyon, France [Google Scholar]
- 10.Zhang Y., Kensler T. W., Cho C. G., Posner G. H., Talalay P. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 3147–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dinkova-Kostova A. T., Holtzclaw W. D., Cole R. N., Itoh K., Wakabayashi N., Katoh Y., Yamamoto M., Talalay P. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 11908–11913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fowke J. H., Chung F. L., Jin F., Qi D., Cai Q., Conaway C., Cheng J. R., Shu X. O., Gao Y. T., Zheng W. (2003) Cancer Res. 63, 3980–3986 [PubMed] [Google Scholar]
- 13.Kwak M. K., Cho J. M., Huang B., Shin S., Kensler T. W. (2007) Free Radic. Biol. Med. 43, 809–817 [DOI] [PubMed] [Google Scholar]
- 14.Kwak M. K., Kensler T. W. (2006) Biochem. Biophys. Res. Commun. 345, 1350–1357 [DOI] [PubMed] [Google Scholar]
- 15.Kwak M. K., Wakabayashi N., Greenlaw J. L., Yamamoto M., Kensler T. W. (2003) Mol. Cell. Biol. 23, 8786–8794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwak M. K., Huang B., Chang H., Kim J. A., Kensler T. W. (2007) Life Sci. 80, 2411–2420 [DOI] [PubMed] [Google Scholar]
- 17.Wu C. (1995) Annu. Rev. Cell Dev. Biol. 11, 441–469 [DOI] [PubMed] [Google Scholar]
- 18.Westerheide S. D., Morimoto R. I. (2005) J. Biol. Chem. 280, 33097–33100 [DOI] [PubMed] [Google Scholar]
- 19.Arrigo A. P. (2007) Adv. Exp. Med. Biol. 594, 14–26 [DOI] [PubMed] [Google Scholar]
- 20.Didelot C., Schmitt E., Brunet M., Maingret L., Parcellier A., Garrido C. (2006) Handb. Exp. Pharmacol. 172, 171–198 [DOI] [PubMed] [Google Scholar]
- 21.Parcellier A., Schmitt E., Gurbuxani S., Seigneurin-Berny D., Pance A., Chantôme A., Plenchette S., Khochbin S., Solary E., Garrido C. (2003) Mol. Cell. Biol. 23, 5790–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parcellier A., Brunet M., Schmitt E., Col E., Didelot C., Hammann A., Nakayama K., Nakayama K. I., Khochbin S., Solary E., Garrido C. (2006) FASEB J. 20, 1179–1181 [DOI] [PubMed] [Google Scholar]
- 23.Arrigo A. P. (2001) IUBMB Life. 52, 303–307 [DOI] [PubMed] [Google Scholar]
- 24.Bruey J. M., Ducasse C., Bonniaud P., Ravagnan L., Susin S. A., Diaz-Latoud C., Gurbuxani S., Arrigo A. P., Kroemer G., Solary E., Garrido C. (2000) Nat. Cell Biol. 2, 645–652 [DOI] [PubMed] [Google Scholar]
- 25.Sakamoto H., Mashima T., Yamamoto K., Tsuruo T. (2002) J. Biol. Chem. 277, 45770–45775 [DOI] [PubMed] [Google Scholar]
- 26.Mi L., Xiao Z., Hood B. L., Dakshanamurthy S., Wang X., Govind S., Conrads T. P., Veenstra T. D., Chung F. L. (2008) J. Biol. Chem. 283, 22136–22146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamada M., So A., Muramaki M., Rocchi P., Beraldi E., Gleave M. (2007) Mol. Cancer Ther. 6, 299–308 [DOI] [PubMed] [Google Scholar]
- 28.Abravaya K., Myers M. P., Murphy S. P., Morimoto R. I. (1992) Genes Dev. 6, 1153–1164 [DOI] [PubMed] [Google Scholar]
- 29.Mosser D. D., Duchaine J., Massie B. (1993) Mol. Cell. Biol. 13, 5427–5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stokoe D., Engel K., Campbell D. G., Cohen P., Gaestel M. (1992) FEBS Lett. 313, 307–313 [DOI] [PubMed] [Google Scholar]
- 31.Yeh C. T., Yen G. C. (2005) Carcinogenesis 26, 2138–2148 [DOI] [PubMed] [Google Scholar]
- 32.Anderson S. P., Howroyd P., Liu J., Qian X., Bahnemann R., Swanson C., Kwak M. K., Kensler T. W., Corton J. C. (2004) J. Biol. Chem. 279, 52390–52398 [DOI] [PubMed] [Google Scholar]
- 33.Vallanat B., Anderson S. P., Brown-Borg H. M., Ren H., Kersten S., Jonnalagadda S., Srinivasan R., Corton J. C. (2010) BMC Genomics 11, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pirkkala L., Nykänen P., Sistonen L. (2001) FASEB J. 15, 1118–1131 [DOI] [PubMed] [Google Scholar]
- 35.Ahn S. G., Thiele D. J. (2003) Genes Dev. 17, 516–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee Y. K., Liu D. J., Lu J., Chen K. Y., Liu A. Y. (2009) J. Cell. Biochem. 106, 267–278 [DOI] [PubMed] [Google Scholar]
- 37.Bruce J. L., Price B. D., Coleman C. N., Calderwood S. K. (1993) Cancer Res. 53, 12–15 [PubMed] [Google Scholar]
- 38.Sarge K. D., Murphy S. P., Morimoto R. I. (1993) Mol. Cell. Biol. 13, 1392–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ialenti A., Grassia G., Di Meglio P., Maffia P., Di Rosa M., Ianaro A. (2005) Mol. Pharmacol. 67, 1620–1628 [DOI] [PubMed] [Google Scholar]
- 40.Manalo D. J., Liu A. Y. (2001) J. Biol. Chem. 276, 23554–23561 [DOI] [PubMed] [Google Scholar]
- 41.Jacobs A. T., Marnett L. J. (2007) J. Biol. Chem. 282, 33412–33420 [DOI] [PubMed] [Google Scholar]
- 42.Liu H., Lightfoot R., Stevens J. L. (1996) J. Biol. Chem. 271, 4805–4812 [PubMed] [Google Scholar]
- 43.Shibahara S., Müller R. M., Taguchi H. (1987) J. Biol. Chem. 262, 12889–12892 [PubMed] [Google Scholar]
- 44.Dong G. Z., Youn H., Park M. T., Oh E. T., Park K. H., Song C. W., Choi E. K., Park H. J. (2009) Int. J. Hyperthermia 25, 477–487 [DOI] [PubMed] [Google Scholar]
- 45.Préville X., Salvemini F., Giraud S., Chaufour S., Paul C., Stepien G., Ursini M. V., Arrigo A. P. (1999) Exp. Cell Res. 247, 61–78 [DOI] [PubMed] [Google Scholar]
- 46.Boelens W. C., Croes Y., de Jong W. W. (2001) Biochim. Biophys. Acta 1544, 311–319 [DOI] [PubMed] [Google Scholar]
- 47.den Engelsman J., Keijsers V., de Jong W. W., Boelens W. C. (2003) J. Biol. Chem. 278, 4699–4704 [DOI] [PubMed] [Google Scholar]
- 48.Mi L., Wang X., Govind S., Hood B. L., Veenstra T. D., Conrads T. P., Saha D. T., Goldman R., Chung F. L. (2007) Cancer Res. 67, 6409–6416 [DOI] [PubMed] [Google Scholar]
- 49.Mi L., Gan N., Cheema A., Dakshanamurthy S., Wang X., Yang D. C., Chung F. L. (2009) J. Biol. Chem. 284, 17039–17051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mi L., Gan N., Chung F. L. (2009) Biochem. Biophys. Res. Commun. 388, 456–462 [DOI] [PMC free article] [PubMed] [Google Scholar]