

Abstract
Maf1, first identified in yeast Saccharomyces cerevisiae, is a general negative regulator of RNA polymerase III (Pol III). Transcription regulation by Maf1 is important under stress conditions and during the switch between fermentation and respiration. Maf1 is composed of two domains conserved during evolution. We report here that these two domains of human Maf1 are resistant to mild proteolysis and interact together as shown by pull-down and size-exclusion chromatography and that the comparable domains of yeast Maf1 interact in a two-hybrid assay. Additionally, in yeast, a mutation in the N-terminal domain is compensated by mutations in the C-terminal domain. Integrity of both domains and their direct interaction are necessary for Maf1 dephosphorylation and subsequent inhibition of Pol III transcription on a nonfermentable carbon source. These data relate Pol III transcription inhibition to Maf1 structural changes.
Keywords: Protein Domains, Protein Phosphorylation, RNA Polymerase III, Transcription Regulation, Transfer RNA (tRNA)
Introduction
In eukaryotic cells, RNA polymerases (Pol)3 I and III are responsible for the synthesis of RNA species involved in ribosome biogenesis and the translation process. RNA synthesis by Pol I and Pol III represents >80% of all yeast nuclear transcription activity and is controlled in a coordinated way in response to various cellular and environmental conditions (1–3).
Pol III is responsible for the transcription of ∼300 different genes in yeast (class III genes), mostly tRNA genes (4–6). Analyses of the Pol III transcription system in yeast have revealed a series of protein-DNA and protein-protein interactions leading to the recruitment of Pol III to its target tRNA genes: binding of the six-subunit TFIIIC factor to the intragenic promoter, TFIIIC-directed recruitment and assembly of the three components of TFIIIB (TBP, Brf1, and Bdp1), and subsequent recruitment of the 17-subunit Pol III enzyme (7). Whereas the essential factors and the basal mechanisms of class III gene transcription are well defined, much less is known about the molecular mechanisms of Pol III regulation.
The unique global negative regulator of Pol III transcription in yeast is the Maf1 protein that mediates several signaling pathways, but is not essential (8, 9). In addition to the down-regulation that normally occurs in the stationary phase and in response to various drugs, also DNA damage, oxidative stress, secretory defects, and respiratory growth require Maf1 to achieve Pol III repression (8, 10–12). The activity of Maf1 is regulated by its phosphorylation, which occurs in favorable conditions. Apart from decreasing direct Maf1 binding to Pol III (13), this phosphorylation acts both to relocate the nuclear pool of Maf1 to the cytoplasm (14) and to prevent import of cytoplasmic Maf1 to the nucleus (15). Diverse unfavorable conditions lead to rapid Maf1 dephosphorylation and its nuclear accumulation, physical association of the dephosphorylated Maf1 with Pol III, and genome-wide Maf1 targeting to Pol III-transcribed genes (13, 16).
The Pol III machinery is remarkably conserved between yeast and human. The most conserved components are those involved in transcription complex assembly: the τ131 subunit of TFIIIC and two components of TFIIIB (TBP, Brf1). The five Pol III-specific subunits in yeast (C31, C34, C37, C53, and C82), all have structural and functional homologs in human cells (17). Also, Maf1 is conserved across eukaryotic organisms from yeast to man (9). This conservation is of particular interest considering that (mis)regulation of Pol III in human has been linked to malignant transformation. Excessive activation of Pol III-directed transcription can lead to tumorgenesis (18–21), and, in line with this observation, two mammalian tumor suppressors, Rb and p53, have been shown to act as global repressors of Pol III transcription (22). Recent results of several groups report Maf1-mediated repression of Pol III transcription in human implicating HsMaf1 ortholog as a new class of mammalian Pol III regulators (23–26). The involvement of HsMaf1 in the aberrant control of Pol III transcription in cancer cells remains to be studied. In the light of the high evolutionary conservation of the Pol III machinery including Maf1, insights into Pol III (mis)regulation by Maf1 gained by studying model organisms, such as yeast, should provide some insight into the role of HsMaf1 in cancer.
Our current interest concerns the relation between Maf1 structure and activity. All members of the Maf1 family have three fairly conserved segments (9) which, however, show no significant homology with protein domains of known function resulting in the striking lack of information on the functional significance of those regions. Point mutations have only highlighted the importance of several serine residues (mostly not phylogenetically conserved) and two nuclear localization sequences (15, 16, 27). We describe here identification of two conserved domains in HsMaf1 and show that the corresponding regions in yeast Maf1 interact. This interaction is crucial for the regulation of Maf1 activity by phosphorylation. Our data provide the first insight into the Maf1 structure in relation to Pol III regulation.
EXPERIMENTAL PROCEDURES
Expression and Purification of HsMaf1 Protein
Human full-length Maf1 (HsMaf1) (aa 1–256) was expressed as a C-terminal His6-tagged protein (HsMaf1-CHis) in insect cells. The protein was purified using cobalt affinity resin (Clontech). As a final purification step, the protein was applied to size-exclusion chromatography equilibrated in buffer I (10 mm Tris, pH 7.4, 150 mm NaCl, 1 mm DTT).
HsMaf1 fragments consisting of aa 1–45, 1–59, 1–63, 1–74, and 85–210 were co-expressed for 15 h at 18 °C in Escherichia coli BL21 Gold (DE3) cells (Stratagene) using expression vector pETMCN-His (C. Romier, IGBMC) coding for tobacco etch virus protease-cleavable N-terminal His-tagged HsMaf1 aa 1–45, 1–59, 1–63, and 1–74 and expression vector pETMCN (as above) coding for nontagged Maf1 aa 85–210. The complex was purified by nickel affinity chromatography (nickel-nitrilotriacetic acid; Qiagen) followed by tobacco etch virus protease cleavage and a second nickel affinity chromatography to remove the His tag. The complex was further purified by anion-exchange chromatography (MonoQ 10/100; GE Healthcare) and size-exclusion chromatography.
HsMaf1 fragment aa 82–236 was expressed from pETM11 (EMBL) as a potentially tobacco etch virus protease-cleavable, N-terminal His-tagged protein that was purified by nickel affinity chromatography (chelating Sepharose; GE Healthcare). However, the purified tagged protein HsMaf1-(aa 82–236) could not be cleaved by tobacco etch virus protease, most likely due to its aggregated state. The tagged protein was further purified by anion-exchange chromatography (5-ml HiTrap Q-Sepharose HP; GE Healthcare) and size-exclusion chromatography (S200 10/300; GE Healthcare).
Limited Proteolysis of HsMaf1 Protein
hsMaf1-CHis protein at a concentration of 1 mg/ml was digested with trypsin for 30 min at 4 °C in buffer I using a protease:protein ratio of 1:150 (w/w). The reaction was stopped by adding PMSF to a final concentration of 1 mm. The proteolysis product was concentrated to 2 mg/ml using an Amicon MWCO 3000 concentrator (Millipore) and subsequently purified using two consecutive Superdex 200 10/300 columns (GE Healthcare) to improve resolution in buffer I. Purified HsMaf1 fragments were unambiguously identified using a combination of Edman degradation and mass spectrometry.
Yeast Saccharomyces cerevisiae Strains and Media
The yeast strains used in this study included wild type YPH500 (MATα, ade2-101, his3-Δ200, leu2-Δ1, lys2-801, trp1-Δ63, ura3-52), maf1-Δ, a derivative of YPH500 (13), and the two-hybrid reporter strain Y190 (MATa, gal4-542, gal80-538, his3, trp1-901, ade2-101, ura3-52, leu2-3, 112, URA3::GAL1-lacZ, LYS2::GAL1(UAS):: HIS3, cyhR) (28). Rich media contained 1% yeast extract, 2% peptone, and 2% glucose (YPD) or 2% glycerol (YPGly). The minimal medium (SC) contained 2% glucose and 0.67% yeast nitrogen base without aa (29). Solid media contained 2% agar. All reagents were from Difco.
Construction of Plasmids to Express Fragments of Yeast Maf1 Protein for Two-hybrid Study
DNA encoding fragments aa 1–12, 1–16, and 1–23 of domain A of yeast Maf1 were synthesized as oligonucleotides. The larger DNA sequences, encoding aa 1–34, 1–39, and 1–42 were amplified using forward primer 5′-TCATCGGGATCCGAATGAAATTTATTGATGAGCTAGATATAGAGAGAGTG-3′ and reverse primers 5′-TCATCGCTCGAGTTTTCTATCTGATGCAACCGC-3′, 5′-TCATCGCTCGAGTGATGCAACCGCCTTTGTTGTG-3′, and 5′-TCATCGCTCGAGTGTTGTGAAAATATCGCAACTGCC-3′, respectively. The intron sequence of the MAF1 gene (localized between bp 7 and 87) was excluded. DNA encoding the amino acid 196–349 fragment of BC domain was amplified with primers 5′-TCATCGGGATCCGATCTGGTACAGCAACCAACAATG-3′ and 5′-TCATCGCTCGAGTTCGCCTGTACTCGAATTTAG-3′. All MAF1 parts were amplified as fragments with BamHI and XhoI termini and inserted into MATCHMAKER GAL4 Two-hybrid Vectors (Clontech), either the pACT2 plasmid carrying the activation domain of Gal4 or the pAS2 plasmid carrying the binding domain of Gal4. The resulting plasmids were named pACT2-Maf1-A(1–12), pACT2-Maf1-A(1–16), pACT2-Maf1-A(1–23), pACT2-Maf1-A(1–34), pACT2-Maf1-A(1–39), pACT2-Maf1-A(1–42), and pAS2-Maf1-BC(196–349). Expression of fusion proteins involving HA-tagged truncated versions of A domain was verified by Western blotting. Each of the derivatives of pACT2 and the single derivative of pAS2 were co-expressed pairwise in the two-hybrid reporter strain Y190. Cells containing these two plasmids were patched on SC medium lacking leucine and tryptophan. The patches were then examined for β-galactosidase activity using an overlay plate assay (30). The intensity of the coloration was calibrated by comparison with a pair of known interactors (τ95/τ55, two TFIIIC subunits) for which the β-galactosidase activity had been measured previously (31). For a β-galactosidase liquid assay, cell lysates were prepared, and the activity was measured colorimetrically as nmol of o-nitrophenyl-β-d-galactopiranoside hydrolyzed per minute per mg of protein. Conversion 0.0045 × A420 = 1 nmol of o-nitrophenyl-β-d-galactopyranoside cleaved was used (30).
Generation of Yeast S. cerevisiae Maf1 Mutant Strains
MAF1 gene was cut from pFL44-MAF1 (32) subcloned in pRS315 (LEU2, CEN) plasmid (33) resulting in pRS315-MAF1. The pAG70, pLM11 and pLM12 plasmids were derived from pRS315-MAF1 using a rapid method for localized mutagenesis (34). For this purpose, MAF1 fragment(aa 1–180) was PCR-amplified from pRS315-MAF1 under mutagenic conditions using 5′-CGAGTTGCTTGTCAATCAGG-3′ and 5′-CTGCTACTGCTCCTTCTTCT-3′ primers and a Diversify PCR Random Mutagenesis kit (Clontech). The product of the low-fidelity PCR was transformed together with gapped linear plasmid pRS315-MAF1 (digested with BclI and BsgI) into the YPH500 maf1-Δ strain (13). Transformants, selected on minimal medium lacking leucine, were subsequently tested for Maf1 activity by replica-plating on YPGly and incubation at 37 °C for 3 days; among 38 independent mutants, pAG70 (maf1-K35E) was selected from colonies that showed defective growth. To isolate pML11 (maf1-K35E/D250E) and pML12 (maf1-K35E/V260D/N344I) plasmids carrying suppressor mutations, pRS315 plasmid pAG70 (carrying the previously isolated maf1 allele with the K35E mutation) was digested with BsaBI and Bsu36I and introduced into the YPH500 maf1-Δ strain together with a MAF1 fragment encoding BC domain (aa 174–375) PCR-amplified under mutagenic conditions using primers 5′-AGAAGAAGGAGCAGTAGCAG-3′ and 5′-CGTATTCTCCTTCGTATTCA-3′. The obtained library of potential suppressor mutants was screened for overcoming the thermosensitivity on YPGly medium caused by the K35E mutation. This screen resulted in identification of pLM11 and pLM12 suppressor mutations in the BC domain of Maf1.
To generate mutations in the two-hybrid plasmids carrying fragments encoding Maf1 domains, the BamHI-XhoI fragment encoding aa 1–42 of Maf1 in the pACT2-Maf1-A(1–42) plasmid was substituted with a PCR-amplified fragment of the maf1 allele K35E from pAG70. Similarly, the mutations found in pML11 and pML12 were introduced in pAS2-Maf1-BC(196–349). The N344I mutation found in the pML12 plasmid was introduced to pAS2-Maf1-BC(196–349) by using a modified reverse primer 3′-TCATCGCTCGAGTTCGCCTGTACTCGAAATTAGACGCGAGC-5′ with a mutation leading to the desired amino acid substitution.
A QuikChange Site-directed Mutagenesis kit (Stratagene) was used to introduce D250E, N344I, V260D, and V260D/N344I to pRS315-MAF1. The sequences of primers used are available upon request.
Northern Blot Analysis
Cells (50 ml of liquid culture, A600 of approximately 0.8) were harvested by centrifugation and resuspended in 50 mm sodium acetate, pH 5.3, 10 mm EDTA. Total RNA was isolated by heating and freezing the cells in the presence of SDS and phenol as described previously (12, 35). RNA (5 μg/sample) was resolved by electrophoresis in 10% PAGE with 8 m urea, transferred to Hybond N+ membrane (Amersham Biosciences) by electroblotting in 0.5× TBE, and cross-linked by UV radiation (1200 mJ/cm2). The membrane was prehybridized in 7% SDS, 0.5 m sodium phosphate, pH 7.2, 1 mm EDTA, pH 7.0, 1% BSA and hybridized at 37 °C in the same solution with oligonucleotide probes labeled with [γ-32P]ATP and T4 polynucleotide kinase (New England Biolabs). The probes were 5′-TATTCCCACAGTTAACTGCGG-3′ for tRNALeu(CAA), 5′-CCTCCAGATGACTTGACCG-3′ for tRNAPhe(GAA), and 5′-GGATTGCGGACCAAGCTAA-3′ for U3 snoRNA. After hybridization, the blots were washed 2 × 10 min with 1× SSC and 1% SDS and 3 × 10 min with 0.5× SSC and 0.1% SDS at 37 °C and exposed to an x-ray film or a PhosphorImager screen (Molecular Dynamics).
Protein Extraction and Immunoblotting
To avoid action of endogenous kinases or phosphatases during cell harvesting and protein extraction, yeast cells were rapidly harvested by centrifugation at 4 °C, and 20% trichloroacetic acid was added to the cell pellet as described earlier (12, 13). Cells were broken with acid-washed glass beads, the supernatant was retained, and trichloroacetic acid-precipitated proteins were pelleted by centrifugation. The pellet was resuspended in sample buffer, pH 8.8, and boiled for 5 min. Protein extracts were separated on SDS-PAGE using a modified acrylamide:bisacrylamide ratio (33.5:0.3). One lane was loaded with protein from 1 absorbance unit of cell culture (10–20 μg). The membrane was blocked for 30 min in TBST (10 mm Tris, 150 mm NaCl, 0.05% Tween 20) containing 5% fat-free dry milk and then incubated for 1 h with Maf1-specific antibody at 1:10,000 dilution (12). The membrane was incubated with secondary anti-rabbit antibody coupled to horseradish peroxidase (DAKO) which was then visualized by chemiluminescence using the ECL detection kit (Millipore).
RESULTS
Two Domains of Maf1 Do Interact
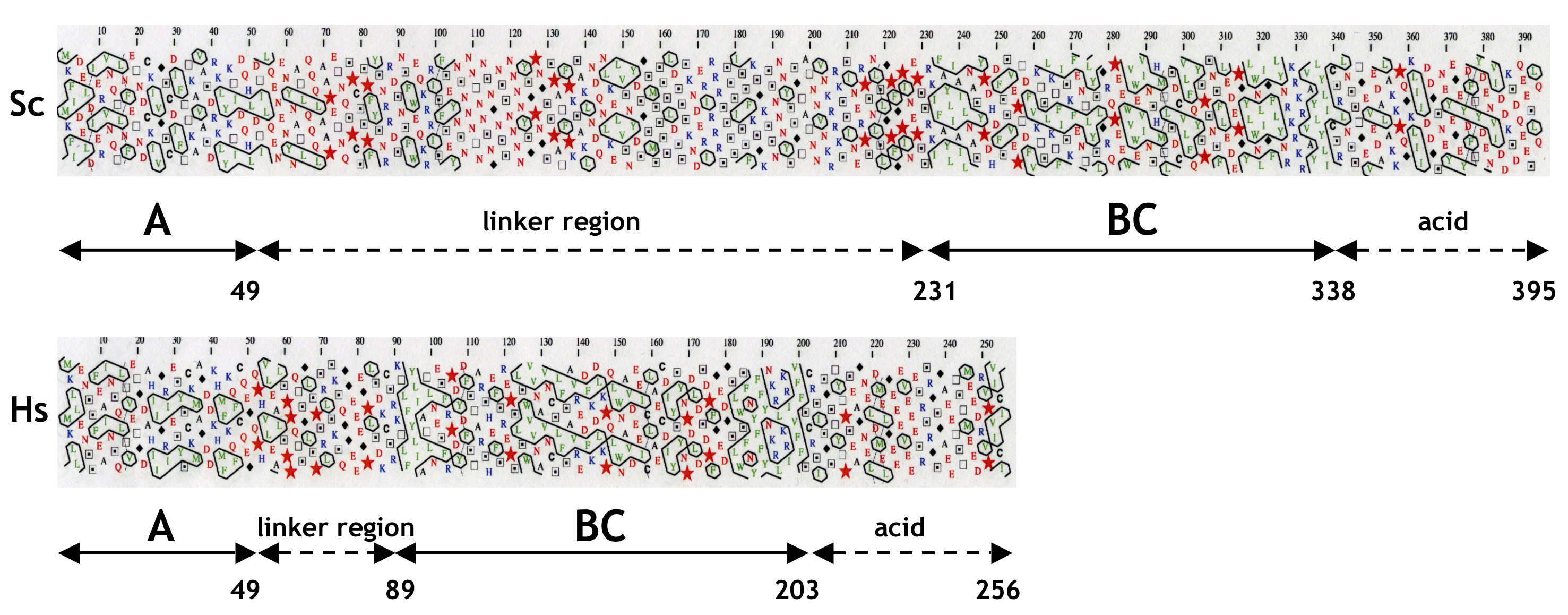
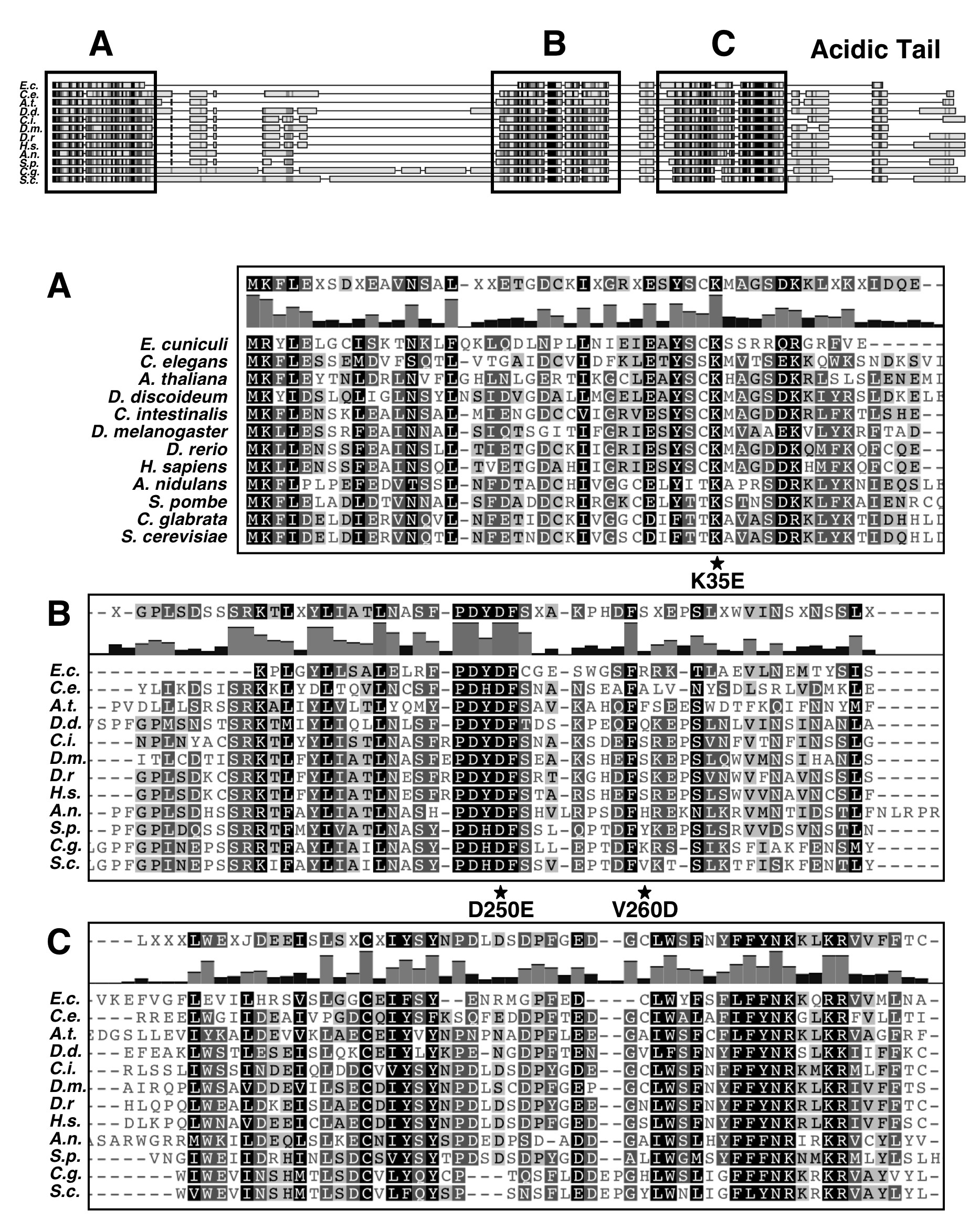
The yeast MAF1 gene encodes a hydrophilic protein of 395 aa rich in serine and asparagine residues, with a predicted molecular mass of 44.7 kDa. Screening of multiple databases with the yeast Maf1 sequence revealed numerous orthologs in other eukaryotes: 1 in human, 50 in animals, 28 in plants, and 27 in lower eukaryotes, but none in prokaryotes. Maf1 proteins contain three phylogenetically conserved sequence regions, labeled A, B, and C (9). The similarity of the yeast and human Maf1 sequences is presented in Fig. 1, and more Maf1 proteins are aligned in supplemental Fig. S1. Because the distance between the B and C segments of ∼10 aa is constant in evolution, with the exception of Aspergillus nidulans (insertion of 15 aa), we consider this region could be a single domain named here as BC. In contrast, the space between regions A and B largely varies between species. The A and BC domains are fused in Encephalitozoon cuniculi whereas in the yeast S. cerevisiae and Candida glabrata they are separated by a long linker of 182 and 174 aa, respectively. Within the BC domain signature sequences for the Maf1 protein family can be identified (PDYDFS and LWSFnYFFYNKklKR; supplemental Fig. S1) (9). These sequence “motifs” are not reported in the PROSITE database. Interestingly, in the majority of Maf1 orthologs, the second motif includes a nuclear targeting signal, which was proved to be functional in S. cerevisiae Maf1 (15).
FIGURE 1.

Similarity of S. cerevisiae (Sc) and H. sapiens (Hs) Maf1 sequences. Alignment of conserved A and BC domains is shown. A and BC domains are boxed, and conserved aa are highlighted. Stars indicate positions of K35E, D250E, and V260D mutations.
To characterize the structure organization of human Maf1 (HsMaf1) experimentally, we carried out limited proteolysis experiments in combination with size-exclusion chromatography as shown in Fig. 2. Proteolytically stable fragments are considered to be structurally well-defined, whereas protease-sensitive sites often correlate with disordered regions of the proteins. Using limited proteolysis with trypsin, HsMaf1 protein (256 aa) was digested into two major stable fragments that were identified as HsMaf1-(aa 1–45) and HsMaf1-(aa 75–234) using a combination of N-terminal sequencing and mass spectrometry. The HsMaf1 linker region between those fragments (aa 46–74) and the C-terminal acidic tail (aa 235–256) was degraded and thus presumably unstructured (Fig. 2A, lane 2). Both fragments were further analyzed by size-exclusion chromatography. Surprisingly, the two HsMaf1 fragments, although of substantially different molecular masses, co-eluted in an apparent 1:1 stoichiometry, suggesting an intramolecular interaction between them (Fig. 2B, red profile).
FIGURE 2.

Domain structure of human Maf1. A, limited proteolysis and resulting proteolytic fragments of HsMaf1. PAGE of full-length human HsMaf1 protein (1, black) that is digested into two stable fragments 1–45 and 75–234 using trypsin are indicated as 2, red. Bacterially co-expressed and co-purified HsMaf1 domains of aa 1–45 and 85–210 are shown as 4, green. HsMaf1 aa 82–236 (3, blue) lacking the N-terminal 45 residues was expressed as a control. B, size-exclusion chromatography profiles. Samples presented in A were separated on single or tandem Superdex 200 10/300 size-exclusion columns (GE Healthcare). Only HsMaf1 construct 82–236 elutes in the void volume of the Superdex 200 size-exclusion column, whereas all other samples are monodisperse and elute approximately at volumes corresponding to monomers. The elution profile of different molecular mass standards is shown as a dashed line. Peaks I, II, III, and IV correspond to ovalbumin (Mr = 44,000), carbonic anhydrase (Mr = 29,000), ribonuclease A (Mr = 13,700), and aprotinin (Mr = 6,500), respectively. Fractions corresponding to the red and green elution profiles were analyzed by denaturing gel electrophoresis and are shown as insets. Fragment 1–45 co-elutes with proteolytic fragments 75–234 and 75–262 (red profile) and with the recombinant fragment 85–210 (green profile).
Taking into account the limited proteolysis and secondary structure prediction results (data not shown), we designed N- and C-terminal constructs of HsMaf1 for co-expression in bacteria. The N-terminal construct aa 1–45 corresponds to the minimal domain defined by proteolysis whereas fragment encoding aa 85–210 was designed slightly shorter than the initial proteolytic fragment. During purification His tag-containing recombinant HsMaf1 fragment 1–45 but also slightly larger constructs aa 1–59, 1–63, and 1–74 co-precipitate the untagged C-terminal construct (85–210) in an apparent 1:1 stoichiometry, suggesting a direct interaction between the N- and C-terminal fragments (supplemental Fig. S2).
Analysis by size-exclusion chromatography supports the results obtained for the proteolytic fragments. Constructs(aa 1–45) and (aa 85–210) co-elute as a single peak at a volume corresponding to the expected molecular mass of ∼20 kDa (Fig. 2B). These results provide further support for a direct interaction between A and BC domains of HsMaf1. Interestingly, the co-expressed complex is considerably more compact compared with the full-length protein, presumably because it is lacking the C-terminal acidic tail. In contrast, an additional construct of aa 82–236 that includes the C-terminal acidic tail eluted as soluble aggregate (Fig. 2B, bottom panel) when expressed in the absence of the N-terminal fragment of aa 1–45. We also tried to express the N-terminal fragment of aa 1–45 as GST fusion protein, but we only obtained minimal amounts presumably because the protein aggregates after tobacco etch virus cleavage (data not shown). Apparently, N- and C-terminal domains of HsMaf1 are both required for the soluble expression of HsMaf1 and co-elute during size-exclusion chromatography, indicating a direct interaction between them.
To investigate whether the interaction between domains of Maf1 is conserved through evolution, we analyzed the proposed interaction in yeast, an organism more amenable to study structure-function relationships of Maf1 using the two-hybrid system. The putative BC domain of S. cerevisiae Maf1 (aa 196–349) was fused to the DNA-binding domain of Gal4 and co-expressed with various Maf1 A domain constructs fused to the Gal4 activation domain in the yeast reporter strain Y190. Interactions between fusion proteins should result in activation of the β-galactosidase reporter gene. Using this approach, we observed a physical interaction between the BC domain of Maf1 encoded by plasmid pAS2-Maf1-BC(196–349) and fragments of domain A of Maf1 encoded by plasmids pACT2-Maf1-A(1–42), pACT2-Maf1-A(1–39), and pACT2-Maf1-A(1–34) (Fig. 3). Domain BC failed to interact with shorter fragments of domain A encoded by pACT2-Maf1-A(1–12), pACT2-Maf1-A(1–16), or pACT2-Maf1-A(1–23). Cells containing the pair pAS2-Maf1-BC and empty pACT2 had no detectable β-galactosidase activity (data not shown in the figure). These results demonstrate the specificity of the two-hybrid interaction between Maf1 domains and define aa 1–34 as the smallest Maf1 A domain still able to bind the BC domain. Reciprocal interactions were impossible to study because the presence of pAS-Maf1-A activates the reporter gene in the absence of a pACT2 fusion. Importantly, pAS2-Maf1-BC was also negative tested with pACT2 encoding fusions of several genes unrelated to Maf1 and encoding components of Pol III complex. Taken together, our results as described above suggest a strong, direct interaction of the A and BC domains of human and yeast Maf1.
FIGURE 3.

Two-hybrid interaction of Maf1 domains. pACT2-Maf1-A(1–12), pACT2-Maf1-A(1–16), pACT2-Maf1-A(1–23), pACT2-Maf1-A(1–34), pACT2-Maf1-A(1–39), and pACT2-Maf1-A(1–42) plasmids were transformed individually together with pAS2 (control plasmid) or pAS2-Maf1-BC(196–349) plasmids into yeast strain Y190. Transformants were assayed for β-galactosidase expression using an overlay plate assay.
Interaction between A and BC Domains Is Important for the Function of Maf1
Limited proteolysis and two-hybrid results show that the A and BC domains of Maf1 together form a stable complex, possibly reflecting an active conformation of Maf1. To evaluate the physiological significance of the interaction between the Maf1 domains, we screened for mutants impaired in Maf1 function that were located in domain A and presumably compromised in domain BC binding. MAF1 fragment aa 1–180 was PCR-amplified under mutagenic conditions and transformed into maf1-Δ cells together with a gapped single-copy plasmid encoding Maf1. Transformants were selected for poor growth at 37 °C on glycerol medium, suggesting a defect of Maf1 function in Pol III repression (12). Sequencing of plasmid pAG70 encoding mutant Maf1 isolated in this manner revealed mutation K35E located in domain A.
To inspect the effect of the K35E mutation in Maf1 on Pol III activity, RNA isolated from cells grown in the presence of glucose and transferred to glycerol medium at 37 °C was analyzed by Northern blotting using probes for pre-tRNALeu and tRNAPhe (Fig. 4A). Following transfer to the medium with the nonfermentable carbon source, pre-tRNA levels were decreased in the wild type but not in maf1-Δ cells (Fig. 4A, compare lanes 1, 5 and 6, 10). Similarly to maf1-Δ, the K35E mutant was defective in its ability to repress pre-tRNA transcription upon transfer to glycerol medium (Fig. 4A, lanes 2 and I7). Thus, the single missense mutation within the Maf1 A domain precluded Pol III repression in maf1-K35E strain.
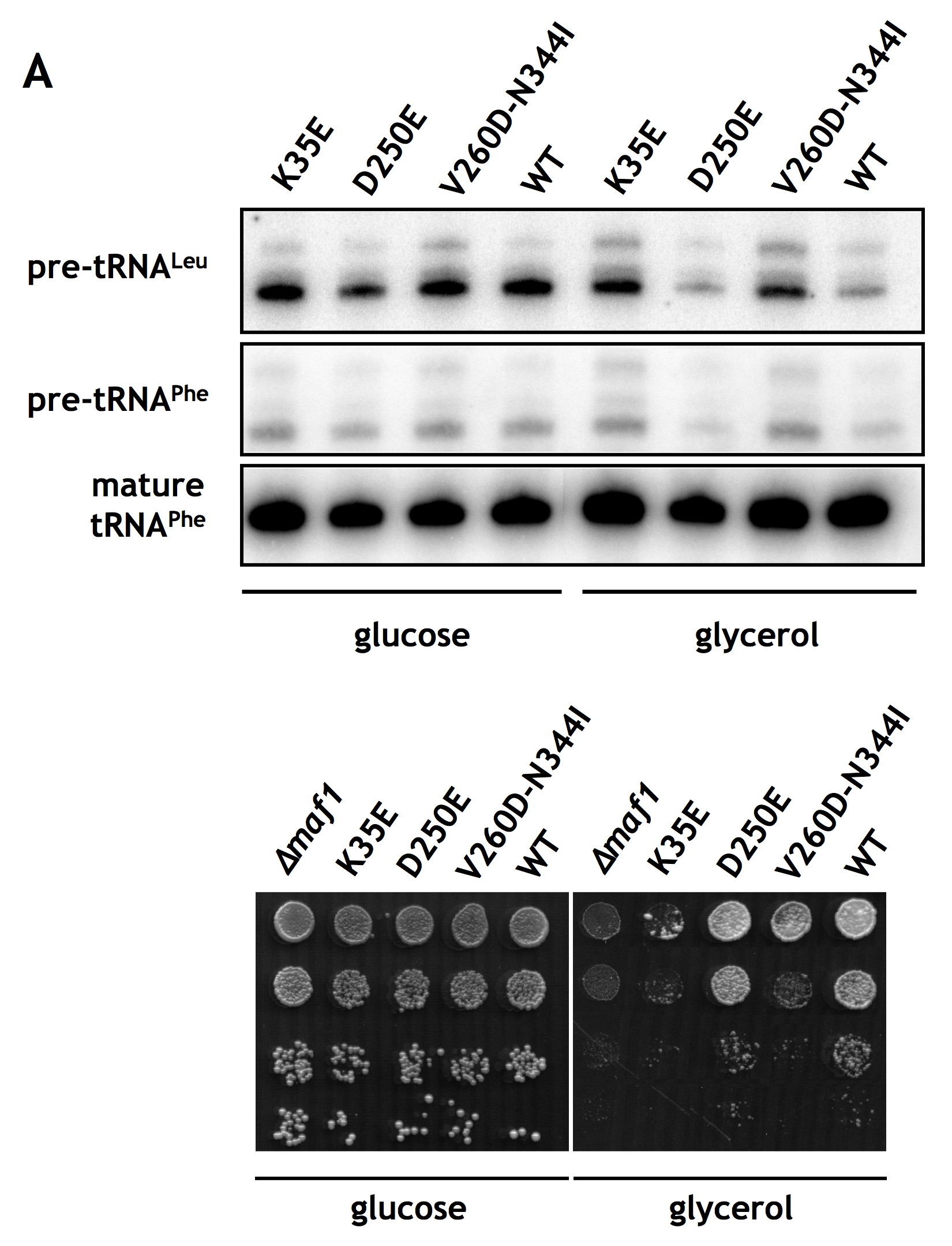
FIGURE 4.

Regulation of Pol III transcription and growth is impaired by the K35E mutation in region encoding A domain and restored by second-site suppressor mutations in BC domain of MAF1. maf1-Δ, maf1-K35E, maf1-K35E/D250E, and maf1-K35E/V260D/N344I mutants and isogenic wild type strain YPH500 (WT) were used. A, cells were grown to exponential phase in glucose medium (YPD) at 30 °C, then transferred to glycerol medium (YPGly) and incubated at 37 °C for 1.5 h. Total RNA isolated from cells was tested by Northern blotting with pre-tRNALeu and tRNAPhe probes. B, 10-fold serial dilutions of cells grown to exponential phase in glucose medium were plated on glucose medium (YPD) and incubated at 30 °C or on glycerol medium (YPGly) and incubated at 37 °C for 2–3 days.
One approach to confirm that the K35E mutation negatively affects the interaction with the BC domain of Maf1 is to identify second-site mutations that compensate for the observed defects. We looked therefore for second-site suppressor mutations within the BC-encoding region of MAF1 that allowed maf1-K35E yeast cells to grow on glycerol medium at 37 °C. DNA encoding aa 174–375 of Maf1 was randomly mutagenized by PCR, and the mutant pool was co-transformed with the pAG70 plasmid containing the primary K35E mutation and gapped within the region of the BC domain. Sequencing of in vivo reconstituted MAF1 from six transformants showing reversion of the original defect identified two plasmids with suppressor mutations: pLM11 (maf1-K35E/D250E) and pLM12 (maf1-K35E/V260D/N344I). The remaining transformants were either back-revertants of K35E mutation or contained suppressor mutations outside the MAF1 gene. Both isolated suppressors of K35E mutation in A domain, single D250E, and combined V260D/N344I were located in the BC domain, thus supporting the interaction between these domains.
Phenotypic characterization of the two suppressor strains showed that they compensate for the defect of growth on glycerol medium at 37 °C of the primary K35E mutation (Fig. 4B). The increased growth capacity of the suppressors on glycerol medium was due to a compensation of the defect in Pol III regulation observed for the K35E mutation. As determined by Northern blotting, the suppressor mutations restored the ability to repress pre-tRNA transcription upon transfer from glucose to glycerol medium (Fig. 4A, lanes 3, 4, 8, and 9). These results indicate that the detected genetic interaction between the two Maf1 domains is a functional one.
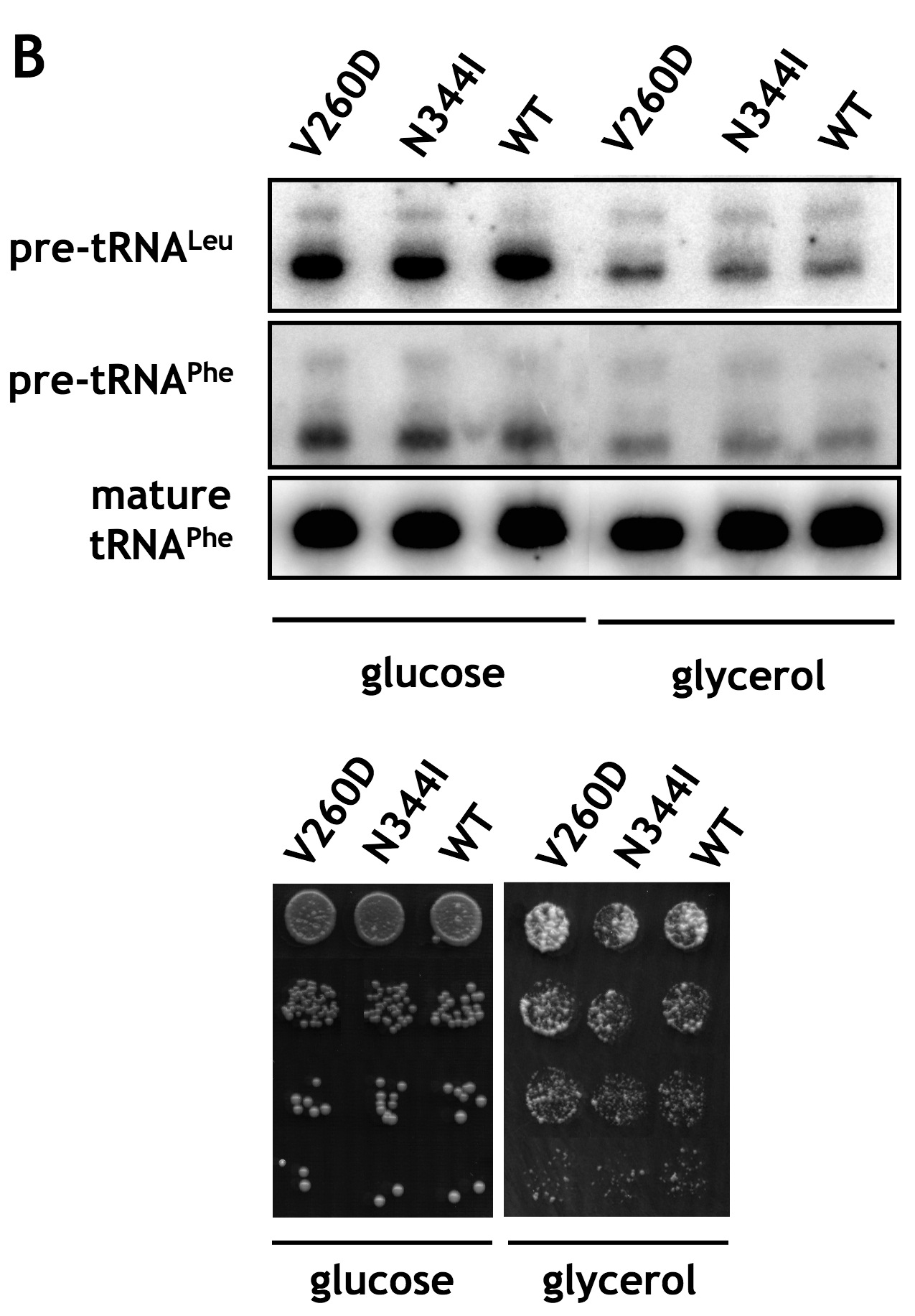
To characterize the association of A and BC domains further, we sought to verify the effects of K35E, D250E, V260D, and N344I mutations, identified in the mutant and suppressor strains, on the interaction of Maf1 domains in the two-hybrid system. The respective mutations were generated in the Maf1 A domain fragment fused to the Gal4 activation domain in the pACT2-Maf1-A(1–42) plasmid or the Maf1-BC domain fused to the DNA-binding domain of Gal4 in pAS2-Maf1-BC(196–349). Various combinations of plasmids, one encoding a wild type Maf1 domain and the other with a mutated domain, both fused to the respective Gal domains, were co-expressed in the yeast reporter strain followed by determination of β-galactosidase activity (Fig. 5). The K35E mutation of domain A reduced the two-hybrid interaction with wild type BC domain by approximately 40% (Fig. 5, compare lanes 2 and 1). Similarly, both the single D250E and the double V260D/N344I mutations in BC domain decreased the interaction with wild type A domain by 22 and 62%, respectively (Fig. 5, compare lanes 7 and 8 with lane 1). Moreover, the double V260D/N344I mutant showed a defect in regulation of tRNA transcription and temperature-sensitive growth in glycerol medium (supplemental Fig. S3).
FIGURE 5.

Two-hybrid interaction of Maf1 domains is impaired by primary mutations in these domains and restored by second-site suppressor mutations. A, pACT2-Maf1-A(1–42) plasmid and its mutated version were transformed individually with pAS2-Maf1-BC(196–349) plasmid or its mutated versions in yeast strain Y190. Transformants were assayed for β-galactosidase expression. Presented profile compares following two-hybrid interactions: wild type A and BC domains of Maf1 (1); mutated A domain K35E with wild type BC domain (2); mutated A domain K35E with four versions of mutated BC domain: D250E (3), V260D/N344I (4), V260D (5), N344I (6); and wild type A domain with mutated BC domain containing single D250E, V260D, or N344I and double N344I/V260D mutations (7-10). B, scheme of Maf1 domain interaction is shown.
Confirming the genetic suppressor results, a combination of mutated Maf1 domains restored their interaction (Fig. 5, compare lanes 3 and 4 with lane 1). Rather unexpectedly, the strongest level of two-hybrid interaction, 80% over the wild type one, was observed for the combination of K35E domain A and domain BC with a single V260D mutation (Fig. 5, compare lane 5 with lane 1). This indicated that in the double suppressor mutation V260D/N344I, identified in genetic screen, the former one is in fact sufficient to restore the interaction with K35E domain A.
Domain Interaction Supports Maf1 Dephosphorylation upon Transfer of Cells to a Nonfermentable Source
As shown before, phosphorylation of Maf1 precluded Pol III repression when yeast cells were grown on glucose medium (14). Transfer of yeast cells to a nonfermentable carbon source resulted in Maf1 dephosphorylation, import of Maf1 into the nucleus, and inhibition of tRNA synthesis (12). Because we showed that the interaction between the Maf1 A and BC domains is crucial for its regulatory action on Pol III, we now asked whether this interaction is also required for efficient regulation of Maf1 activity.
To investigate whether the altered domain interactions in the Maf1 mutant proteins were correlated with changes in the phosphorylation of Maf1, differentially phosphorylated forms of Maf1 were resolved by SDS-PAGE and identified by immunoblotting at various times after culture transfer from glucose to glycerol medium (Fig. 6). As reported previously, wild type Maf1 was quickly dephosphorylated upon this transition. Remarkably, the K35E mutation in domain A, preventing its interaction with the BC domain, appeared to preclude the dephosphorylation of Maf1. Significantly, restoring the domain interaction with D250E (pLM11) and V260D/N344I (pLM12) suppressor mutations reestablished rapid dephosphorylation of Maf1 following transfer from glucose to a nonfermentable carbon source. Maf1 dephosphorylation in the suppressor mutants was only a little slower than in the wild type strain (supplemental Table S1). Interestingly, double V260D/N344I mutation in the context of wild type A domain precluded interaction between domains (Fig. 5) and slowed the rate of dephosphorylation of Maf1 upon transfer of yeast to glycerol medium (supplemental Fig. S4). Altogether, these results suggest that the interaction of A and BC domain greatly facilitates the dephosphorylation of Maf1.
FIGURE 6.

Mutations altering interaction of Maf1 domains affect kinetics of Maf1 dephosphorylation upon transfer of yeast from glucose to medium with nonfermentable carbon source. maf1-K35E, maf1-K35E/D250E, and maf1-K35E/V260D/N344I mutant strains and isogenic wild type strain YPH500 (WT) were grown to exponential phase in YPD glucose medium (Exp), then transferred to glycerol YPGly medium, incubated at 37 °C, and harvested as indicated. Protein extracts of lysed cells were analyzed by Western blotting with polyclonal anti-Maf1 antibodies.
DISCUSSION
In this paper we describe the importance of the interaction between two Maf1 domains for its activity as a repressor of Pol III transcription. Limited proteolysis of HsMaf1 resulted in two stable fragments aa 1–45 and aa 75–234 corresponding to the evolutionarily conserved A and BC domains. Size-exclusion chromatography of the proteolytic fragments corresponding to the two domains showed their co-elution. Similar fragments were co-expressed in bacteria, where they co-purified and co-migrated during size-exclusion chromatography as a soluble complex. In contrast, both fragments behaved poorly when individually expressed. Our results suggest that Maf1 A and BC domains form modules that do not fold independently but rather need to be co-expressed to form a stable and soluble entity. In contrast, the connecting linker (aa 46–74) and the C-terminal acidic tail (aa 235–256) appear solvent-exposed and unstructured as suggested by hydrophobic cluster analysis (supplemental Fig. S6).
Considering the high conservation of Maf1 in eukaryotes, the domain interaction and its functional role were probed in the model organism yeast S. cerevisiae. Using the yeast two-hybrid system we confirmed the physical interaction between A and BC domains of Maf1 in yeast and identified the first 34 residues as the shortest fragment of the A domain sufficient for interaction with the BC domain. Lack of interaction between further truncated fragments of the A domain (aa 1–12, 1–16 or 1–23) with the BC domain shows the specificity of the method used and emphasizes the importance of structural features of the A domain for its interaction with the BC domain.
To investigate further the role of domain A we isolated single-point mutant K35E (on plasmid pAG70). In accordance with its growth characteristics, this mutant failed to repress Pol III activity. Moreover, as shown by the two-hybrid system, the K35E mutation reduced the interaction between the A and BC Maf1 domains by 40%. In support of structural and/or functional interactions between the Maf1 domains, we isolated second-site suppressor mutations within the BC domain that compensated for the defect caused by the K35E mutation in domain A. We found two such suppressors: D250E and V260D/N344I. Further phenotypic characterization of the suppressor strains revealed that these additional mutations in the BC domain restored not only the ability of the K35E-mutated Maf1 to grow on glycerol medium at 37 °C, but also its ability to repress Pol III activity. Interestingly, residue 260 in domain BC seems to be crucial for the interaction with domain A. Although the V260D mutant BC domain interacted with K35E mutant domain A >80% stronger than did wild type domain BC, its interaction with the wild type domain A was decreased by almost 60% (Fig. 5, fifth and ninth lanes). All of these results indicated that the genetic interaction identified between Maf1 domains corresponds to a physical interaction indispensable for the Maf1 function.
Bioinformatic analysis using the protein structure prediction server (PSIPRED version 2.6) (36, 37) provides further support for our biochemical analysis. In the yeast and human Maf1 A domain, two α-helices (aa 7–16 and 40–56) separated by two adjacent β-strands (aa 24–27 and 29–35) are predicted, whereas for the BC domain five α-helices and concomitant four β-strands are predicted (supplemental Fig. S5). No secondary structure elements are predicted for the linker region. Notably, the K35E mutation is located at the end of the second β-strand of the A domain. Our attempts to minimize the Maf1 A domain identified aa 1–34 as the smallest fragment still enabling interaction with the domains. We hypothesize that the interaction between domains A and BC requires the first α-helix and two adjacent β-strands. We also applied hydrophobic cluster analysis, a tool to investigate protein stability and folding that uses two-dimensional helical representation of protein sequences to identify possible hydrophobic cores formed by several residues (38). The hydrophobic cluster analysis revealed the presence of two regions rich in hydrophobic cores corresponding to the A and BC domains of S. cerevisiae Maf1 (supplemental Fig. S6). Between them a region poor in hydrophobic residues was found which, according to the alignment of Maf1 eukaryotic sequences, corresponds to the linker region between the A and BC domains. Similar analysis in human Maf1 revealed the same organization of two clusters of hydrophobic cores separated by a short region free of hydrophobic clusters consistent with our limited proteolysis results.
We found the interaction between the A and BC domains, facilitating Maf1 dephosphorylation, to be necessary for the full repression of Pol III activity. One might hypothesize that in the same time the interaction between domains of Maf1 is influenced by its phosphorylation state. Accordingly, different forms of Maf1 observed on polyacrylamide gels, interpreted as phosphorylated and subsequently dephosphorylated form of Maf1, might represent different conformational states depending on the interaction between A and BC domains. Note that the S90A, S101A, and S177A/S178A mutations in the linker were found to change the proportion between slow and fast migrating Maf1 forms in favor of the former, without strongly affecting the phosphorylation status of Maf1, thus indicating that residues in the linker could strongly modify the Maf1 shape (39).
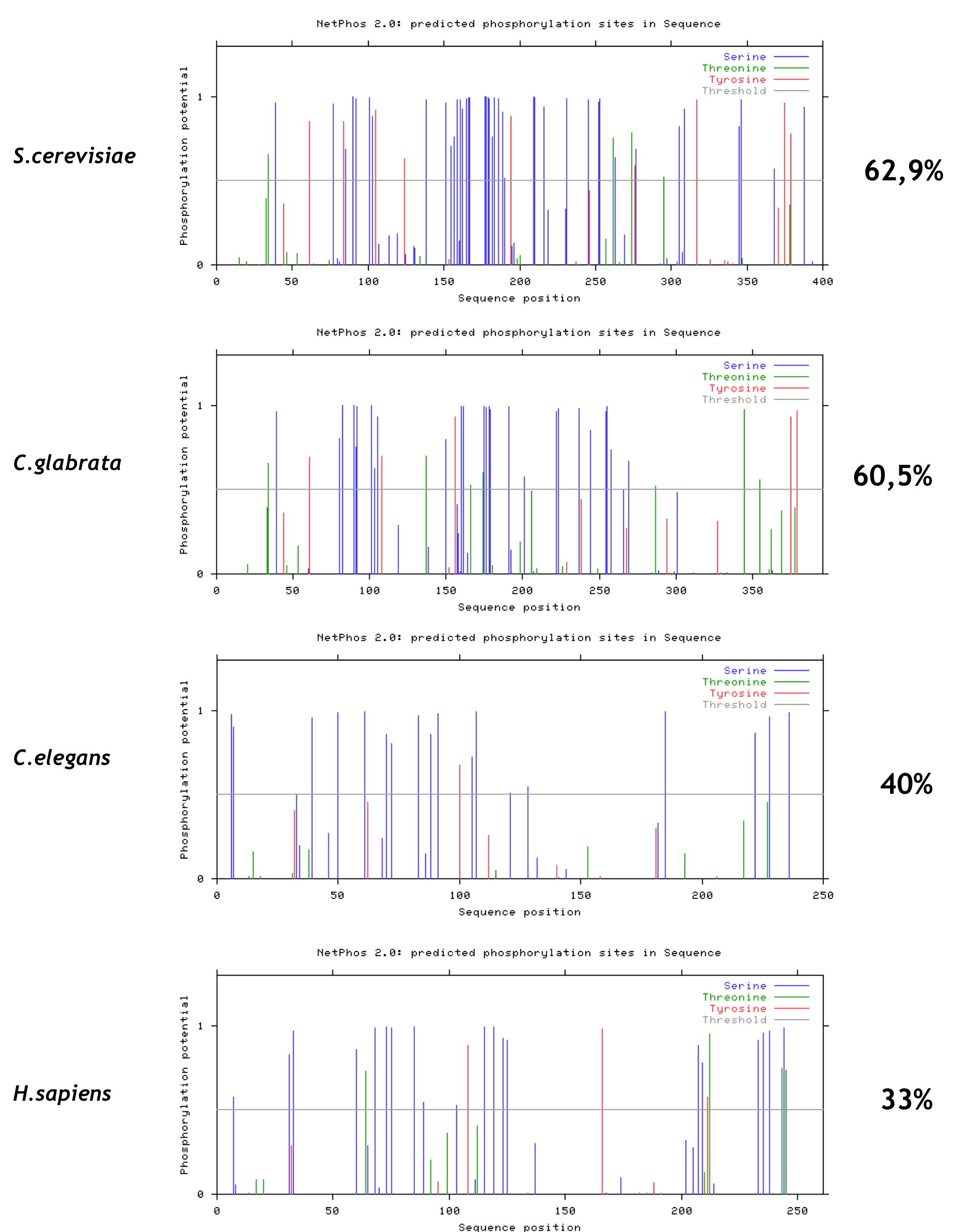
Considering that Maf1 contains two conserved domains, we propose the unstructured linker to become phosphorylated and thus modulating the conformational state of Maf1. To investigate this question further we used the NetPhos 2.0 server (40) to predict possible phosphorylation sites in four Maf1 orthologs containing linkers of different lengths. Predicted phosphorylation sites are frequently found in the long linkers between the Maf1 A and BC domains of S. cerevisiae and C. glabrata relatively, whereas they are more uniformly distributed along the Maf1 sequences of Caenorhabditis elegans and Homo sapiens, although they are also present in their shorter linkers (supplemental Fig. S7). We therefore speculate that phosphorylation and dephosphorylation of the exposed linker evoke a specific conformation of the protein that changes the distance between domains modulating their interaction and Pol III repression (supplemental Fig. S8). The interaction of the domains, mediated by dephosphorylation of the linker may affect both the import of Maf1 into the nucleus and its interaction with Pol III. This conjecture is supported by data of Ref. 15, showing that PKA-mediated phosphorylation of six residues located in linker inhibited nuclear import of Maf1 and Pol III repression.
Supplementary Material
Acknowledgments
Mass spectrum analysis was carried out at the Proteomics Core Facility at EMBL Heidelberg. We are grateful to Andre Sentenac, Joël Acker, Christine Conesa, Danuta Oficjalska-Pham, Raphaël Guerois, and Isabelle Callebaut for stimulating discussions. We also thank Gudrun von Scheven for technical assistance.
This work was supported by Ministry of Science and Higher Education, Poland, Grants N301 023 32/1117 and N301 243236, French National Research Agency Grant ANR-07-BLAN-0039-01, and Association pour la Recherche contre le Cancer Grant 1078.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S8 and Table S1.
- Pol
- polymerase
- aa
- amino acids.
REFERENCES
- 1.Willis I. M., Desai N., Upadhya R. (2004) Prog. Nucleic Acids Res. Mol. Biol. 77, 323–353 [DOI] [PubMed] [Google Scholar]
- 2.Warner J. R. (1999) Trends Biochem. Sci. 24, 437–440 [DOI] [PubMed] [Google Scholar]
- 3.Warner J. R., Vilardell J., Sohn J. H. (2001) Cold Spring Harbor Symp. Quant. Biol. 66, 567–574 [DOI] [PubMed] [Google Scholar]
- 4.Harismendy O., Gendrel C. G., Soularue P., Gidrol X., Sentenac A., Werner M., Lefebvre O. (2003) EMBO J. 22, 4738–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts D. N., Stewart A. J., Huff J. T., Cairns B. R. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 14695–14700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moqtaderi Z., Struhl K. (2004) Mol. Cell. Biol. 24, 4118–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geiduschek E. P., Kassavetis G. A. (2001) J. Mol. Biol. 310, 1–26 [DOI] [PubMed] [Google Scholar]
- 8.Upadhya R., Lee J., Willis I. M. (2002) Mol. Cell 10, 1489–1494 [DOI] [PubMed] [Google Scholar]
- 9.Pluta K., Lefebvre O., Martin N. C., Smagowicz W. J., Stanford D. R., Ellis S. R., Hopper A. K., Sentenac A., Boguta M. (2001) Mol. Cell. Biol. 21, 5031–5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boisnard S., Lagniel G., Garmendia-Torres C., Molin M., Boy-Marcotte E., Jacquet M., Toledano M. B., Labarre J., Chédin S. (2009) Eukaryot. Cell 8, 1429–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desai N., Lee J., Upadhya R., Chu Y., Moir R. D., Willis I. M. (2005) J. Biol. Chem. 280, 6455–6462 [DOI] [PubMed] [Google Scholar]
- 12.Ciesla M., Towpik J., Graczyk D., Oficjalska-Pham D., Harismendy O., Suleau A., Balicki K., Conesa C., Lefebvre O., Boguta M. (2007) Mol. Cell. Biol. 27, 7693–7702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oficjalska-Pham D., Harismendy O., Smagowicz W. J., Gonzalez de Peredo A., Boguta M., Sentenac A., Lefebvre O. (2006) Mol. Cell 22, 623–632 [DOI] [PubMed] [Google Scholar]
- 14.Towpik J., Graczyk D., Gajda A., Lefebvre O., Boguta M. (2008) J. Biol. Chem. 283, 17168–17174 [DOI] [PubMed] [Google Scholar]
- 15.Moir R. D., Lee J., Haeusler R. A., Desai N., Engelke D. R., Willis I. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 15044–15049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts D. N., Wilson B., Huff J. T., Stewart A. J., Cairns B. R. (2006) Mol. Cell 22, 633–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schramm L., Hernandez N. (2002) Genes Dev. 16, 2593–2620 [DOI] [PubMed] [Google Scholar]
- 18.Marshall L. (2008) Cell Cycle 7, 3327–3329 [DOI] [PubMed] [Google Scholar]
- 19.Marshall L., Kenneth N. S., White R. J. (2008) Cell 133, 78–89 [DOI] [PubMed] [Google Scholar]
- 20.Marshall L., White R. J. (2008) Nat. Rev. Cancer 8, 911–914 [DOI] [PubMed] [Google Scholar]
- 21.Johnson S. A., Dubeau L., Johnson D. L. (2008) J. Biol. Chem. 283, 19184–19191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White R. J. (2008) Trends Genet. 24, 622–629 [DOI] [PubMed] [Google Scholar]
- 23.Goodfellow S. J., Graham E. L., Kantidakis T., Marshall L., Coppins B. A., Oficjalska-Pham D., Gérard M., Lefebvre O., White R. J. (2008) J. Mol. Biol. 378, 481–491 [DOI] [PubMed] [Google Scholar]
- 24.Reina J. H., Azzouz T. N., Hernandez N. (2006) PLoS ONE 1, e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson S. S., Zhang C., Fromm J., Willis I. M., Johnson D. L. (2007) Mol. Cell 26, 367–379 [DOI] [PubMed] [Google Scholar]
- 26.Rollins J., Veras I., Cabarcas S., Willis I., Schramm L. (2007) Int. J. Biol. Sci. 3, 292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huber A., Bodenmiller B., Uotila A., Stahl M., Wanka S., Gerrits B., Aebersold R., Loewith R. (2009) Genes Dev. 23, 1929–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harper J. W., Adami G. R., Wei N., Keyomarsi K., Elledge S. J. (1993) Cell 75, 805–816 [DOI] [PubMed] [Google Scholar]
- 29.Sherman F. (2002) Methods Enzymol. 350, 3–41 [DOI] [PubMed] [Google Scholar]
- 30.Werner M., Chaussivert N., Willis I. M., Sentenac A. (1993) J. Biol. Chem. 268, 20721–20724 [PubMed] [Google Scholar]
- 31.Manaud N., Arrebola R., Buffin-Meyer B., Lefebvre O., Voss H., Riva M., Conesa C., Sentenac A. (1998) Mol. Cell. Biol. 18, 3191–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boguta M., Czerska K., Zoładek T. (1997) Gene 185, 291–296 [DOI] [PubMed] [Google Scholar]
- 33.Sikorski R. S., Hieter P. (1989) Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muhlrad D., Hunter R., Parker R. (1992) Yeast 8, 79–82 [DOI] [PubMed] [Google Scholar]
- 35.Schmitt M. E., Brown T. A., Trumpower B. L. (1990) Nucleic Acids Res. 18, 3091–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones D. T. (1999) J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 37.Bryson K., McGuffin L. J., Marsden R. L., Ward J. J., Sodhi J. S., Jones D. T. (2005) Nucleic Acids Res. 33, W36–W38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Callebaut I., Labesse G., Durand P., Poupon A., Canard L., Chomilier J., Henrissat B., Mornon J. P. (1997) Cell Mol. Life Sci. 53, 621–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee J., Moir R. D., Willis I. M. (2009) J. Biol. Chem. 284, 12604–12608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blom N., Gammeltoft S., Brunak S. (1999) J. Mol. Biol. 294, 1351–1362 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}