

Abstract
Several mammalian forkhead transcription factors have been shown to impact on cell cycle regulation and are themselves linked to cell cycle control systems. Here we have investigated the little studied mammalian forkhead transcription factor FOXK2 and demonstrate that it is subject to control by cell cycle-regulated protein kinases. FOXK2 exhibits a periodic rise in its phosphorylation levels during the cell cycle, with hyperphosphorylation occurring in mitotic cells. Hyperphosphorylation occurs in a cyclin-dependent kinase (CDK)·cyclin-dependent manner with CDK1·cyclin B as the major kinase complex, although CDK2 and cyclin A also appear to be important. We have mapped two CDK phosphorylation sites, serines 368 and 423, which play a role in defining FOXK2 function through regulating its stability and its activity as a transcriptional repressor protein. These two CDK sites appear vital for FOXK2 function because expression of a mutant lacking these sites cannot be tolerated and causes apoptosis.
Keywords: CDK (Cyclin-dependent Kinase), Cell Cycle, Gene Transcription, Protein Phosphorylation, Transcription Factors, Forkhead, FoxK2
Introduction
The forkhead transcription factors are an evolutionarily conserved family of proteins that can be found in organisms ranging from yeasts through to humans (reviewed in Refs. 1 and 2). There are more than 40 different human forkhead transcription factors. All forkhead transcription factors contain the forkhead winged helix DNA binding domain but can be further subgrouped according to the presence of additional domains and overall sequence conservation. One such subfamily contains FOXK1 and FOXK2, which both possess an FHA3 domain in addition to a highly conserved forkhead DNA binding domain.
Forkhead transcription factors control a wide range of cellular processes, and many have been shown to have developmental roles (reviewed in Ref. 2) and be deregulated in cancer (reviewed in Ref. 3). However, one fundamental process that is controlled by forkhead transcription factors is the cell cycle. This was first elucidated in Saccharomyces cerevisiae, where the forkhead proteins Fkh1p and Fkh2p were shown to play a key role in the periodic control of gene expression at the G2-M phase of the cell cycle (4–7). Since then, forkhead transcription factors have been associated with controlling transcription during M-G1 in Schizosaccharomyces pombe (8, 9) and more recently linked to cell cycle transcription in mammalian systems (reviewed in Refs. 10 and 11). Members of the FOXO subfamily have been linked to both G1-S (12) and G2-M control (13) (reviewed in Ref. 10), whereas FOXM1 has been associated mainly with controlling gene expression at the G2-M boundary (reviewed in Refs. 11 and 14).
In addition to their role in regulating transcription during the cell cycle, forkhead transcription factors have themselves been shown to be controlled by components of the core cell cycle regulatory machinery, including cell cycle-regulated kinases. In S. cerevisiae, Fkh2p is a target for CDK·cyclin B-type kinases, and this triggers its activation (15). Furthermore, Fkh2p recruits the coactivator Ndd1p through a phosphodependent interaction with its FHA domain. This interaction is triggered by CDK·cyclin B-dependent phosphorylation of Ndd1p (16, 17). Further activation of this complex is then achieved through phosphorylation of Ndd1p by the polo kinase Cdc5p (18). In mammalian cells, FOXM1 appears to play a role analogous to that of Fkh2p in controlling G2-M transcription and is activated in a similar manner by a combination of CDK·cyclin complexes (19–21) and the polo kinase Plk-1 (22), which converge on FOXM1 rather than also targeting a co-activator protein. Moreover, FOXO transcription factors have also been shown to be targets of CDK·cyclin kinases, demonstrating the widespread nature of connections between cell cycle regulatory kinases and forkhead transcription factors (reviewed in Ref. 10).
FOXK1 and FOXK2 have domain structures that resemble yeast Fkh2p in that they all contain a FHA domain. However, little is known about FOXK2 and its regulatory activities, other than its initial discovery as a regulator of IL-2 transcription (23). The mouse FOXK1 homologue, MNF, has been linked to cell cycle control because its loss in myogenic stem cells causes proliferative defects (24), in part through up-regulation of p21 expression (25). More recently, FOXK1 has also been linked to SRF-dependent gene regulation, which shows parallels with the interactions between yeast Fkh2p and the SRF-like protein Mcm1p (26).
Here, we have investigated the potential role of FOXK2 in the cell cycle. We demonstrate that FOXK2 is phosphorylated in a cell cycle-dependent manner. This phosphorylation peaks during M phase and is mediated by CDK·cyclin complexes. We have identified two sites of modification that play a role in controlling the activity of FOXK2. Our results therefore indicate that FOXK2 is linked to the cell cycle regulatory machinery.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
The following plasmids were used in mammalian cell transfections. pCH110 (Amersham Biosciences) and p21-luc (kindly provided by Neil Perkins). pAS2252 (pCMV-driven construct encoding full-length FLAG-tagged human FOXK2) was constructed by a three-step procedure. The NcoI/BglII fragment of pAS1191 (Gal-FOXK2, kindly provided by Richard Goodman (27)) was first ligated into the same sites in the pBs-based vector pAS728 to create pAS1199. Next, a BglII/XbaI-cleaved PCR fragment (primer pair ADS1303/ADS1304 with pAS1191 as a template) was ligated into the same sites in pAS1199, creating pAS2251. The KpnI/XbaI full-length encoding the FLAG-tagged FOXK2 fragment of pAS2251 was then ligated into pCMV5 using KpnI/XbaI to create pAS2252. pAS1424 (encoding FOXK2(S368A/S423A)) was constructed by a two-step QuikChange mutagenesis strategy (Stratagene) using the primer·template combinations ADS1716/ADS1717·pAS2252 to create pAS1422 (encoding FOXK2(S368A)) and then ADS1718/ADS1719·pAS1422. Similarly, pAS2550 (encoding FOXK2(S368D/S423D)) was constructed using primer·template combinations of ADS2372/ADS2373·pAS2252 to create pAS2549 (encoding FOXK2(S368D)) and then ADS2374/ADS2375· pAS2549.
The plasmids used for creating stable cell lines with inducible GFP-FOXK2 fusions, pAS1430 and pAS1431 (encoding N-terminal EGFP-tagged FOXK2(WT) or FOXK2(S368A/S423A), respectively) were constructed by a two-step procedure. First, the NheI (Klenow blunt-ended)/XbaI fragments from pAS2516 or pAS2520 (encoding EGFP-FOXK2(WT) or EGFP-FOXK2(S368A/S423A)) were cloned into the SmaI and XbaI sites in pUC19 (New England Biolabs) to create pAS1432 or pAS1433, respectively. Next, the KpnI/SalI fragments from pAS1432 or pAS1433 were ligated into the KpnI and XhoI sites in pCDNA5-FRT/TO (Invitrogen) to create pAS1430 and pAS1431, respectively. For creating stable cell lines with His and triple FLAG-tagged FOXK2, pAS2523 was constructed by inserting full-length FOXK2 amplified from pAS2252 by PCR using ADS1305 (5′-GCATGGATCCATGGCGGCGGCCGCGGCGGCGCTC-3′) and ADS2037 (5′-GGCTGCGTCGACGTTCTGGACACCCTTTTCCCTTAC-3′) followed by BamHI and SalI digestion into the same sites in pBRIT-LoxP-CTAP (kindly provided by M. Rudnicki) (28).
For bacterial expression, pGEX-FOXK2(189–660), encoding the C-terminal part of FOXK2, was created by direct cloning of the NcoI/SacI fragment from pAS2251 into pGEX-KG (pAS363) to create pAS2195. pGEX-FOXK2(189–660)(T389A) (pAS2648), pGEX-FOXK2(189-660)(S398A) (pAS2649), and pGEX-FOXK2(189-660)(T389A/S398A) (pAS2650) containing mutations in the Ser/Thr phosphorylation sites, were made by QuikChange mutagenesis using the primer·template combinations ADS1667/1668·pAS2195, ADS1669/1670·pAS2195, and ADS1669/1670·pAS2648, respectively. Similarly, further mutants were created, pGEX-FOXK2(189-660)(S423A) (pAS2646) and pGEX-FOXK2(189-660)(S368A/S423A) (pAS2647) using the primer·template combinations ADS1718/1719·pAS2195 and ADS1716/1717·pAS2646.
Tissue Culture, Flow Cytometry, Cell Transfection, Reporter Gene Assays, RT-PCR, and RNA Interference
HEK293 and U2OS cells were grown in DMEM supplemented with 10% fetal bovine serum. To construct HeLa cell lines inducibly expressing EGFP-FOXK2 derivatives, pAS1430 and pAS1431 vectors were cotransfected with the Flp recombinase encoding plasmid pOG44 (Invitrogen) into Flp-in TRex tetracycline transactivator HeLa cells (kindly provided by Stephen Taylor). Hygromycin-resistant colonies were pooled and expanded, and the transgene expression was induced with 1 μg/ml doxycycline (Sigma). To construct U2OS cell lines stably expressing EGFP-FOXK2 derivatives, pAS2516 and pAS2520 vectors were transfected into U2OS cells and subjected to selective pressure using neomycin. Single cells were then expanded into colonies to generate individual clonal cell lines. To construct U2OS cell lines stably expressing His-FLAG-tagged FOXK2 (U2OS-FOXK2-HF cells) or empty vector control (U2OS(HF) cells), the retroviral vector pAS2523 and pBRIT-LoxP-CTAP vectors were transfected into Phenix amphotropic packaging cells (Orbigen), and U2OS cells were infected by produced retrovirus and subjected to selective pressure using 1.5 μg/ml puromycin. Single cells were then expanded into colonies to generate individual clonal cell lines. Clonal line 16 was subsequently used because it expressed levels of epitope-tagged FOXK2 similar to those of endogenous FOXK2.
For cell cycle analysis, cells were synchronized at different times during the cell cycle by serum starvation for 24–72 h (G0 arrest), hydroxyurea (5 mm) or mimosine (50 μmol) (early S phase arrest) for 24–40 h, or nocodazole (100 ng/ml) for 16 h (G2-M). Nocodazole-treated cells were further divided into adherent cells (G2) and loosely adherent “shake off” cells (M phase). The CDK inhibitors were added 30 min before harvesting (roscovitine, 25 μm; purvalanol A, 10 μm; alsterpaullone 10 μm).
For flow cytometry, U2OS cells were fixed in 70% cold ethanol for a minimum of 30 min. The DNA was stained by treatment with 50 μg of propidium iodide and 50 μg of RNase A in a total volume of 400 μl of PBS and incubated at 37 °C for 30 min. Samples were examined on a CYAN-Calibur flow cytometer, and data were analyzed using the ModFit software (ModFit LT).
Transfections were performed with Polyfect (Qiagen) for HEK293 cells, Fugene HD (Roche Applied Science) for HeLa cells, and either reagent for U2OS cells according to the manufacturers' instructions. For reporter gene assays, typically 0.2 μg of reporter plasmid and 10 ng of pCH110 were co-transfected with 0–0.1 μg of expression plasmids. Cell extracts were prepared, and equal amounts of protein were used in luciferase and β-galactosidase assays as described previously with the exception of smaller sample and buffer volumes appropriate for use of the Orion microplate reader (Berthold Detection Systems) (29).
Real-time RT-PCR was carried out as described previously (26). The following primer pairs were used for RT-PCR experiments: CLNB1, ADS1728 (5′-GGCCAAAATGCCTATGAAGA-3′) and ADS1729 (5′-AGATGTTTCCATTGGGCTTG-3′); FOXK2, ADS1745 (5′-GCTGACAACTCACAGCCTGA-3′) and ADS1746 (5′-TCCGCAGTCCTGTAGTAGGG-3′); and 18 S internal control, ADS4005 (5′-TCAAGAACGAAAGTCGGAGGTT-3′) and ADS4006 (5′-GGACATCTAAGGGCATCACAG-3′).
SMARTpool siRNA duplexes against GAPDH, CDK1, CDK2, CLNA, CLNB were purchased from Dharmacon. To carry out RNA interference (RNAi), transfections were performed using LipofectamineTM RNAiMAX transfection reagent (Invitrogen) according to the manufacturer's instructions.
ChIP Analysis
ChIP analysis was performed as described previously (30). Briefly, for each ChIP experiment, 1–2 × 106 U2OS cells were cross-linked with 1% formaldehyde for 10 min at room temperature, harvested, and rinsed with 1× PBS. Cell nuclei were isolated, pelleted, and sonicated. DNA fragments were enriched by immunoprecipitation using anti-FLAG antibody (F3165, Sigma-Aldrich). After heat reversal of the cross-links, the enriched DNA was purified and analyzed by real-time PCR using the primer pair ADS2383 (5′-ATCAGAACTGCCCTCCAGTG-3′) and ADS2384 (5′-CGGCGATGTCTTTATGGAGT-3′) for amplifying a FOXK2 binding region in the MCM3 promoter.
Fluorescence Microscopy
U2OS cells were fixed with 4% paraformaldehyde, and the following antibodies were used: anti-FOXK2 (1:100, ILF1, ab5298, Abcam) and an FITC-labeled secondary rabbit anti-goat antibody (Zymed Laboratories Inc.). Cells were mounted employing a combined nuclear stain method (DAPI, Vectashield). Images were obtained with an OlympusBX51 upright microscope using a ×100/0.30 Plan Fln objective attached to a CCD Coolsnap-ES camera (Photometrics). Specific band pass filter sets for DAPI and FITC were used to prevent bleed-through from one channel to the next. Images were then processed and analyzed using ImageJ (available from the National Institutes of Health Web site). Confocal microscopy was analyzed using a Leica Microsystems (Mannheim, Germany) TCS SP2 confocal laser-scanning microscope.
For time-lapse imaging, 20,000 inducible HeLa cells expressing EGFP-FOXK2 (WT) were seeded into 24-well glass bottom plates and then induced by 1 μg/ml doxycycline for 24 h. Live cell images were taken every 5 min for 24 h using a Leica ASMDW work station. The microscope setup consisted of an inverted microscope (DMIRE2) and multipoint time lapse stage controlled by ASMDW software. The deconvoluted images were edited by ImageJ software or processed to AVI movies by ASMDW software (Adobe, San Jose, CA).
Apoptosis was measured by detecting the presence of the active cleaved form of caspase 3. U2OS cells (105 cells/24-mm-diameter dish) cultured on glass coverslips were transiently transfected as described above. 24 h after transfection, the cells were washed, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 1% BSA for 30 min. The cells were incubated for 60 min with rabbit polyclonal antibody against cleaved caspase 3 (1:200; 9661S, Cell Signaling Technology). The cells were then washed and incubated with Alexa Fluro594-conjugated anti-rabbit IgG (1:250; A11012, Invitrogen). The cell nuclei were counterstained, and fluorescence was examined as described above. The results presented are representative of at least three experiments.
Western Blot, Immunoprecipitation, and GST Pull-down Analysis
Western blotting was carried out with the primary antibodies, FOXK2 (ab5298, Abcam), GAPDH (ab9485, Abcam), antibodies supplied by Santa Cruz Biotechnology (cyclin E (sc-247), CLNA (sc-751), CLNB1 (sc-245), CDK2 (sc-163), CDK1 (sc-54), and ERK2 (sc-154)) and FLAG (F3165, Sigma-Aldrich), essentially as described previously (26). Rabbit polyclonal anti-phospho-Ser368 FOXK2 antibody was generated by Eurogentec and was raised in rabbits injected with a synthetic phosphopeptide corresponding to amino acids 360–373 of human FOXK2 (SSRSAPApSPNHAG). For demonstrating phosphorylation-dependent mobility shifts on Western blots, 10 μg of total protein from cell lysates were treated with 20 units of λ-phosphatase for 30–50 min at 37 °C prior to loading on a gel. To analyze protein stability, the protein synthesis inhibitor cycloheximide (50 μg/ml) was added to cells for the times indicated in the figure legends. Protein levels were analyzed by Western analysis and quantified using Bio-Rad Quantity One software.
For immunoprecipitation, cells were washed twice with ice-cold phosphate-buffered saline and then lysed in buffer containing 20 mm Tris, pH 7.4, 137 mm NaCl, 25 mm β-glycerophosphate, 2 mm sodium pyrophosphate, 2 mm EDTA, 1% Triton X-100, 10% glycerol, 1 mm sodium orthovanadate, and protease inhibitor mixture (Roche Applied Science). Lysates were centrifuged at 14,000 × g for 20 min to remove insoluble material. FOXK2 proteins were immunoprecipitated from lysates by incubation with FOXK2 antibodies and protein G-Sepharose beads (Amersham Biosciences) for 3 h at 4 °C. Beads were washed four times in lysis buffer, and bound proteins were eluted by adding SDS loading buffer. GST pull-down analysis was performed essentially as described previously (31) with purified bacterially expressed recombinant GST-FOXK2(189–660) and whole cell extracts of U2OS cells.
In Vitro Kinase Assays
In vitro kinase assays were carried out using recombinant GST-FOXK2 proteins purified from bacteria and either recombinant CDK·cyclin complexes purified from overexpressing baculovirus-infected insect cells (Fig. 4, B and D) or from commercial sources (Figs. 4E and 5B) (CDK1·CLNB and CDK2·CLNA, Upstate Biotechnology, Inc.). Assays were generally performed in k-buffer (50 mm Tris-Cl, pH 7.5, 10 mm MgCl2, 1 mm DTT, 100 μm ATP) as described previously (32) with 1 μg of substrate and 1 unit/50 ng kinase for 30 min at 30 °C. Phosphorylated forms were either detected by incorporating radioactive phosphate from [γ-32P]ATP, Western blotting with anti-phosphospecific antibodies, or mass spectrometry.
FIGURE 4.

CDK·cyclin-dependent FOXK2 phosphorylation. A, U2OS cells were grown asynchronously (Asy) or in the presence of nocodazole (Noc) for 16 h and either left untreated or treated with 10 μm purvalanol A (PuA) or alsterpaullone (Alst) for 30 min before harvesting. Cell lysates were subjected to immunoprecipitation (IP) using anti-FOXK2 antibody followed by immunoblot (IB) with FOXK2 antibody. B, U2OS cells were transfected with siRNA duplexes against GAPDH, CDK2, cyclin A, or cyclin B where indicated. 30 h after transfection, cells were grown in the presence (Noc) or absence (Asy) of nocodazole for 16 h before harvesting. The expression of FOXK2, cyclin B, cyclin A, CDK2, and GAPDH was detected by Western analysis. C, cell cycle profiles of U2OS cells following treatment with the indicated siRNA constructs for 30 h prior to a further 16-h incubation in the presence or absence of nocodazole.
FIGURE 5.

Mapping CDK·cyclin complex phosphorylation sites in FOXK2 in vitro. A, schematic of the FOXK2 domain structure and the GST-FOXK2(189–660) construct used in in vitro assays. The conserved FHA and forkhead domains (FOX) are shaded in gray; highly conserved regions are marked in black. B, in vitro kinase assay with the indicated cyclin·CDK complexes and FOXK2(189–660) (top) and histone H1 (bottom) substrates. Phosphorylated products were detected by autoradiography. Truncated fragments of FOXK2(189–660) are marked with asterisks. C, GST pull-down assay using GST or GST-FOXK2(189–660) and total cell extracts from U2OS cells. CLNA and CDK2 were detected by immunoblot. 10% input is shown. D, in vitro kinase assay with the indicated cyclin·CDK complexes and FOXK2(189–660) and histone H1 substrates. Phosphorylated products were detected by autoradiography (top), and input substrate proteins were detected by Coomassie staining (bottom). Truncated fragments of FOXK2(189–660) are marked with asterisks. E, phosphorylated peptides by mass spectrometry. Phosphorylated serine residues are boxed. F, in vitro kinase assay with the indicated GST-FOXK2 substrates and CDK2·CLNA. Phosphorylation was detected by phosphorimaging (top), and total FOXK2 levels were detected by Coomassie staining (bottom).
Identification of Phosphorylation by Mass Spectrometry
Enzymatic in-gel digestion with trypsin followed by nano-LC-MS/MS analysis was performed as described previously (33). In brief, excised protein bands were washed with 50% (v/v) acetonitrile in 25 mm ammonium bicarbonate, shrunk by dehydration in acetonitrile, and dried in a vacuum centrifuge. The dried gel pieces were incubated with 50 ng of trypsin (sequencing grade; Promega, Madison, WI) in 20 μl of 50 mm ammonium bicarbonate at 37 °C overnight. To extract the peptides, 20 μl of 0.5% (v/v) trifluoroacetic acid (TFA) in acetonitrile was added, and the separated liquid was taken to dryness under vacuum. For MS analysis, the peptide mixture was dissolved in 6 μl of 0.1% TFA in acetonitrile/water (5:95, v/v). MS and tandem MS (MS/MS) experiments were performed on a quadrupole time-of-flight mass spectrometer, Q-Tof Ultima (Micromass, Manchester, UK). A Micromass CapLC liquid chromatography system was used to deliver the peptide solution to the electrospray source. The sample was injected, and peptides were separated using an analytical column (Atlantis dC18, 3 μm, 100 Å, 150 mm × 75-μm inner diameter; Waters GmbH, Eschborn, Germany) at an eluent flow rate of 150 nl/min. Mobile phase A was 0.1% formic acid (v/v) in acetonitrile/water (5:95, v/v), and B was 0.1% formic acid in acetonitrile/water (8:2, v/v). Runs were performed using a gradient of 4–50% B in 90 min. Data-dependent acquisition (survey scanning) was performed using one MS scan followed by MS/MS scans of the most abundant peak. The MS survey range was m/z 300–1500, and the MS/MS range was m/z 100–1999. Data analysis was performed by use of MassLynx software (Micromass-Waters). The processed MS/MS spectra and MASCOT server version 2.0 (Matrix Science Ltd., London, UK) were used to search in house against a protein data base containing the FOXK2 sequence. A maximum of three missed cleavages was allowed, and the mass tolerance of the precursor and sequence ions was set to 100 ppm and 0.1 Da, respectively. The search includes variable modifications of cysteine with acrylamide, methionine oxidation, and phosphorylation of serine, threonine, or tyrosine. To identify phosphorylated FOXK2 peptides, probability-based scoring (MASCOT identity score, p < 0.05) was used. In addition, tandem mass spectra of phosphopeptides were manually verified and compared with the theoretical fragment ions of FOXK2.
RESULTS
FOXK2 Regulation in the Cell Cycle
To establish potential links to the cell cycle, we first determined whether FOXK2 levels fluctuate in a cyclical manner in synchronized cell populations. FOXK2 is expressed at a similar level in a range of different asynchronously growing cell types, including human U2OS osteosarcoma cells (Fig. 1A). We focused on U2OS cells and examined FOXK2 mRNA levels in cells released from a nocodazole-induced cell cycle block in G2-M phase. Little change in FOXK2 mRNA levels was seen during the subsequent cell cycle, although CLNB1 levels fluctuated in the expected manner, with high levels in cells in the G2-M phase (Fig. 1B). Next, we examined endogenous FOXK2 protein levels in nocodazole-synchronized U2OS cells. In these cells, FOXK2 migrates as two prominent bands in G2-M phase (Fig. 1C, lane 1). The upper band becomes more prominent 4 h after release, but this reverts to a faster mobility band after 8–12 h as cells begin to accumulate in G1 phase (Fig. 1C, lanes 3 and 4). A transient reduction in protein levels is then observed as cells pass through G1-S (Fig. 1C, lanes 5 and 6). The change in mobility of the bands corresponding to FOXK2 is indicative of potential phosphorylation changes. We therefore treated samples from the 0 and 4 h time points with λ-phosphatase and monitored the mobility of the FOXK2 proteins. In both cases, the upper slower mobility band was lost, and a single higher mobility band was obtained (Fig. 1D), thereby demonstrating that the upper band did indeed correspond to a phosphorylated form of FOXK2.
FIGURE 1.

FOXK2 expression during the cell cycle. A, Western blot analysis of FOXK2 expression levels in the indicated cell types. FOXK2 and ERK2 expression were detected by immunoblot (IB) with the indicated antibodies. B, real-time RT-PCR analysis of FOXK2 and CLNB1 expression in U2OS cells synchronized in nocodazole (Noc) and released for the indicated times. Experiments were performed in duplicate, and the data are the average of two independent experiments. C, Western blot analysis of FOXK2 expression U2OS cells synchronized in nocodazole and released for the indicated times. The expression of endogenous FOXK2, cyclin E (CLNE), CLNB, and GAPDH (loading control) was determined by immunoblot. DNA content profiles of cells at the indicated time points determined by propidium iodide (PI) staining and approximate cell cycle stages are shown in the panel below. D, immunoblot of FOXK2 after λ-phosphatase (λ-Pase) treatment of cell lysates from the indicated time points after nocodazole release. Bands corresponding to phosphorylated (P-FOXK2) and non-phosphorylated FOXK2 are indicated.
FOXK2 is therefore continually expressed through the cell cycle, but it is regulated at the protein level. In particular, the levels of FOXK2 phosphorylation fluctuate during the cell cycle.
FOXK2 Localization in the Cell Cycle
Because several FOX transcription factors change their localization during the cell cycle (reviewed in Refs. 10 and 11), we determined the subcellular localization of endogenous FOXK2. FOXK2 is found in the nucleus of the majority of asynchronously growing U2OS cells, and this does not change overtly through the cell cycle (Fig. 2A) (data not shown). However, in cells that had apparently just undergone cell division, the FOXK2 and DNA localization patterns did not completely overlap (Fig. 2A, arrows). To further investigate this phenomenon, we created stable HeLa cell lines that inducibly express EGFP-tagged FOXK2 and monitored the localization of EGFP-FOXK2 over a 24-h period (supplemental Movie 1). Generally, FOXK2 appears to be located in the nucleus, but as cells divide and the nuclear envelope breaks down, FOXK2 becomes dispersed throughout the cell and then reappears in the nucleus following cytokinesis (Fig. 2B and supplemental Movie 1). Interestingly, confocal analysis showed a relocalization of FOXK2 away from the DNA during mitosis, which is most clearly seen in cells going through anaphase and telophase (Fig. 2C). FOXK2 then relocalizes with the DNA following cytokinesis (Fig. 2C). This is in contrast to the continual association of another forkhead transcription factor, FoxI1, with DNA, even during telophase (34).
FIGURE 2.

Localization of FOXK2 in the cell cycle. A, localization of endogenous FOXK2 in asynchronously (Asy) growing U2OS cells. Cells were stained to detect endogenous FOXK2 (green) and DNA (blue). Whole cells are shown in brightfield detections in the first panels. The arrowheads indicate cells where DNA and FOXK2 localization do not completely overlap. B and C, localization of FOXK2 throughout the mitotic cell cycle in HeLa cells. B, images chosen from time lapse images of live, logarithmically growing HeLa cells inducibly expressing EGFP-FOXK2 (see supplemental Movie 1). Whole cells are shown in brightfield detections in the first panels. C, images from fixed cells imaged using confocal microscopy. GFP-FOXK2 (green), DNA (DAPI stain; blue), and merged images are shown. D, ChIP analysis of FOXK2 binding to the MCM3 promoter in asynchronous cell populations or mitotic cells (M phase) using anti-FLAG antibodies in the U2OS(FOXK2-HF) or control U2OS(HF) cell lines. Data are shown relative to levels in asynchronous cell populations in control U2OS(HF) cells (taken as 1). Error bars, S.E.
To further examine the exclusion of FOXK2 from DNA during mitosis, we performed ChIP analysis on a FOXK2 target gene, MCM3, identified by ChIP-chip analysis using a stably transfected U2OS cell line expressing FLAG-tagged FOXK2.4 Binding of FOXK2 to this target gene could be detected in asynchronously growing cells, but binding levels decreased markedly in mitotic cells isolated by shake off from nocodazole-treated populations (Fig. 2D). Little binding to the MCM3 gene locus was seen in control cells lacking an epitope-tagged version of FOXK2. FOXK2 is therefore predominantly localized to the nucleus with little evidence for cytoplasmic localization during the cell cycle, although during mitosis it is transiently excluded from the DNA.
Cell Cycle-dependent FOXK2 Phosphorylation
Nocodazole-treated cells arrest in both G2 and M phases. To establish more precisely where FOXK2 becomes maximally phosphorylated, we isolated M phase cells by mitotic shake off following nocodazole treatment. Although cells in G2 contained a small amount of the upper band corresponding to phosphorylated FOXK2 protein, M phase cells exclusively contained this hyperphosphorylated form (Fig. 3A). As an alternative method of cell cycle synchronization, we used mimosine, which blocks progression through S phase. Little FOXK2 phosphorylation was seen upon release from a mimosine block, until cells began to accumulate in G2 and M phases, when a slow mobility upper FOXK2 band was observed (Fig. 3, B and C). Again, this lower mobility band was demonstrated to be a phosphorylated form by treatment with λ-phosphatase (Fig. 3D). Hence, these experiments confirm that FOXK2 is phosphorylated during the late cell cycle stages and more precisely define maximal phosphorylation occurring during mitosis.
FIGURE 3.
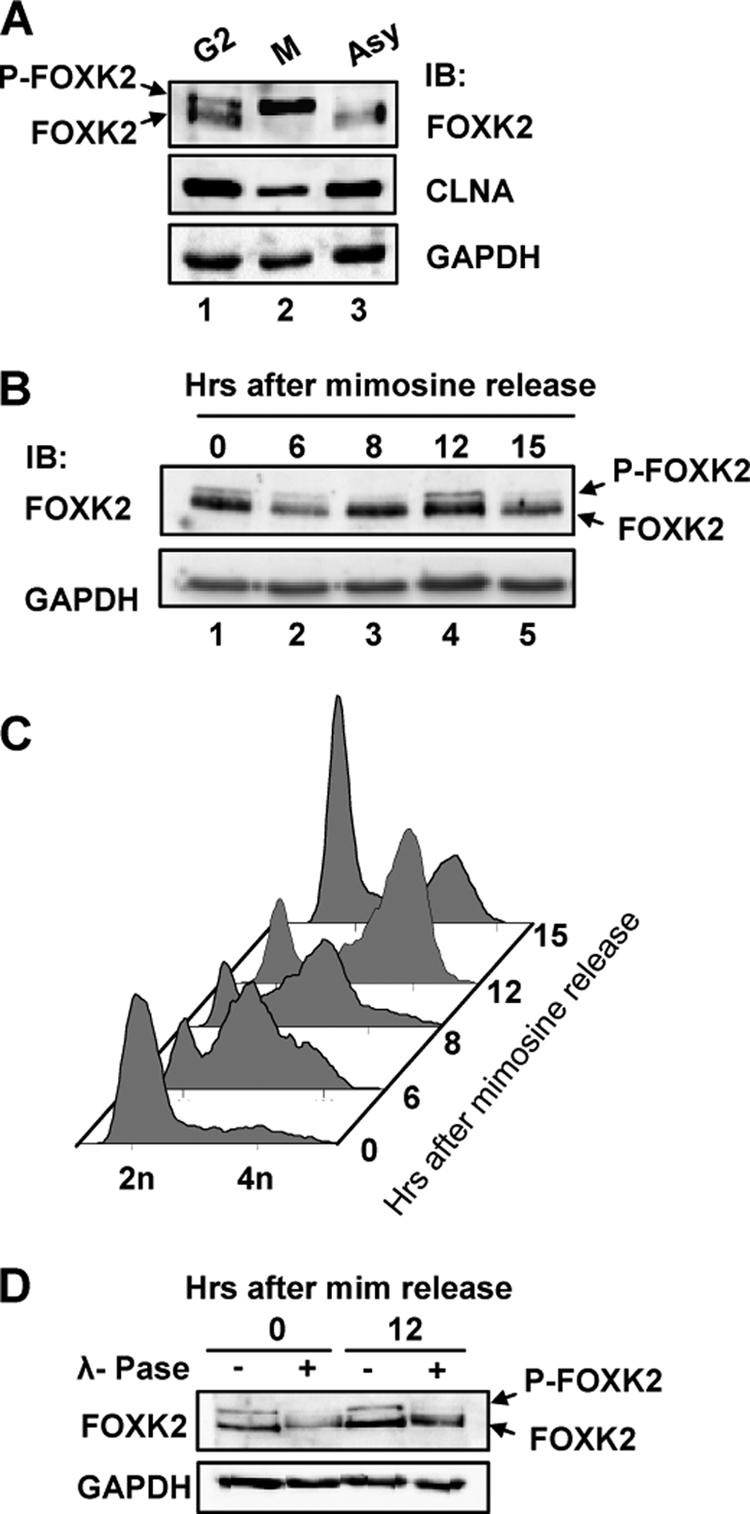
Cell cycle-dependent FOXK2 phosphorylation. A, Western blot analysis of FOXK2 during G2 and M phases. Nocodazole-synchronized mitotic U2OS cells were spilt into attached cells (G2) and mitotic cells (M) by mitotic shake off, and FOXK2 protein expression was compared with asynchronously growing cells (Asy). Protein bands corresponding to non-phosphorylated and phosphorylated FOXK2 (P-FOXK2) are indicated by arrows. Immunoblot was performed with the indicated antibodies. B and C, U2OS cells were synchronized with mimosine and released for the indicated times. The expression of endogenous FOXK2 and GAPDH were detected by immunoblot (IB) (B), and DNA content was analyzed (C). D, cell lysates from the indicated time points were treated with λ-phosphatase (λ-Pase) and probed for FOXK2 and GAPDH levels by Western blot analysis. mim, mimosine.
CDK·cyclin-dependent FOXK2 Phosphorylation
The majority of cell cycle-dependent phosphorylation events have been attributed to cyclin-dependent kinase activity (reviewed in Ref. 35). To determine whether this is the case for FOXK2, we first treated cells with two different CDK inhibitors, purvalanol A and alsterpaullone, and monitored FOXK2 phosphorylation events. Treatment with either CDK inhibitor caused a reduction in the generation of the lower mobility phosphorylated form of FOXK2 in nocodazole-treated cells (Fig. 4A, top, lanes 4–6).
To establish more clearly which CDK·cyclin complex might be acting on FOXK2, we used an siRNA approach to selectively deplete individual CDKs or cyclins. We focused on CDK2 and cyclins A and B1 because these cyclins have been shown to function during G2 and M phases (reviewed in Ref. 36). Depletion of either one of these cyclins caused a loss of the upper phosphorylated form of FOXK2 in nocodazole-treated cells (Fig. 4B, lanes 11 and 12). However, depletion of CDK2 caused only partial loss of FOXK2 phosphorylation (Fig. 4B, lane 10), suggesting the involvement of additional CDKs. Indeed, depletion of CDK1 resulted in a more substantial loss of FOXK2 phosphorylation (see Fig. 6F, first panel, lanes 3 and 4). To ensure that the effects of the siRNAs could be attributed directly to phosphorylation of FOXK2 rather than an indirect effect caused by arresting the cell cycle prior to mitotic entry, we examined the cell cycle profiles of siRNA-treated cells. Little effect on the percentage of cells in the G2/M population was seen in either asynchronous or nocodazole-treated cell populations upon siRNA-mediated depletion of individual CDKs and cyclins (Fig. 4C), making it unlikely that the effects of CDK·cyclin depletion on FOXK2 phosphorylation were indirect. Indeed, this is reinforced by our CDK inhibitor studies, where cells were allowed to accumulate in G2/M before adding the inhibitors. Together, these results therefore demonstrate that CDK·cyclin complexes, in particular in combination with CLNA or CLNB, are important for generating the hyperphosphorylated form of FOXK2 at G2-M.
FIGURE 6.

FOXK2 is phosphorylated by CDK·cyclin complexes at serine 368 in vivo. A, gel mobility shifts in the migration of WT and phosphodeficient mutant S368A/S423A GFP-FOXK2 proteins. FOXK2 expression was induced by doxycycline treatment of stably transfected HeLa cells. Cells were grown in the presence or absence of nocodazole (Noc) for 16 h and treated with or without roscovitin (Rosc) for 2 h before harvesting. Cell lysates were subjected to immunoprecipitation (IP) using anti-FOXK2 antibody followed by immunoblot (IB) with FOXK2 antibody. B, in vitro kinase assays using GST-FOXK2(189–660) and recombinant CDK1·cyclin B or CDK2·/cyclin A, Western blots (top) using an anti-phospho-Ser368 FOXK2 antibody and a Coomassie-stained gel of the samples (bottom) are shown. The asterisks represent degradation products. C, HeLa cells containing stably integrated doxycycline (dox)-inducible WT and S368A/S423A mutant forms of EGFP-tagged FOXK2 were either left uninduced (−) or induced (dox) for 24 h. FOXK2 was isolated by immunoprecipitation with anti-FOXK2 antibody and detected by immunoblot using an anti-phospho-Ser368 FOXK2 antibody or anti-FOXK2 antibody. Input levels of FOXK2 and ERK2 were detected by immunoblot. D, endogenous FOXK2 was isolated by immunoprecipitation from asynchronous (Asy) or U2OS cells synchronized in G1, S, G2, or M phases. Precipitated proteins detected by immunoblot using anti-phospho-Ser368 FOXK2 or FOXK2 antibody and input proteins by direct immunoblot with the indicated antibodies. E, endogenous FOXK2 phosphorylation was analyzed as in D from asynchronous U2OS cells or cells treated with nocodazole for 16 h, with or without purvalanol A (PuA) treatment. Mitotic cells were collected by manual shake off (lanes 3 and 4). F, U2OS cells were transfected with siRNA duplexes against either CDK1 (left) or cyclin B (right) where indicated. 30 h after transfection, cells were grown in the absence (Asy) or presence (Noc) of nocodazole for 16 h before harvesting. Cell lysates were analyzed as in D.
Ser368 and Ser423 Are Phosphorylated by CDK·CLNA in Vitro
To identify whether CDK·cyclin complexes could directly phosphorylate FOXK2 and to identify potential phosphorylation sites, we carried out in vitro kinase assays using recombinant kinase complexes and a GST-FOXK2 fusion protein. We focused on the C-terminal part of FOXK2 (amino acids 189–660) because this contains the majority of the potential CDK phosphorylation sites corresponding to SP or TP motifs. The CDK2·CLNA complex could efficiently phosphorylate FOXK2 (Fig. 5B, lanes 2 and 3), but little phosphorylation was observed with CDK2·cyclin E kinase, despite this kinase efficiently phosphorylating a control histone H1 substrate (Fig. 5B, lane 1). This suggested that CDK·CLNA complexes might bind to FOXK2, and this was confirmed using a GST pull-down assay (Fig. 5C). Because CLNB depletion also causes a loss of FOXK2 phosphorylation in vivo (Fig. 4), we also compared the ability of CDK1·CLNB with that of CDK2·CLNA to phosphorylate FOXK2(189–660) in vitro. Both kinase complexes phosphorylated FOXK2 with similar efficiencies (Fig. 5D).
Mass spectrometry was then used to identify the sites of CDK·cyclin phosphorylation in vitro. Because CDK2·CLNA was the most active kinase against FOXK2 that we initially tested, we used this kinase to produce phosphorylated FOXK2 for mass spectrometry analysis. The mass spectra of the tryptic digest revealed a distinct triply charged mass peak at m/z 899.41 and m/z 755.05 corresponding to the phosphorylated tryptic sequences 364SAPASPNHAGVLSAHSSGAQTPESLSR390 and 416FAQSAPGSPLSSQPVLITVQR436, respectively. MS/MS spectra of the phosphorylated peptides 364–390 and 416–436 (Fig. 5E and supplemental Fig. 1) show fragment ions that clearly confirm the presence of tryptic peptides containing phosphorylation at Ser368 and Ser423. To establish the relative contribution of these two sites to FOXK2 phosphorylation, we mutated these two amino acids along with two other evolutionarily conserved potential CDK sites, threonine 389 and serine 398, and tested the resulting proteins as CDK2·CLNA substrates (Fig. 5F). Decreases in phosphorylation efficiency were seen with the individual T389A, S398A, and S423A mutants. However, a more substantial loss of phosphorylation was seen with the double S368A/S423A mutant (Fig. 5E, lane 6), consistent with the identification of these sites by mass spectrometry analysis. Mutation of S368A alone had only slight effects on CDK2·CLNA-mediated FOXK2 phosphorylation (data not shown). Together, these results therefore identify serine 368 and serine 423 in combination as major sites of FOXK2 phosphorylation by CDK·CLNA complexes in vitro.
FOXK2 Is Phosphorylated at Serine 368 in Vivo
To establish whether serine 368 and serine 423 are phosphorylated in vivo, we first compared the mobility of bands corresponding to wild type or the S368A/S423A mutant version of FOXK2 in mitotic cells following nocodazole treatment. A slower mobility band corresponding to phosphorylated FOXK2 was induced in both cases, which was inhibited by treatment with the CDK inhibitor roscovitine (Fig. 6A). However, the efficiency of generation of this species was much reduced in the S368A/S423A mutant (Fig. 6A, compare lanes 2 and 5). This indicates an important contribution of serine 368 and serine 423 to generating the hyperphosphorylated FOXK2 species but also suggests that additional phosphorylation events take place in vivo. Indeed, a recent global proteomic study indicated that FOXK2 is phosphorylated at Ser423 but in addition at Ser393 in mitotic cells (37).
To provide more definitive proof of CDK-mediated FOXK2 phosphorylation in vivo, we generated a phosphospecific antibody against serine 368. First, we tested the specificity of the antibody for the phosphorylated form of FOXK2 in vitro. Recombinant GST-FOXK2(189–660) was subjected to in vitro kinase assays with CDK1·CLNB or CDK2·CLNA complexes, and the generation of phosphorylated serine 368 was determined by Western blotting. As expected, CDK2·CLNA was able to phosphorylate serine 368, however, CDK1·CLNB was also able to efficiently phosphorylate serine 368, in keeping with a potential role in mitotic cells and its ability to phosphorylate FOXK2 in vitro (Fig. 6B, lanes 2 and 3). In contrast, no signal was detected using the phosphospecific antibody in the absence of prior CDK-mediated phosphorylation (Fig. 6B, lane 1). As a second test of antibody specificity, we examined the phosphorylation status of wild-type FOXK2 and a S368A/S423A mutant form in vivo. Stable cell lines were created that contain doxycycline-inducible EGFP-FOXK2 constructs. Equal amounts of EGFP-FOXK2 fusion proteins were produced in both cell lines (Fig. 6C, top), but phosphorylated serine 368 was only detected in cells expressing wild-type FOXK2 and not in cells expressing the S368A/S423A mutant version (Fig. 6C, second panel).
Next, we utilized the anti-phospho-Ser368 antibody to further probe the CDK-mediated phosphorylation events impacting FOXK2 in vivo. First, we blocked cells at various points of the cell cycle and analyzed serine 368 phosphorylation. Weak phosphorylation was observed throughout the cell cycle, but a clear enrichment was seen in mitotic cells (Fig. 6D, top). This corresponds to when FOXK2 becomes hyperphosphorylated and migrates with slower mobility (Fig. 6D, second panel). Interestingly, FOXK2 phosphorylation peaks in mitotic cells where there are high levels of CLNB and little CLNA, suggesting that CLNB might be a relevant cyclin involved in FOXK2 phosphorylation. To probe whether this mitotic phosphorylation event involves CDKs in vivo, we treated cells with purvalanol A following nocodazole treatment. This CDK inhibitor efficiently blocked the phosphorylation of serine 368 (Fig. 6E), in keeping with the more general effects in blocking hyperphosphorylation of FOXK2. Finally, we probed the role of the CDK1·CLNB complex in phosphorylating serine 368 in vivo through siRNA knockdown approaches. Depletion of either CDK1 (Fig. 6F, lane 4) or CLNB1 (Fig. 6F, lane 8) resulted in a loss of serine 368 phosphorylation in mitotic cells. Collectively, these experiments therefore further provide a link between CDK·cyclin complexes and FOXK2 phosphorylation in vivo.
FOXK2 Phosphorylation Promotes Its Degradation and Reduced Repressive Activity
One possible mechanism by which phosphorylation might affect FOXK2 function is in affecting its stability. We therefore compared the stability of wild-type FOXK2 with the CDK site mutant version, FOXK2(S368A/S423A) by blocking protein synthesis with cycloheximide, followed by monitoring the decay in protein levels over time. Wild-type FOXK2 showed a half-life of about 5 h. In contrast, the FOXK2(S368A/S423A) mutant was stable over this time period with little loss of protein observed even after 6 h (Fig. 7A). We also tested an additional FOXK2 protein that contained phosphomimicking amino acids at serines 368 and 423 (FOXK2(S368D/S423D)). This protein had a similar stability to the wild-type protein and, if anything, was even less stable (Fig. 7B). These results are therefore consistent with a role for CDK-mediated phosphorylation of serines 368 and 423 in promoting FOXK2 turnover.
FIGURE 7.

Role of CDK phosphorylation sites in FOXK2 stability and transcriptional repression. A and B, U2OS cells were transfected with FLAG-tagged WT, S368A/S423A, or S368D/S423D mutants of FOXK2 for 24 h. The cells were then treated with cycloheximide (CHX) for the indicated times (A) or for 6 h (B). FOXK2 levels were detected by immunoblot (IB) using anti-FLAG antibody and normalized against ERK2 levels. Data are the average of three independent experiments. Error bars, S.E. C, luciferase reporter assay in U2OS cells with a p21 promoter-driven luciferase reporter plasmid in the presence of increasing amounts (0, 5, 20, 50, and 100 ng) of FOXK2(WT) (white bars) or FOXK2(S368D/S423D) (black bars). The data are presented relative to the reporter activity in the absence of FOXK2 (taken as 1). Data represent the mean of duplicate samples. The levels of FOXK2 protein expression were determined by immunoblot alongside control ERK2 levels (right).
By analogy with FOXK1, FOXK2 probably acts as a transcriptional regulatory protein, through repressing transcription. FOXK1 has previously been implicated in repressing the expression of the cell cycle inhibitory protein p21 (25), so we first probed whether FOXK2 might also share this function. Overexpression of FOXK2 reduced the activity of a luciferase reporter gene driven by the p21 promoter (Fig. 7C), making this a useful marker for FOXK2 transcriptional activity. Control reporters were unaffected by FOXK2 overexpression (data not shown). The mutant FOXK2(S368A/S423A) protein repressed transcription to an equivalent level as the wild-type protein (data not shown), suggesting that the non-phosphorylated protein was the repressive form. Moreover, we also made a triple mutant, incorporating the S393A mutation of the potential CDK phosphorylation site identified by others (37), but incorporation of this additional mutation had little effect on the repressive activity of FOXK2 (data not shown). We therefore instead compared wild-type FOXK2 and the phosphomimetic form FOXK2(S368D/S423D) in activating the p21 promoter-driven reporter construct. In comparison with the wild-type protein, this form was less repressive under each of the concentrations tested (Fig. 7C), although complete loss of repression was not observed. Importantly, this did not reflect gross changes in protein expression because steady state levels of protein were equivalent (Fig. 7C, left). The CDK phosphorylation sites that we have identified therefore appear functionally important in controlling FOXK2 stability and its transcriptional repressive activities.
FOXK2(S368A/S423A) Promotes Apoptosis
To establish whether CDK-mediated FOXK2 phosphorylation plays a role in cell function, we attempted to make stable U2OS cell lines expressing either wild-type FOXK2 or FOXK2(S368A/S423A). However, although we were able to readily make clones that stably expressed EGFP fusions with wild-type FOXK2, we were unable to make any lines that stably expressed EGFP-FOXK2(S368A/S423A) fusion proteins. Indeed, in the cell lines that detectably expressed EGFP, the EGFP portion was still present but contained additional variable length sequences, none of which corresponded to the full-length EGFP-FOXK2 fusion protein (Fig. 8A, lanes 1–4). We repeated this experiment with similar consequences for isolating clonal cell lines; however, we simultaneously maintained polyclonal cell populations in selective media and monitored EGFP-FOXK2 levels. After 10 days of growth, we were still able to readily detect the expression of the fusion to wild-type FOXK2, but the levels of EGFP-FOXK2(S368A/S423A) were much reduced (Fig. 8B, right). In contrast, in transient transfection assays, FOXK2(WT) and FOXK2(S368A/S423A) were expressed to equivalent levels (Fig. 8B, left).
FIGURE 8.

FOXK2(S368A/S423A) promotes apoptosis. A, Western analysis of EGFP-FOXK2 expression in clonal U2OS cell lines selected for stable expression of the indicated WT and mutant (C9, C12, C16, and C17) FOXK2 derivatives. Fusion proteins were detected by immunoblot (IB) with either FOXK2 or GFP antibodies. The positions of molecular mass markers (kDa) are shown on the left. The asterisk represents endogenous FOXK2. B, Western analysis of EGFP-FOXK2 expression in transiently transfected (24 h; left) or stably transfected (10 days; right) pools of U2OS cells. C, caspase 3 activity in transiently transfected U2OS cells. The arrows indicate the locations of cells with active caspase 3. Quantification of the data is shown on the right and is the mean of four experiments.
The inability to generate clonal cell lines stably expressing FOXK2(S368A/S423A) indicated that this mutant protein might be detrimental to cell growth and survival. We therefore examined whether FOXK2(S368A/S423A) expression caused apoptosis and hence eliminated cells expressing this fusion protein. Few cells expressing EGFP or EGFP-FOXK2(WT) contained the apoptosis marker, active caspase 3. In contrast, significantly more cells expressing FOXK2(S368A/S423A) exhibited staining for active caspase 3 (25% of cells compared with 10% of WT cells) (Fig. 8C). Together, these results point to an important role for CDK-mediated FOXK2 phosphorylation because, when this phosphorylation is prevented in the FOXK2(S368A/S423A) mutant, cell viability is affected and apoptosis ensues.
DISCUSSION
Several forkhead transcription factors have been associated with controlling periodic gene expression during the mammalian cell cycle, including FOXM1 and FOXO family members (reviewed in Refs. 10 and 11). Moreover, both FOXM1 and FOXO transcription factors act as direct targets for CDK·cyclin complexes, the key drivers of the cell cycle. Here we have demonstrated that an additional forkhead transcription factor, FOXK2, is also subject to regulatory phosphorylation by CDK·cyclin complexes.
FOXK2 phosphorylation was revealed by the appearance of a slower mobility species on denaturing gels. Phosphorylation peaks in mitotic cells, where all of the FOXK2 migrates as a lower mobility form. In agreement with this, the mitotically active CDK1·CLNB appears to be the major kinase complex acting on FOXK2 in vivo. Importantly, siRNA knockdown experiments suggest that CDK2 and CLNA also influence FOXK2 phosphorylation, indicating that phosphorylation might be initiated prior to mitosis. In fact, low levels of serine 368 phosphorylation can be seen throughout the cell cycle but with a distinct peak in mitotic cells. This behavior closely resembles the sequential integration of CDK1·CLB5 (an S phase cyclin) and CDK1·CLB2 (a G2-M phase cyclin) on the activity of the forkhead transcription factor Fkh2p in S. cerevisiae (15–17).
The large mobility shift in FOXK2 upon phosphorylation suggests that the slower mobility species probably represents a hyperphosphorylated form containing multiple phosphorylation events. Indeed, we have identified two sites, serines 368 and 423, that are targets for CDK·cyclin complexes in vitro and contribute to phosphorylation and the generation of the slower mobility species in vivo. However, there are clearly more CDK-dependent sites phosphorylated in vivo. Whereas CDK inhibitors and CDK·cyclin knockdown efficiently reduce FOXK2 phosphorylation (Fig. 4), in comparison, FOXK2 phosphorylation is much less affected in the FOXK2(S368A/S423A) mutant (Fig. 6A). Importantly, this latter experiment reveals additional intermediary bands that are indicative of partial phosphorylation. Thus, serines 368 and 423 are probably contributory rather than the sole determinants of CDK-mediated phosphorylation of FOXK2. The two CDK-dependent serines identified in this study were obtained by a combination of in vitro kinase assay and mass spectrometry, which may not reflect the total phosphorylation events on FOXK2 in vivo. Indeed, by using stable isotope labeling along with a two-step strategy for phosphopeptide enrichment and high mass accuracy mass spectrometry, Ser393 was identified as an additional phosphorylation site of FOXK2 in HeLa cells arrested in the mitotic phase of the cell cycle (37). However, it remains unknown whether these sites are directly phosphorylated by CDKs or other kinases activated during mitosis. Furthermore, although the results in this paper focus on a C-terminal fragment of FOXK2, we have also detected phosphorylation of the N-terminal region of FOXK2 by CDK·cyclin complexes in vitro (data not shown). This further suggests a multisite phosphorylation mechanism whereby phosphorylation of different CDK sites would occur over time, potentially through the activity of different CDK·cyclin complexes, but ultimately result in complete multisite phosphorylation at a precise point of the cell cycle (i.e. in mitosis). Such switchlike mechanisms are operative at other points in the cell cycle (reviewed in Ref. 38). It is also possible that phosphorylation of FOXK2 might be important at other times during the cell cycle because phosphorylation increases in cells blocked during early S phase (Figs. 3B and 6D) albeit to a lesser extent than in mitotic cells. Thus, although we clearly show the functional importance of the C-terminal phosphorylation events, additional regulatory events triggered through alternative phosphorylation sites will require further investigation.
FOXK2 seems to function as a transcriptional repressor protein, and one target appears to be the cell cycle regulator p21. By using a phosphomimetic version of FOXK2, our data indicate that one role of phosphorylation is likely to be a reduction in p21 repression. Because maximal FOXK2 phosphorylation occurs during mitosis, this loss of repression might contribute to increasing p21 levels and thereby enhance the activity of CDK4·CLND complexes during G1 phase. We also find that phosphorylation of FOXK2 appears to be important in regulating its stability, with the phosphorylated form being less stable. FOXK2 instability is also apparent in cell release from nocodazole block (Fig. 1C), but the timing of this is variable, and we have been unable to demonstrate this effect with transiently expressed proteins, most likely due to overexpression (data not shown). This precludes a comparative analysis of the FOXK2(S368A/S423A) mutant protein, so we are unable to make a definitive connection between the changes caused by CDK·cyclin-mediated phosphorylation and the effects seen on FOXK2 levels in cells released from nocodazole block. The changes in FOXK2 levels could, however, contribute to the loss of repression of key target genes as cells exit mitosis. Although we have focused on serines 368 and 423, it is likely that other CDK sites will contribute to both of these regulatory activities and hence produce more profound effects on FOXK2 activity. However, it is clear that phosphorylation at serines 368 and 423 is vitally important because loss of phosphorylation at these sites cannot be tolerated, because the mutant FOXK2(S368A/S423A) promotes apoptosis, and stable cell lines containing this mutant cannot be generated. It is unclear how this is manifested at the transcriptional level and whether p21 might be a critical target of FOXK2 in this context or whether other targets might be more relevant. Further studies will be directed to uncovering the function of FOXK2 during the cell cycle and its repertoire of target genes in this context.
Supplementary Material
Acknowledgments
We thank Anne Clancy, Karren Palmer, and Michael Schümann for excellent technical assistance; Mike Jackson for help with the flow cytometry and both Jane Kott and Peter March for help with the microscopy; Alan Whitmarsh and members of our laboratories for comments on the manuscript and stimulating discussions; and Stephen Taylor, Michael Rudnicki, Cecilie Freddie, Carole Beachill, Richard Gaynor, and Neil Perkins for reagents.
This work was supported by grants from the Biotechnology and Biological Sciences Research Council (to D. J. M.), by the Wellcome Trust and a Royal Society-Wolfson award (to A. D. S.), and by an overseas research scholarship of Interdisziplinäres Zentrum für Klinische Forschung at the University of Leipzig (D02) (to A. M.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1 and Movie 1.
Z. Ji and A. D. Sharrocks, unpublished data.
- FHA
- forkhead-associated
- CDK
- cyclin-dependent kinase
- EGFP
- enhanced green fluorescent protein
- CLNA
- cyclin A
- CLNB
- cyclin B.
REFERENCES
- 1.Carlsson P., Mahlapuu M. (2002) Dev. Biol. 250, 1–23 [DOI] [PubMed] [Google Scholar]
- 2.Hannenhalli S., Kaestner K. H. (2009) Nat Rev Genet. 10, 233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myatt S. S., Lam E. W. (2007) Nat. Rev. Cancer 7, 847–859 [DOI] [PubMed] [Google Scholar]
- 4.Koranda M., Schleiffer A., Endler L., Ammerer G. (2000) Nature 406, 94–98 [DOI] [PubMed] [Google Scholar]
- 5.Kumar R., Reynolds D. M., Shevchenko A., Shevchenko A., Goldstone S. D., Dalton S. (2000) Curr. Biol. 10, 896–906 [DOI] [PubMed] [Google Scholar]
- 6.Pic A., Lim F. L., Ross S. J., Veal E. A., Johnson A. L., Sultan M. R., West A. G., Johnston L. H., Sharrocks A. D., Morgan B. A. (2000) EMBO J. 19, 3750–3761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu G., Spellman P. T., Volpe T., Brown P. O., Botstein D., Davis T. N., Futcher B. (2000) Nature 406, 90–94 [DOI] [PubMed] [Google Scholar]
- 8.Buck V., Ng S. S., Ruiz-Garcia A. B., Papadopoulou K., Bhatti S., Samuel J. M., Anderson M., Millar J. B., McInerny C. J. (2004) J. Cell Sci. 117, 5623–5632 [DOI] [PubMed] [Google Scholar]
- 9.Bulmer R., Pic-Taylor A., Whitehall S. K., Martin K. A., Millar J. B., Quinn J., Morgan B. A. (2004) Eukaryot. Cell. 3, 944–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho K. K., Myatt S. S., Lam E. W. (2008) Oncogene 27, 2300–2311 [DOI] [PubMed] [Google Scholar]
- 11.Laoukili J., Stahl M., Medema R. H. (2007) Biochim. Biophys. Acta 1775, 92–102 [DOI] [PubMed] [Google Scholar]
- 12.Medema R. H., Kops G. J., Bos J. L., Burgering B. M. (2000) Nature 404, 782–787 [DOI] [PubMed] [Google Scholar]
- 13.Alvarez B., Martínez-A C., Burgering B. M., Carrera A. C. (2001) Nature 413, 744–747 [DOI] [PubMed] [Google Scholar]
- 14.Laoukili J., Kooistra M. R., Brás A., Kauw J., Kerkhoven R. M., Morrison A., Clevers H., Medema R. H. (2005) Nat Cell Biol. 7, 126–136 [DOI] [PubMed] [Google Scholar]
- 15.Pic-Taylor A., Darieva Z., Morgan B. A., Sharrocks A. D. (2004) Mol. Cell. Biol. 24, 10036–10046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darieva Z., Pic-Taylor A., Boros J., Spanos A., Geymonat M., Reece R. J., Sedgwick S. G., Sharrocks A. D., Morgan B. A. (2003) Curr. Biol. 13, 1740–1745 [DOI] [PubMed] [Google Scholar]
- 17.Reynolds D., Shi B. J., McLean C., Katsis F., Kemp B., Dalton S. (2003) Genes Dev. 17, 1789–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darieva Z., Bulmer R., Pic-Taylor A., Doris K. S., Geymonat M., Sedgwick S. G., Morgan B. A., Sharrocks A. D. (2006) Nature 444, 494–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Major M. L., Lepe R., Costa R. H. (2004) Mol. Cell. Biol. 24, 2649–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lüscher-Firzlaff J. M., Lilischkis R., Lüscher B. (2006) FEBS Lett. 580, 1716–1722 [DOI] [PubMed] [Google Scholar]
- 21.Chen Y. J., Dominguez-Brauer C., Wang Z., Asara J. M., Costa R. H., Tyner A. L., Lau L. F., Raychaudhuri P. (2009) J. Biol. Chem. 284, 30695–30707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu Z., Malureanu L., Huang J., Wang W., Li H., van Deursen J. M., Tindall D. J., Chen J. (2008) Nat. Cell Biol. 10, 1076–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li C., Lai C. F., Sigman D. S., Gaynor R. B. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 7739–7743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garry D. J., Meeson A., Elterman J., Zhao Y., Yang P., Bassel-Duby R., Williams R. S. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5416–5421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hawke T. J., Jiang N., Garry D. J. (2003) J. Biol. Chem. 278, 4015–4020 [DOI] [PubMed] [Google Scholar]
- 26.Freddie C. T., Ji Z., Marais A., Sharrocks A. D. (2007) Nucleic Acids Res. 35, 5203–5212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nirula A., Moore D. J., Gaynor R. B. (1997) J. Biol. Chem. 272, 7736–7745 [DOI] [PubMed] [Google Scholar]
- 28.McKinnell I. W., Ishibashi J., Le Grand F., Punch V. G., Addicks G. C., Greenblatt J. F., Dilworth F. J., Rudnicki M. A. (2008) Nat. Cell Biol. 10, 77–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang S. H., Yates P. R., Whitmarsh A. J., Davis R. J., Sharrocks A. D. (1998) Mol. Cell. Biol. 18, 710–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X., Guo C., Chen Y., Shulha H. P., Schnetz M. P., LaFramboise T., Bartels C. F., Markowitz S., Weng Z., Scacheri P. C., Wang Z. (2008) Nat. Methods 5, 163–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shore P., Sharrocks A. D. (1994) Mol. Cell. Biol. 14, 3283–3291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Järviluoma A., Child E. S., Sarek G., Sirimongkolkasem P., Peters G., Ojala P. M., Mann D. J. (2006) Mol. Cell. Biol. 26, 2430–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klemm C., Otto S., Wolf C., Haseloff R. F., Beyermann M., Krause E. (2006) J. Mass Spectrom. 41, 1623–1632 [DOI] [PubMed] [Google Scholar]
- 34.Yan J., Xu L., Crawford G., Wang Z., Burgess S. M. (2006) Mol. Cell. Biol. 26, 155–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan D. O. (1997) Annu. Rev. Cell Dev. Biol. 13, 261–291 [DOI] [PubMed] [Google Scholar]
- 36.Fung T. K., Poon R. Y. (2005) Semin. Cell Dev. Biol. 16, 335–342 [DOI] [PubMed] [Google Scholar]
- 37.Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper K. (2006) Oncogene 25, 5228–5232 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.