

Abstract
CYP3A4 is a dominant human liver cytochrome P450 enzyme engaged in the metabolism and disposition of >50% of clinically relevant drugs and held responsible for many adverse drug-drug interactions. CYP3A4 and its mammalian liver CYP3A orthologs are endoplasmic reticulum (ER)-anchored monotopic proteins that undergo ubiquitin (Ub)-dependent proteasomal degradation (UPD) in an ER-associated degradation (ERAD) process. These integral ER proteins are ubiquitinated in vivo, and in vitro studies have identified the ER-integral gp78 and the cytosolic co-chaperone, CHIP (C terminus of Hsp70-interacting protein), as the relevant E3 Ub-ligases, along with their cognate E2 Ub-conjugating enzymes UBC7 and UbcH5a, respectively. Using lentiviral shRNA templates targeted against each of these Ub-ligases, we now document that both E3s are indeed physiologically involved in CYP3A ERAD/UPD in cultured rat hepatocytes. Accordingly, specific RNAi resulted in ≈80% knockdown of each hepatic Ub-ligase, with a corresponding ≈2.5-fold CYP3A stabilization. Surprisingly, however, such stabilization resulted in increased levels of functionally active CYP3A, thereby challenging the previous notion that E3 recognition and subsequent ERAD of CYP3A proteins required ab initio their structural and/or functional inactivation. Furthermore, coexpression in HepG2 cells of both CYP3A4 and gp78, but not its functionally inactive RING-finger mutant, resulted in enhanced CYP3A4 loss greater than that in corresponding cells expressing only CYP3A4. Stabilization of a functionally active CYP3A after RNAi knockdown of either of the E3s, coupled with the increased CYP3A4 loss on gp78 or CHIP coexpression, suggests that ERAD-associated E3 Ub-ligases can influence clinically relevant drug metabolism by effectively regulating the physiological CYP3A content and consequently its function.
Keywords: Cytochrome P450, Drug Metabolism, E3 Ubiquitin Ligase, endoplasmic Reticulum (ER), Enzyme Inactivation, Enzyme Turnover, Liver, Proteasome, Protein Degradation, ERAD
Introduction
The CYP3A subfamily of hepatic cytochrome P450 hemoproteins includes CYP3A4, the dominant human liver P450 enzyme responsible for the metabolism of more than 50% of clinically relevant drugs and other xenobiotics (1). The CYPs 3A,2 in common with many hepatic P450s, are excellent examples of integral endoplasmic reticulum (ER) membrane-anchored monotopic proteins, with their N termini embedded in the ER and their catalytic domains exposed to the cytosol. Using various in vivo and in vitro reconstituted eukaryotic systems, we have shown that both native3 and structurally inactivated CYPs 3A incur ubiquitin (Ub)-dependent proteasomal degradation (UPD), in a typical ER-associated degradation (ERAD) process involving phosphorylation, ubiquitination, ER membrane extraction into the cytosol, and subsequent degradation by the 26S proteasome (2–13). Indeed mechanism-based CYP3A inactivation often results in active site structural lesions within their cytosolic domain (2, 6, 7), thereby qualifying these proteins as bona fide ERAD-C substrates.
The pathways of P450 degradation appear to be highly conserved in all eukaryotes from yeast to man (11–22). Accordingly, our studies of heterologous expression of CYP3A4 in wild type Saccharomyces cerevisiae and mutants containing defined genetic lesions in various ERAD components have enabled us to characterize CYP3A ERAD/UPD by identifying several participants such as the ERAD-associated soluble E2 Ub-conjugating enzyme Ubc7p and its membrane anchor Cue1p, Cdc48p-Ufd1p-Npl4 AAA-ATPase complex (homologous to the mammalian p97 complex), and Rpn1p (Hrd2p), an essential 26S proteasomal cap subunit, as important participants in CYP3A ERAD (13, 14, 17). Despite the unambiguous finding that CYP3A4 is indeed degraded in an Ubc7p/Cue1p- and Rpn1p-dependent ERAD process (14, 17), none of the canonical yeast E3 Ub-ligases such as Doa10p or even Hrd1p/Hrd3p involved in the ERAD of several integral and lumenal ER proteins and/or cytosolic proteins (19–27) could be implicated (13, 14, 17). Because this could reflect subtle differences in the recognition of a mammalian CYP3A protein by these yeast Ub-ligases, we examined in vitro CYP3A4 ubiquitination systems functionally reconstituted with purified, recombinant mammalian E2-E3 enzymes. These analyses, although enabling us once again to exclude HRD1 and TEB4, the mammalian homologs of yeast Hrd1p and Doa10p (26, 28–31), revealed MmUBC7 (Ube2g2)/gp78/AMFR and UbcH5a/CHIP as functional E2-E3 enzyme complexes in in vitro CYP3A4 ubiquitination (10).
The E3 Ub-ligase gp78/AMFR (“glycoprotein” 78/autocrine motility factor receptor) is a polytopic protein N-terminally anchored to the ER membrane with its intrinsic RING-finger Ub-ligase, Cue1-like, UBC7/Ube2g2-binding, substrate recognition, and p97-binding regions all situated in its cytosolic C-terminal (residues 309–643) domain (gp78C), which is the functionally active E3 domain (32–39). Its ER topology, UBC7 dependence, and functional p97 association are consistent with the known requirements for CYP3A4 ERAD and its plausible role in this process. On the other hand, UbcH5a-dependent cytosolic E3 CHIP (C terminus of Hsp70-interacting protein) is a U-box E3 Ub-ligase that also functions as a co-chaperone (40–45). CHIP contains three tandem tetratricopeptide repeats in its N-terminal domain, which bind Hsc70 and Hsp90, and a C-terminal U-box, which is RING-finger-like and functions as a chaperone-dependent Ub-ligase that links the client substrates of the cytosolic chaperone machinery to their UPD (41–51). Support for its involvement in CYP3A4 ERAD could not be gleaned from our yeast analyses because S. cerevisiae lacks CHIP (20). However, our preliminary findings of CYP3A4 stabilization in a temperature-sensitive Hsp70-defective yeast mutant (ssa1-45), coupled with the reported ability of CHIP to ubiquitinate not only CYP3A4 (10) but also CYP2E1, another hepatic ER-anchored P450 (49), in in vitro functionally reconstituted ubiquitination systems, underscored its plausible involvement in CYP3A4 ERAD.
Given the ability of both gp78 and CHIP to ubiquitinate CYP3A4 in vitro, and the compelling preliminary evidence supporting a plausible role for each E3 Ub-ligase in CYP3A ERAD, we sought to establish their relative physiological relevance to this process in cultured primary rat hepatocytes through in vivo gp78 and/or CHIP knockdown via RNA interference (RNAi). Our findings described herein document that both E3 Ub-ligases are relevant to CYP3A4 ERAD, as specific knockdown of each enzyme results in an immunochemically detectable CYP3A stabilization that is functionally relevant and has clinical implications for drug-drug interactions (DDIs).
EXPERIMENTAL PROCEDURES
Materials
7-Hydroxy-4-trifluoromethylcoumarin (HFC) and 7-benzyloxy-4-trifluoromethylcoumarin (BFC) were obtained from BD Gentest (Woburn, MA). β-Glucuronidase/arylsulfatase was purchased from Roche Applied Science. Lipofectamine 2000 was obtained from Invitrogen. Common cell culture media, supplements, culture plasticware, and commercial sources of protease inhibitors and dexamethasone (Dex) have been reported previously (52, 53). Goat polyclonal IgGs were raised commercially against purified recombinant rat hepatic CYP3A23 and purified by Hi-Trap® protein A-Sepharose affinity chromatography.
Plasmids pCIneo-gp78-(1–643) encoding full-length human gp78, pCIneo-gp78Rf-m, its double RING-finger double mutant (gp78RM), and pCIneo-gp78C encoding the cytosolic C-terminal gp78 domain (residues 309–643) (35, 36) were provided by Dr. A. M. Weissman. The cytosolic C-terminal gp78 domain was digested from pGEX4T2-gp78C and cloned into pcDNA6 vector to make the eukaryotic overexpression plasmid. All other buffers and reagents were of the highest commercial grade. pcDNA3.1-CHIP plasmid encoding the full-length 303-residue human CHIP was provided by Dr. Cam Patterson.
Lentiviral shRNA Constructs
shRNA target sequences were selected based on the Gene LinkTM prediction program. The shRNA primers were amplified using pTZu6 + 1 followed by cloning into the pHRCMVpuroWsin18 vector. For gp78 (XM_341644.4), the sequences targeted were GCCTACCGCGCCCTCAGCCAGCCC (shRNA1, targeting exon 3, 300–318 nt) and CAAGACACCTCTTGTCCAACATGC (shRNA2, targeting exon 9, 1318–1336 nt). In the case of CHIP (NM_001025625.2), the sequences targeted were GAGAGTTATGATGAGGCCATT (shRNA1, targeting exon 2, 415–435 nt), CAAGGAGCAGCGACTCAACTT (shRNA2, targeting exon 10, 1410–1430 nt), GAGAGTGAGCTGCACTCCTAT (shRNA3, targeting exon 11, 1558–1578 nt), and CCCTTCGCATTGCTAAGAAGA (shRNA4, targeting exon 12, 1852–1872 nt). The control (CT) shRNA sequence was a “scrambled” GACTTCATAAGGCGCATG sequence that through BLAST analyses was verified not to be similar or to target any sequence in the rat genome.
Lentiviral Packaging in HEK293T and Quantitation
HEK293T cells were grown in Dulbecco's modified Eagle's medium (DMEM-high glucose) containing 10% v/v fetal bovine serum (FBS) and penicillin/streptomycin (50 μg/ml) to 80–90% confluency on 6-well collagen-coated plates. On the day of the transfection, the medium was changed to serum-free Opti-MEM. The packaging vesicular stomatitis virus G expression vector pMD.G1, rev expression vector pCMVΔR8.91, and an appropriate lentiviral pHRCMVPURO-Wsin18-shRNA expression plasmid were mixed with Lipofectamine 2000 in Opti-MEM. The cells were incubated at 37 °C with this mixture for 4–6 h. The medium was then changed to serum containing DMEM. The medium from the infected cells was collected on day 2 and fresh medium added. On day 4, the medium was collected again and pooled with that from day 2. These pooled media containing secreted virus were filtered through a 0.45-μm filter, and the virus stock was stored at −80 °C. For quantitation of the viral stock, a 5–10-ml aliquot of the pooled virus-containing media was centrifuged over a 10-ml cushion of 20% w/v sucrose at 100,000 × g at 4 °C in a Beckman Ultracentrifuge. The supernatant was removed, the glassy pellet was resuspended in lysis buffer, and viral RNA was extracted. This viral RNA was used for absolute quantification by quantitative real-time PCR (qRT-PCR) analyses (see below) using the following primers and probes. The gp78 primers were ACCTCATGCACCACATTCACATGC (forward) and CAAGACACCTCTTGTCCAACATGC (reverse), and the probe was 5′-/56-FAM/TAATGCGGTGTCTTGGGCTGTGTGAA/36-TAMSp/-3′. The CHIP primers were ACCCGGAACCCACTTGTGGCAGTG (forward) and CTGGATGGGCAGTCTGTGAAGGCG (reverse), and the probe was 5′-/FAM/ATCTGAAGATGCAGCAGCCTGAA/36-TAMSp/-3′.
Each viral stock was quantified using a standard curve. Thereafter, the entire volume of viral supernatant was centrifuged and the pellet resuspended in Williams' Medium E to yield a final concentration of 500 ng/ml. This stock was aliquoted and stored at −80 °C until used for rat hepatocyte infection.
Rat Hepatocyte Infection and Culture
Male Sprague-Dawley rats (4–6 weeks old) purchased from Simonsen Laboratories (Gilroy, CA) were fed and given water ad libitum and handled according to the Institutional Animal Care and Use Committee guidelines. Hepatocytes were isolated from rats by in situ liver perfusion with collagenase (liver digest medium). Hepatocytes (3.5 × 106) were mixed with the packaged lentiviral shRNA containing medium (1 μg of viral RNA/plate), seeded onto 60-mm Permanox culture dishes precoated with type I rat tail collagen, and incubated at 37 °C. After 3 h, the medium was removed, and fresh medium containing 1 μg of virus/plate was added to the hepatocytes for overnight infection. After 18–20 h, the medium was changed, and cells were overlaid with 0.25 mg/ml Matrigel. Cells were cultured as described (4, 52–54) in Williams' E medium containing insulin-transferrin-selenium G, 0.1 μm Dex, 50 units/ml penicillin/streptomycin, 2 mm l-glutamine, and 0.1% w/v BSA. Cells were maintained for 2 days with a daily change of medium to enable the recovery and restoration of hepatic function (55). At 72 h of culture, puromycin (5 μg/ml) was added to the medium for selection of cells that had been successfully infected by the shRNA-containing virus. The medium was replaced daily, and cells were cultured for a further 4–5 days with daily light microscopic examination for any signs of cell death or cytotoxicity. In some cases, cells were treated with vehicle (dimethyl sulfoxide) or the CYP3A inducer Dex (10 μm) from days 3 to 7. In other cases, on day 7, Dex (10 μm)-pretreated cells were treated with DDEP (3,5-dicarbethoxy-2,6-dimethyl-4-ethyl-1,4-dihydropyridine; 30 μm) for 3 h before harvesting.
In preliminary experiments, the optimal time for ≥80% knockdown of gp78 or CHIP was determined to be ≥7 days. At this time, cells were harvested in lysis buffer consisting of Tris-HCl (20 mm, pH 7.5), 1% v/v Triton, NaCl (150 mm), 10% v/v glycerol, EDTA (1 mm), EGTA (1 mm), NaF (100 mm), tetrabasic sodium pyrophosphate (10 mm), β-glycerophosphate (17.5 mm), N-ethylmaleimide (5 mm), Na3VO4 (1 mm), and protease inhibitors PMSF (1 mm), leupeptin (20 μm), aprotinin (1.5 μm), E-64 (50 μm), pepstatin (10 μm), antipain (10 μm), 4-2(aminoethyl)benzenesulfonyl fluoride (AEBSF; 1 mm), and bestatin (60 μm). The cells were lysed using an OMNITM-TH homogenizer and sonicated for 40 s. Lysates were clarified by sedimentation at maximum speed in a tabletop microcentrifuge at 4 °C for 15 min. Lysate supernatants were subjected to Western immunoblotting analyses and densitometric quantification using ImageJ software (see below).
In parallel, some shRNA-infected cell cultures were also harvested in an RNA-stabilizing reagent (Qiagen) and used for total RNA extraction and qRT-PCR analyses (see below).
HepG2 Cell Culture and Expression
HepG2 cells were grown in minimal Eagle's medium (MEM) containing 10% v/v FBS and supplemented with nonessential amino acids in 6-well collagen-coated plates till they reached 80–90% confluency. gp78, CHIP, and/or CYP3A expression plasmids were mixed with Lipofectamine 2000 and Opti-MEM. The mixture was added and the cells incubated at 37 °C for 4–6 h, following which the medium was changed to serum-containing MEM. Protein expression was verified by harvesting cells at 0–48 h from the time of transfection by Western immunoblotting analyses of each protein using a specific polyclonal antibody.
CYP3A Immunoprecipitation
CYP3A was routinely immunoprecipitated before the extent of its ubiquitination was determined as described previously (4, 52). For CYP3A immunoprecipitation, 1 mg of cell lysate protein was immunoprecipitated with 2 mg of goat anti-CYP3A antibody. Similarly, 500 μg of protein from the sodium carbonate-washed microsomal subfraction, 2 mg of protein from the corresponding cytosolic subfraction, and 200 μg of the solubilized “trichloroacetic acid pellet” were immunoprecipitated with 3 mg, 2 mg, and 500 μg, respectively, of goat anti-CYP3A antibody.
Immunoblotting Analyses
CYP3A and ubiquitinated CYP3A immunoprecipitates were immunoblotted as described (4, 12, 52). For gp78 and CHIP immunoblotting analyses, lysate protein (50 μg) was subjected to 10% SDS-PAGE followed by electroblotting onto a nitrocellulose membrane. The gp78 and CHIP proteins were immunoblotted with primary rabbit polyclonal antibodies (sc-33541 and sc-66830, respectively, from Santa Cruz Biotechnology, Inc., Santa Cruz, CA). The gp78 antibody (sc-33541) was raised against an epitope corresponding to gp78 residues 236–345 in the internal and C-terminal domain, thus recognizing both full-length gp78 and gp78C. The CHIP antibody (sc-66830) was raised against an epitope corresponding to its C-terminal residues 73–303. This was followed by goat anti-rabbit HRP-conjugated secondary antibody (catalog No. 170-6515) from Bio-Rad Laboratories. Unless otherwise indicated, 5% w/v nonfat milk in 0.1% v/v Tween TBS (TTBS) was used for blocking and to make all primary and secondary antibody dilutions. All immunoblots were developed with the SuperSignal West maximum sensitivity Femto or Pico chemiluminescent substrate (product No. 34095) from Pierce. Actin immunoblotting analyses were routinely conducted with each lysate (10 μg of protein) to ensure equivalent protein loading. Densitometrically derived arbitrary units of individual immunoblots were normalized against the corresponding values of the actin loading controls and then expressed as percent of control/basal values.
Densitometric Quantification
Direct quantification of the immunoblots was performed by ImageJ software (National Institutes of Health) analyses.
qRT-PCR Analyses
Total RNA was extracted with the Ambion RNAqueous-Micro kit (Ambion Inc., Austin, TX; catalog No. AM1931) and treated with DNase to free it from DNA contamination using the Ambion DNA-free kit. This was followed by reverse transcription to cDNA by Moloney murine leukemia virus reverse transcriptase (Invitrogen) exactly as described (53, 54). Universal PCR Master Mix (catalog No. 430447) and TaqMan primer-probe mixes were purchased from Applied Biosystems Inc. for the detection of rat mRNA sequences for CYP3A23 (catalog No. Rn01412959_g1), cytochrome b5 (catalog No. Rn01483963_m1), and β-glucuronidase (GUS; catalog No. Rn00566655_m1). The gp78 primers were: ACCTCATGCACCACATTCACATGC (forward) and CAAGACACCTCTTGTCCAACATGC (reverse). The CHIP primers were: ACCCGGAACCCACTTGTGGCAGTG (forward) and CTGGATGGGCAGTCTGTGAAGGCG (reverse). The PCR reaction mixture (10 μl) was made as follows (final concentrations): 1x PCR master mix containing 1x TaqMan buffer, 200 μm of each dNTP, 1.5 units of AmpliTaq Gold DNA polymerase, 200 nm probes, 900 nm primers, nuclease-free water, 10 ng of cDNA (from the reverse transcriptase reaction). PCR was performed in a MicroAmp ABI Prism 384-well clear optical reaction plate using the ABI Prism 7900 detector system. The initial denaturation cycle was at 95 °C for 10 min followed by 40 cycles with denaturation at 95 °C for 20 s, and annealing/extension at 60 °C for 1 min. The Ct (cycle number at which the fluorescent signal reaches the threshold level) value reflecting the expression of each gene was normalized to that of the endogenous control β-glucuronidase (GUS) gene. Relative gene expression was calculated as 2−ΔCt where ΔCt is defined as Ct for the gene of interest − Ct for GUS. All values are expressed as percent increase/decrease with respect to the RNA value in untreated hepatocytes.
The specificity of the knockdown was verified by parallel qRT-PCR analyses of the RNA sample with the appropriate primers and probes to detect CYP3A23, gp78, CHIP, or cytochrome b5 mRNA.
Immunofluorescence Staining
shRNA-infected cells were fixed in methanol for 15 min at −20 °C and incubated for 1 h with 2% v/v nonimmune rabbit serum. This was followed by incubation with goat anti-CYP3A antibody (1:500, v/v) for 1 h at room temperature. After washing with PBS, cells were incubated with Alexa Fluor 488 rabbit anti-goat antibody (1:3000, v/v). Cells were observed with a Zeiss Axiovert 200M, LSM 510 Meta confocal microscope at ×20 magnification. Images were collected at 1024 × 1024 frame resolution with a pinhole of 0.75 Airy unit. The relative immunofluorescence intensity was quantitated as the mean intensity of a cytoplasmic area (4 μm × 5 μm) excluding the nucleus within each cell, using the dedicated Zeiss confocal software. Intensity values (mean ± S.D.; n = 50 cells) ranged from 1 to 256, corresponding to the fluorescence dynamic range of 1 (no signal) and 256 maximal (saturation) immunofluorescence signal, respectively, of the 20-μm2 area.
Preparation of Microsomal and Cytosolic Subfractions
To determine whether the ER localization and/or subsequent extraction into the cytosol of the parent and ubiquitinated CYP3A species was affected by the 7-day gp78 or CHIP knockdown, on the 8th day the protein was radiolabeled by 35S pulse-chase (60 μCi of l-[35S]Met/Cys for 1 h at 37 °C). This was followed by chase with cold methionine/cysteine as detailed (52). Two h following cold chase, microsomes were isolated from rat hepatocytes as described (54). The supernatant obtained after the first 100,000 × g ultracentrifugation was used as the cytosolic subfraction and subjected to CYP3A immunoprecipitation as described above. To determine the content of CYP3A normally ER-integrated relative to that dislocated but still loosely associated with the external ER surface, microsomes were subjected to a 0.1 m sodium carbonate wash. Accordingly, the resuspended microsomes from 35S-pulse-chase experiments were treated with 0.1 m sodium carbonate and then incubated on ice for 30 min followed by ultracentrifugation at 180,000 × g for 1 h. This treatment is expected to solubilize and “wash” out CYP3A protein loosely associated with the ER membranes into the sodium carbonate fraction (supernatant from the 180,000 × g ultracentrifugation). The resulting 180,000 × g pellet, representing firmly integrated microsomal proteins, was resuspended and used for CYP3A immunoprecipitation. The 180,000 × g supernatant was further treated with 100% trichloroacetic acid, incubated on ice for 30 min to precipitate constituent proteins, and then sedimented at 21,000 × g. The 21,000 × g supernatant was discarded after it was deemed free of any nonprecipitated radiolabeled proteins including CYP3A. The trichloroacetic acid-precipitated protein pellet (“trichloroacetic acid pellet”) was washed with 100% acetone, solubilized in immunoprecipitation buffer, and subjected to CYP3A immunoprecipitation as described above. This immunoprecipitate represents any CYP3A proteins dislocated from the ER but remaining loosely associated with the ER membranes.
BFC 7-O-Debenzylation Assays
CYP3A function was assessed by a fluorescence-based assay performed as described (56) by direct incubation of rat hepatocytes cultured in 60-mm Permanox plates as described above and infected with gp78 shRNA 1 + 2, CHIP-4 shRNA, or control shRNA for 7 days. A volume of 500 μl of incubation medium (1 mm Na2HPO4, 137 mm NaCl, 5 mm KCl, 0.5 mm MgCl2, 2 mm CaCl2, 10 mm glucose, and 10 mm Hepes, pH 7.4, buffered solution) containing the substrate BFC (100 μm) was added to each plate. At defined time points between 0 and 60 min at 37 °C, reactions were stopped by aspiration of the incubation medium. Any HFC conjugates formed during the assay were hydrolyzed by incubation of the medium supernatants with β-glucuronidase/arylsulfatase (150 Fishman units/ml and 1200 Roy units/ml, respectively) for 2 h at 37 °C. Samples were diluted (1:2, v/v) in the quenching solution (0.25 mm Tris, 60% acetonitrile), and HFC metabolite formation was quantified fluorimetrically (excitation wavelength 410 nm and emission wavelength 510 nm) using a SpectraMax M5e fluorescence microplate reader (Molecular Devices, Sunnyvale, CA). An HFC standard curve was used to express the results as mean ± S.D. μmol of HFC metabolite formed/3.5 × 106 hepatocytes of three individual cultures. Microsomes were isolated from cultures similarly treated in parallel, and their BFC O-debenzylase activity was determined after 30 min of incubation at 37 °C (56).
Statistical Analyses
Experiments were performed in triplicate. Data were compared by analysis of variance, and p values of <0.05 were considered statistically significant.
RESULTS
Effects of Lentiviral shRNAi-mediated gp78 Knockdown on CYP3A Content in Cultured Rat Hepatocytes
In preliminary experiments we determined the relative efficiency individually and in combination of shRNA-1 and -2 directed against gp78 on CYP3A content of Dex-treated cultured hepatocytes after 7 days of infection (Fig. 1A). We found that shRNA-1 and shRNA-2 yielded a comparable gp78 knockdown, as revealed by the relative CYP3A stabilization (≈270%) relative to basal CYP3A levels (100%) in cells infected with a shRNA known to target no known rat gene, henceforth referred to as “control” (CT) shRNA. However, concomitant infection with both shRNA-1 and shRNA-2 consistently led to a higher extent (325 ± 7.02%) of CYP3A stabilization relative to control levels (Fig. 1A). Accordingly, this shRNA combination (termed gp78 1/2) was used in all subsequent studies. qRT-PCR analyses of total hepatic RNA isolated from cultured hepatocytes revealed that after 7 days of infection, gp78 1/2 was found to knock down mRNA levels by 78 ± 1.5% of basal values (Fig. 1B). qRT-PCR analyses also indicated no corresponding mRNA knockdown of CYP3A23, cytochrome b5 (another integral ER protein), CHIP, or GUS (a housekeeping gene), thereby attesting to the specificity of the gp78 knockdown (Fig. 1C). Parallel gp78 immunoblotting analyses of lysates from hepatocytes cultured in parallel revealed a corresponding 77 ± 1.6% hepatic gp78 protein knockdown from basal values in control shRNA-infected cells (Fig. 1D). No CHIP protein knockdown was seen when aliquots of these lysates were subjected to CHIP immunoblotting analyses (Fig. 1D). CYP3A stabilization was associated with consistently lowered ubiquitination as monitored by ubiquitin immunoblotting analyses of the CYP3A immunoprecipitates from these cell lysates (Fig. 1E).
FIGURE 1.

Effects of RNAi-mediated gp78 knockdown on CYP3A content in cultured rat hepatocytes. Effects are shown of shRNA-1 and shRNA-2 targeted against hepatic gp78, individually or in combination (gp78 1/2), on hepatic CYP3A content derived from each shRNA-infected cell culture. Hepatocytes were infected with shRNA-1, shRNA-2, shRNA-1 + shRNA-2 (gp78 1/2) directed against hepatic gp78, or an shRNA containing a sequence known not to target any known rat gene (CT) and then treated with the CYP3A inducer Dex. A, a representative example of CYP3A Western immunoblotting analyses of these hepatocyte lysates (50 μg of protein) is shown at the top, with corresponding aliquots used for actin immunoblotting analyses as loading controls. Densitometric quantification of hepatic CYP3A content from three individual experiments is shown at the bottom. Statistical analyses revealed significant differences in hepatic CYP3A content between CT and shRNA-1-, shRNA-2-, or shRNA gp78 1/2-infected cells at p < 0.01. No statistically significant differences were observed between shRNA 1- or shRNA 2-infected cells. B, the effects of gp78 shRNA 1/2 on hepatic gp78 mRNA. C, evidence of the target specificity of gp78 shRNA 1/2 against gp78 mRNA and that of CHIP-4 shRNA against CHIP mRNA by qRT-PCR analyses are documented. D, a representative example of gp78 Western immunoblotting analyses of these hepatocyte lysates (50 μg of protein) is shown at the top, with corresponding aliquots used for actin immunoblotting analyses as loading controls. Densitometric quantification of hepatic gp78 content from three individual experiments is shown at the bottom. Statistical analyses revealed significant differences in hepatic gp78 content between CT and shRNA gp78 1/2-infected cells at p < 0.01. E, effects of gp78 shRNA 1/2 on hepatic ubiquitinated CYP3A species, detected after ubiquitin immunoblotting (IB) analyses of CYP3A immunoprecipitates (IP) derived from each shRNA-infected cell culture are shown. Densitometric quantification of hepatic ubiquitinated CYP3A (area between 65 kDa and top of the gel) from three individual experiments is shown at the right. Statistical analyses revealed significant differences in hepatic CYP3A content between CT and shRNA gp78 1/2-infected cells at p < 0.01.
Stabilization of the constitutive CYP3A (CYP3A2) on the order of 173 ± 7.67% relative to corresponding control values was also observed on parallel CYP3A immunoblotting analyses of lysates from non-Dex-treated cells (Fig. 2). This was further enhanced on Dex-mediated CYP3A induction to 278 ± 14.2%. Treatment of Dex-pretreated cells with the CYP3A mechanism-based inactivator DDEP for 6 h, as expected, significantly lowered the basal levels of CYP3A (Fig. 2). However, this CYP3A loss was significantly abrogated by hepatic gp78 knockdown, and CYP3A was stabilized to levels almost as high (256 ± 12.7%) as those observed in cells treated with Dex alone (Fig. 2). Together these findings verify that DDEP-mediated acceleration of hepatic CYP3A ERAD is blocked on hepatic gp78 knockdown, consistent with a significant role for gp78 in CYP3A ERAD.
FIGURE 2.
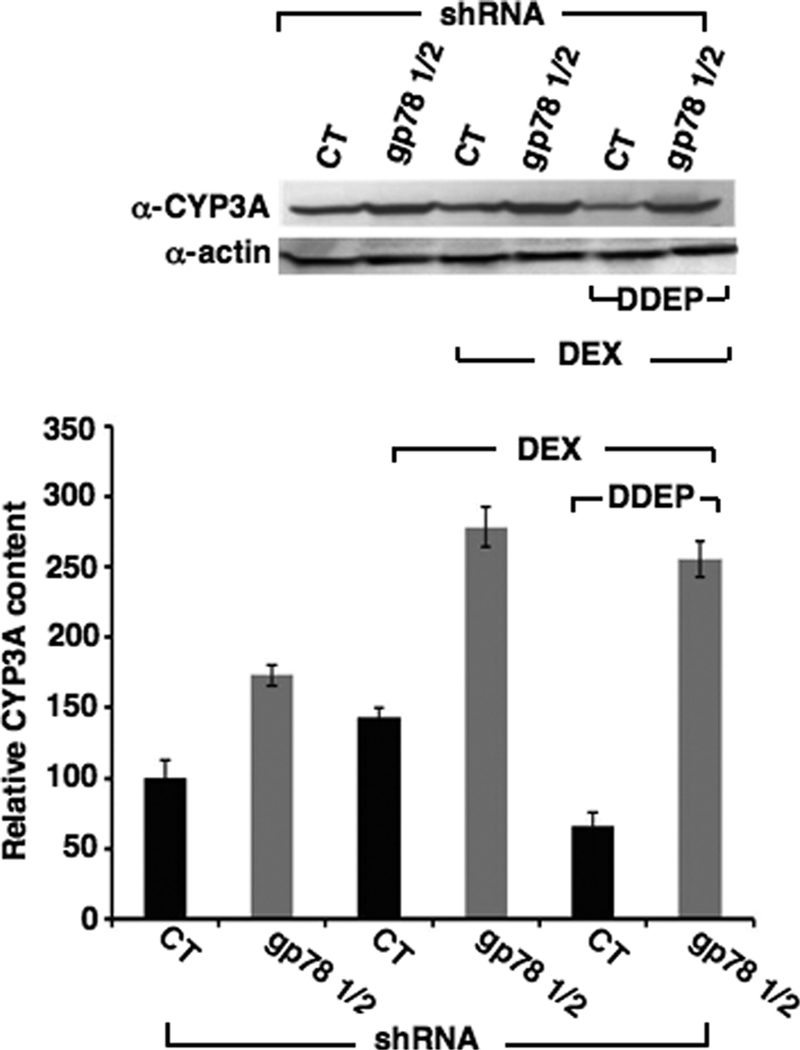
Stabilization of native constitutive, Dex-inducible, and DDEP-inactivated CYP3A content in cultured rat hepatocytes following RNAi-mediated gp78 knockdown. Hepatocytes were infected with shRNA gp78 1/2 or the control shRNA (CT). Combined effects of shRNA gp78 1/2 on hepatic CYP3A content of untreated (first 2 lanes) and Dex-pretreated hepatocytes (next 4 lanes) are shown. Some of the Dex-pretreated cultures were also treated with DDEP, a mechanism-based CYP3A inactivator for 6 h (last 2 lanes). A representative example of CYP3A Western immunoblotting analyses of these hepatocyte lysates (50 μg of protein) is shown at the top, with corresponding aliquots used for actin immunoblotting analyses as the loading controls. Densitometric quantification of hepatic CYP3A content from three individual experiments is shown at the bottom. Statistical analyses revealed significant differences in hepatic CYP3A content between CT and corresponding shRNA gp78 1/2-infected cells at p < 0.01.
Coexpression of Hepatic gp78 and CYP3A4 in HepG2 Cells Enhances CYP3A4 Turnover
In preliminary studies, the time course of expression of the pcDNA6-His A vector encoding full-length gp78 (gp78-WT), its full-length ring mutant of gp78 (gp78RM), or the transmembrane anchor-deleted C-terminal domain of gp78 (gp78C) was found to be comparable at 48 h (Fig. 3A). This time point was therefore used to examine the individual effects of coexpressing each of these vectors with that of CYP3A4 (Fig. 3B). Coexpression of gp78-WT and CYP3A4 led to a significant decrease in CYP3A4 levels in HepG2 cells as compared with control cells expressing CYP3A4 alone (None) or cells coexpressing CYP3A4 and the empty pcDNA6-His A vector (Mock) (Fig. 3B). By contrast, coexpression of the inactive RING-finger mutant, gp78RM, had no significant effect on CYP3A4 levels in HepG2 cells (Fig. 3B). Surprisingly, coexpression of just the active C-terminal domain, gp78C, led to a significantly enhanced loss of coexpressed CYP3A4, even though the levels of gp78 protein were nearly comparable by immunoblotting analyses (Fig. 3B). These findings revealed that the soluble gp78C was more efficient than its full-length ER-anchored counterpart in supporting CYP3A4 ERAD4 (Fig. 3B).
FIGURE 3.

Effects of coexpression of CYP3A4 and gp78 in cultured HepG2 cells. A, HepG2 cells were grown to confluence and then co-infected with a vector expressing CYP3A4 and a vector expressing full-length functionally active gp78 (gp78-WT), its RING-finger mutant (gp78-RM), or just its ER membrane anchor-deleted C-terminal domain (gp78-C) for 0–48 h. The time course of the densitometrically quantified gp78 protein in cells expressed by each of these vectors is shown. B, HepG2 cells coexpressing CYP3A4 and a vector expressing gp78-WT, its inactive gp78-RM, or gp78-C and harvested at 48 h are shown. Control HepG2 cells expressing just CYP3A4 by itself (no coexpression vector) or along with the empty vector (Mock) were cultured in parallel. A representative example of CYP3A Western immunoblotting analyses of these hepatocyte lysates (30 μg of protein) is shown at the top, with corresponding aliquots used for actin immunoblotting analyses as the loading controls. Densitometric quantification of hepatic CYP3A content (mean ± S.D.) from three individual experiments is shown at the bottom. Statistical analyses revealed significant differences in hepatic CYP3A4 content of cells expressing CYP3A4 alone and corresponding gp78-WT or gp78-C expressing cells at p < 0.01. Significant differences were also observed in the CYP3A4 content of gp78-WT- or gp78-C-expressing cells and those expressing gp78-RM at p < 0.01, and between the CYP3A4 content of gp78-WT and gp78-C expressing cells at p < 0.01. No significant differences in CYP3A4 content were observed between cells expressing CYP3A4 alone (None) and cells expressing the empty vector (Mock).
Effects of Lentiviral shRNAi-mediated Hepatic CHIP Knockdown on CYP3A Content in Cultured Rat Hepatocytes
Four different lentiviral shRNA templates targeted against specific CHIP exons were used to knock down hepatic CHIP content in cultured rat hepatocytes (Fig. 4). Although individually all four shRNAs significantly knocked down CHIP relative to the control shRNA targeting no known rat gene, shRNA construct CHIP-4 was the most effective, exhibiting a CHIP mRNA knockdown of ∼80.6 ± 1.13% (Fig. 4A) and a corresponding reduction of 73.3 ± 0.84% basal hepatic CHIP protein content (Fig. 4B). The specificity of CHIP-4 was documented by qRT-PCR analyses indicating no corresponding mRNA knockdown of CYP3A23, cytochrome b5, gp78, or GUS, thereby attesting to the specificity of the CHIP knockdown (Fig. 1C). Corresponding CYP3A immunoblotting analyses showed a statistically significant hepatic CYP3A stabilization ranging from 168 ± 6.11 to 237 ± 2.90% with shRNA-1, -2, and -3 and an even greater CYP3A stabilization with CHIP shRNA-4 of ≈ 250 ± 2.90% (Fig. 4C). Corresponding ubiquitin immunoblotting analyses of CYP3A immunoprecipitates from the corresponding lysates faithfully reflected the relative extent of hepatic CHIP protein knockdown (Fig. 4D). The extent of CYP3A stabilization after CHIP knockdown was comparable with that seen with a similar extent of hepatic gp78 knockdown, thereby revealing that both E3 Ub-ligases were involved in CYP3A ERAD in vivo. Our attempts to simultaneously knock down both gp78 and CHIP resulted in considerable cytotoxicity and cell death and, thus, unreliable CYP3A stabilization.
FIGURE 4.

Effects of RNAi-mediated CHIP knockdown on CYP3A content in cultured rat hepatocytes. Hepatocytes were infected with shRNA templates CHIP-1, CHIP-2, CHIP-3, or CHIP-4 targeted against various exons of the rat CHIP gene or the control shRNA template (CT) and then treated with the CYP3A inducer Dex. Individual effects of shRNAs CHIP-1–4 on hepatic CHIP mRNA (A) and CHIP protein content (B) are shown. B, a representative example of CHIP Western immunoblotting analyses of these hepatocyte lysates (50 μg of protein) is shown at the top, with corresponding aliquots used for actin immunoblotting analyses as loading controls. Densitometric quantification of hepatic CHIP content from three individual experiments is shown at the bottom. Statistical analyses revealed significant differences in hepatic CHIP content between CT and cells infected with each CHIP-shRNA at p < 0.01. C, individual effects of shRNAs CHIP-1–4 on hepatic CYP3A content derived from each shRNA-infected cell culture are shown. A representative example of CYP3A Western immunoblotting analyses of these hepatocyte lysates (50 μg of protein) is shown at the top, with corresponding aliquots used for actin immunoblotting analyses as loading controls. Densitometric quantification of hepatic CYP3A content (mean ± S.D.) from three individual experiments is shown at the bottom. Statistical analyses revealed significant differences in hepatic CYP3A content between CT and shRNA CHIP-1, CHIP-2, CHIP-3, and CHIP-4 at p < 0.01, p < 0.01, p < 0.01, and p < 0.01, respectively. No statistically significant differences were observed between CHIP-1- or CHIP-2-infected cells. D, individual effects of shRNAs CHIP-1–4 on hepatic ubiquitinated CYP3A species are indicated, as detected after Ub immunoblotting (IB) analyses of CYP3A immunoprecipitates (IP) derived from each shRNA-infected cell culture. Densitometric quantification of hepatic ubiquitinated CYP3A (area between 65 kDa and top of the gel) from three individual experiments is shown at the right. Statistical analyses revealed significant differences in hepatic CYP3A content between CT and shRNA CHIP-1, CHIP-2, CHIP-3, and CHIP-4-infected cells at p < 0.05, p < 0.01, p < 0.01 and p < 0.01, respectively.
Effects of CHIP Coexpression on CYP3A4 Degradation in HepG2 Cells
In preliminary studies, the time course of expression of the vector encoding full-length CHIP was found to be maximal between 24 and 48 h (Fig. 5A). The 48-h time point was therefore selected to examine the individual effects of coexpressing this vector with CYP3A4 (Fig. 5B). Coexpression of CHIP and CYP3A4 led to a significant decrease in CYP3A4 levels in HepG2 cells, as compared with control cells expressing CYP3A4 alone or cells coexpressing CYP3A4 and the empty vector (Mock) (Fig. 5B). These findings are consistent with a significant role of CHIP in CYP3A4 ERAD (Fig. 5B).
FIGURE 5.

Effects of coexpression of CYP3A4 and CHIP in cultured HepG2 cells. A, HepG2 cells were grown to confluence and then co-infected with expression vectors for CYP3A4 and CHIP for 0–48 h. The time course of the densitometrically quantified CHIP protein expressed by the CHIP vector is shown. B, HepG2 cells coexpressing CYP3A4 and CHIP vectors and harvested at 48 h are shown. Control HepG2 cells expressing just CYP3A4 by itself (no coexpression vector (None)) or along with the empty vector (Mock) were cultured in parallel. A representative example of CYP3A Western immunoblotting analyses of these hepatocyte lysates (30 μg of protein) is shown at the top, with corresponding aliquots used for actin immunoblotting analyses as the loading controls. Densitometric quantification of hepatic CYP3A content (mean ± S.D.) from three individual experiments is shown at the bottom. Statistical analyses revealed significant differences in hepatic CYP3A4 content between cells expressing CYP3A4 by itself and corresponding CHIP-expressing cells at p < 0.01. No significant differences in hepatic CYP3A4 content were observed between cells expressing CYP3A4 by itself and those expressing the empty vector (mock).
In Situ Verification of CYP3A Stabilization after gp78 or CHIP Knockdown
Confocal immunofluorescence microscopic analyses of cells infected with CT, gp78 1/2, or CHIP-4 shRNA showed no significant effects on cell morphology at 7 days of infection. However, the CYP3A content, consistent with the immunoblotting analyses, was visibly and markedly increased in gp78 1/2- or CHIP-4 shRNA-infected cells over the controls (Fig. 6). Accordingly, quantification of the relative immunofluorescence intensity of a 20-μm2 cytoplasmic area (excluding the nucleus) of CT and gp78 1/2- and CHIP-4 shRNA-infected cells yielded 47.2 ± 4.8, 84.5 ± 4.3, and 69.4 ± 3.6 arbitrary units (mean ± S.D., n = 50 cells), respectively.
FIGURE 6.

RNAi-mediated gp78 or CHIP knockdown with in situ verification of CYP3A stabilization in cultured rat hepatocytes by confocal immunofluorescence microscopy. Rat hepatocyte cultures were infected with CT shRNA, shRNA gp78 1/2, or shRNA CHIP-4 for 7 days. shRNA-infected rat hepatocyte cultures were fixed and stained with antibodies to CYP3A (green). Data from a representative experiment showing CYP3A accumulation are shown.
Relative Intracellular Localization of the Parent and/or Ubiquitinated CYP3A Species after gp78 or CHIP Knockdown
The above confocal immunofluorescence analyses clearly indicated CYP3A stabilization after either gp78 or CHIP knockdown relative to corresponding controls. However, at the magnification used, it provided no clues as to the precise intracellular CYP3A localization following its stabilization and/or whether gp78 or CHIP knockdown affected its ER extraction. To gain better insight into this trafficking, CYP3A was subjected to 35S-pulse-chase analyses. In CT shRNA infected cells, in the presence of functionally active gp78, CHIP Ub-ligases, and p97 AAA ATPase following the 2-h chase period, [35S]-CYP3A was found as parent (55 kDa) and HMM-ubiquitinated species in both the ER and the cytosol (Fig. 7). This is consistent with a fully functional CYP3A ubiquitination process, its extraction into the cytosol, and its subsequent proteasomal degradation. By contrast, after gp78 knockdown, the parent CYP3A content in the ER was increased relative to that of the CT cells. However, the corresponding level of the HMM CYP3A species was considerably decreased in the ER and also even further decreased in the cytosol relative to corresponding levels in CT cells. These findings are consistent with a significant role of gp78 in CYP3A ubiquitination and its recruitment of p97 to the ER (see “Discussion”). The finding that HMM-ubiquitinated [35S]-CYP3A species were still detected in the ER, albeit at a reduced level, is consistent with unaltered CHIP and/or residual gp78 activity in these cells (Fig. 7A).
FIGURE 7.

Relative intracellular localization of parent and ubiquitinated HMM CYP3A species after gp78 or CHIP RNAi in cultured hepatocytes. Rat hepatocyte cultures were infected with CT shRNA, shRNA gp78 1/2, or shRNA CHIP-4 for 7 days. On the 8th day, they were subjected to 35S-pulse-chase analyses. Two h after cold chase, cells were harvested, and homogenates were subfractionated into cytosol and microsomes. CYP3A immunoprecipitates (45 μl) from the cytosol, sodium carbonate-washed microsomes, and a trichloroacetic acid (TCA) pellet derived from the sodium carbonate wash were obtained as described under “Experimental Procedures” and subjected to SDS-PAGE analyses. The gels were dried and then exposed to PhophorImaging screens and visualized using a Typhoon scanner. A, a typical SDS-polyacrylamide gel is shown as a representative of corresponding CYP3A immunoprecipitates from pooled hepatocyte cultures. B, the relative [35S]-CYP3A intensity of the parent CYP3A (55-kDa band) and the ubiquitinated CYP3A species between 65 and 250 kDa in each lane were quantified using ImageQuant software. Values are mean ± S.D. of three individual determinations. The color wheel intensity code is as follows: white > magenta > red > orange > yellow > green > light blue > dark blue > black.
On the other hand, following CHIP knockdown, the levels of ER-anchored parent CYP3A species significantly increased, whereas the levels of detectable HMM-ubiquitinated [35S]-CYP3A species were further reduced, despite a functional gp78 (Fig. 7A). This finding suggests a significant role of CHIP in CYP3A ubiquitination. Moreover, very little CYP3A was extracted into the cytosol relative to the corresponding levels in CT cells or even those after gp78 knockdown (Fig. 7A). The corresponding quantification of the parent (55 kDa) and HMM (65–250 kDa) [35S]-CYP3A content from three separate experiments using ImageQuant software verifies this assessment (Fig. 7B).
Is the Observed Hepatic CYP3A Stabilization Functionally Relevant?
Although our RNAi analyses revealed that both gp78 and CHIP were involved in CYP3A ubiquitination, it was unclear whether the stabilized hepatic CYP3A represented a functionally active CYP3A species or a structurally inactive species already marked for cellular disposal. In the former case, such stabilization would be pharmacologically/clinically relevant but in the latter case inconsequential except for its cellular “garbage” accumulation factor and consequent contribution to ER stress induction. We therefore examined the functional relevance of the CYP3A stabilized after gp78 or CHIP knockdown in cultured rat hepatocytes “in vivo” and in hepatic microsomes isolated after such knockdown relative to corresponding controls (Fig. 8). A fluorescence-based assay for monitoring the CYP3A-dependent O-debenzylation of BFC, a relatively selective diagnostic probe for CYP3A4 function (56), was used after preliminary optimization for intact hepatocytes in monolayer cultures as well as hepatic microsomes derived from these cultures. A significant time-dependent increase of HFC, the O-debenzylated BFC metabolite, was detected in the culture medium after both gp78 and CHIP knockdown relative to the corresponding controls (Fig. 8A). No corresponding increase in HFC formation was detected in the cell lysates, thereby revealing that most of the HFC formed was exported extracellularly into the culture medium. Corresponding BFC assays with microsomes isolated from these gp78- and CHIP-knocked down hepatocytes showed a significant increase of 2.2- and 2.8-fold over corresponding control values for HFC formation at 7 days post-knockdown (Fig. 8B). These findings in both intact cells and microsomes isolated from these cells indicate that the CYP3A stabilization observed on knockdown of either of the Ub-ligases is functionally relevant.
FIGURE 8.

Functional relevance of hepatic CYP3A stabilization after gp78 or CHIP RNAi. Rat hepatocyte cultures were infected with the control shRNA, shRNA gp78 1/2, or shRNA CHIP-4 for 7 days. On the 7th day, the functional activity of CYP3A was assayed in intact hepatocytes or microsomal preparations isolated from these cells by assessing their ability to catalyze the 7-O-debenzylation of BFC, a diagnostic CYP3A functional probe, to HFC. A, the time course of HFC formation (μmol of HFC formed/3.5 × 106 cells) in the medium is assayed following treatment with β-glucuronidase + arylsulfatase to convert any in vivo conjugated HFC metabolites to the free unconjugated HFC species. The time course includes experimental values (mean ± S.D.) from three individual experiments. Statistical analyses revealed significant differences in hepatic CYP3A function between CT and shRNA gp78 1/2 or CHIP-4-infected cells at p < 0.05 and p < 0.01, respectively. Statistically significant differences in hepatic CYP3A function between shRNA gp78 1/2- and shRNA CHIP-4-infected cells were also observed at p < 0.01. B, values for HFC formation (μmol of HFC formed/mg of protein/30 min) in BFC assays catalyzed by microsomes derived from CT, shRNA gp78 1/2- or CHIP-4-infected cells are also shown. Statistical analyses revealed significant differences in hepatic CYP3A function between CT and shRNA gp78 1/2- and CHIP-4-infected cells at p < 0.01 and p < 0.01, respectively. Statistically significant differences in hepatic CYP3A function between shRNA gp78 1/2- and shRNA CHIP-4-infected cell microsomes were also observed at p < 0.01.
DISCUSSION
The E3 Ub-ligases gp78 and CHIP are known to participate in the ERAD/UPD of several heterologous cellular substrates (32–51). Although gp78 apparently also ubiquitinates itself (36, 57, 58), CHIP ubiquitinates Hsp70, an essential functional partner in these UPD-relevant CHIP-chaperone complexes (43, 45, 59). This Hsp70 ubiquitination reportedly occurs after all of the aberrant proteins generated during a heat shock/proteotoxic stress response have been duly cleared via UPD, thereby signaling the restoration of cellular normalcy (45). Our findings detailed above clearly reveal that both gp78 and CHIP individually are involved in CYP3A ERAD in cultured primary hepatocytes and thus support the findings from our in vitro reconstituted CYP3A4 ubiquitination systems (9, 10). Thus, the CYP3A proteins appear to be quite promiscuous in their choice of cellular E2-E3 complexes summoned for their turnover. Although CHIP is known to ubiquitinate CYP2E1 (49), another hepatic P450, our preliminary findings indicate that in vitro CYP2E1 is also a gp78 substrate.5
Our finding that RNAi knockdown of either E3 results in the stabilization of a functionally active CYP3A protein suggests that each Ub-ligase has an important role in the regulation of its physiological levels and, apparently, these individual roles are complementary rather than redundant. Accordingly, any genetic polymorphisms or defects that impair the function of each of the Ub-ligases in humans could significantly increase both basal and Dex-inducible hepatic CYP3A4 content and result in clinically relevant DDIs due to enhanced drug metabolism. Conversely, conditions that enhance E3 expression would also lower CYP3A content and influence clinically relevant DDIs due to impaired drug metabolism. This was indeed the case when functionally active full-length gp78 or its C-terminal domain, but not its functionally inactive RING mutant, was expressed in HepG2 cells (Fig. 3). This CYP3A decrease may be clinically relevant because the E3 gp78 gene is a prometastatic oncogene that is overexpressed in certain malignant tumors and human cancers of the lung, gastrointestinal tract, breast, liver, thymus, and skin (60–63). It apparently is not only one of the 189 most mutated genes in breast and colon cancers, but also its expression correlates well with the progression and poor prognosis for these diseases (61, 63). This prometastatic action of gp78 is mediated by its targeting of the ER transmembrane metastasis suppressor KAI1/CD82 for ERAD (62, 63). Interestingly, African American breast cancer patients show a significantly higher expression of gp78 in their tumors relative to those of their European American cohorts (64). This genetic predisposition for higher gp78 expression, if extended to surrounding normal liver tissue and coupled with an inherently low hepatic CYP3A4/CYP3A5 function, may constitute a significant risk factor for clinically relevant DDIs in the African American population.
At a molecular level, our findings of increased levels of functionally active CYP3A following the knockdown of each Ub-ligase are both intriguing and surprising. They reveal that not all of the CYP3A species targeted to ERAD are ab initio irrevocably inactivated, structurally damaged, and/or aberrant, thereby qualifying as fatally damaged proteins requiring cellular disposal. Although, in the case of irreversibly inactivated P450s, it is predictable that structural damage would mark the protein for removal by ERAD/UPD, this is much less obvious in the case of a native functional P450. Our findings thus also raise the specific issue of the critical timing during the cellular life span of a functional P450 when it is first committed to triage and the specific mechanism whereby this occurs. They suggest either of two possibilities: 1) that in addition to targeting an inactive, structurally damaged P450 species, each E3 Ub-ligase also acts directly in the recognition and targeting to ERAD of a native, structurally and functionally intact P450 species, as recently proposed for other “wild type” proteins (20); or 2) that when its ERAD process is disrupted, the native/functional CYP3A species accumulates in the ER essentially because its forward progression to the particular cellular intermediate that is specifically recognized by either the gp78 or CHIP E3 Ub-ligase is blocked. Our preliminary findings indicate that this intermediate species may be a multisite phosphorylated P450 protein (9).5
Additional mechanistic possibilities also exist. First, it is plausible that with the knockdown of either gp78 (which actually contains a p97/VCP-interacting motif (VIM) in its C-terminal domain (36–38)) or CHIP,6 p97 recruitment to the ER is impaired, and consequently CYP3A extraction out of the ER is reduced leading to its ER accumulation. Our findings reveal that indeed the ER extraction of parent and ubiquitinated CYP3A species into the cytosol is markedly reduced after gp78 knockdown and even more so after CHIP knockdown (Fig. 7). Although impaired CYP3A ubiquitination is most likely responsible, it remains to be determined whether impaired p97 recruitment also contributes to some extent to CYP3A stabilization.
Second, that an “abnormal” CYP3A4 species is a substrate for both E3-ligases and refolding chaperones. If the E3-ligases and chaperones were to compete for abnormal CYP3A4, then down-regulation of the E3-ligases would be expected to increase the levels of normal CYP3A4 through refolding (as discussed below). Other conceivable indirect mechanisms by which E3 knockdown could increase functionally active CYP3A4 levels include elevation of signaling proteins or chaperone levels that might have a “protective” effect on CYP3A4. However, our parallel immunoblotting analyses of four relevant hepatic cytosolic and ER chaperones, Hsp70, Hsp90, Grp78, and Grp94, following gp78 or CHIP knockdown argue against such a cellular chaperone elevation (supplemental Fig. S1).
CHIP-Hsp70 chaperone complexes are involved in key cellular protein triage decisions (40). As such, CHIP is known to attenuate both Hsp70 ATPase activity and client substrate affinity, thereby reducing its protein refolding capabilities (40). Given that Hsp70-mediated corralling of the P450 substrate would be required for its recognition by CHIP, the intriguing possibility exists that CHIP knockdown may actually promote the refolding of the Hsp70-bound CYP3A species into a functionally active protein by slowing its degradation and thus diverting it away from UPD. Consistent with this possibility, we find it noteworthy that: 1) despite the otherwise comparable extent (≈2.5-fold) of immunochemically detectable CYP3A stabilization, the level of functionally active CYP3A stabilized after CHIP knockdown is significantly greater than that detected after gp78 knockdown (Fig. 8); and 2) the stabilization of parent [35S]-CYP3A species is relatively greater while HMM-ubiquitinated [35S]-CYP3A species are minimally detected in the hepatic ER after CHIP knockdown (Fig. 7). Furthermore, after gp78 knockdown, the relative stabilization of parent [35S]-CYP3A species was less, whereas the level of HMM-ubiquitinated [35S]-CYP3A species was relatively higher in the hepatic ER than the corresponding values observed after CHIP knockdown (Fig. 7).
Although each of the two E3 Ub-ligases can directly ubiquitinate CYP3A4 in vitro (10) and apparently operate concurrently in vivo, it is presently unclear whether gp78 and CHIP function independently or cooperatively in CYP3A ERAD/UPD. Indeed, gp78 has been proposed to function as an E4 Ub-ligase (38, 65). As such it could function after the protein is first modified by CHIP and thus targeted for ERAD/UPD. This possibility may account for the lower extent of functionally active CYP3A stabilization observed after gp78 knockdown, if CYP3A were indeed to be initially ubiquitinated by CHIP. The markedly reduced ER-anchored HMM-ubiquitinated [35S]-CYP3A species after hepatic CHIP knockdown, despite a functional gp78 (Fig. 7), strengthens this notion. Our studies attempting to target both Ub-ligases for simultaneous knockdown not surprisingly resulted in cytotoxicity and cell death, consistent with their critical role in the ERAD/UPD of important cellular proteins and protein quality control.
We find it interesting that CHIP−/− mice exhibit premature aging and various pathologies associated with disrupted protein quality control (66). Among these is the widespread oxidative damage as monitored by tissue 8-isoprostane levels, excellent indices of intracellular lipid peroxidation. The marked increase in hepatic 8-isoprostane levels in 3-month-old CHIP−/− mice relative to those of age-matched wild type controls suggests early oxidative damage to the livers of these young mice that within 12 months not only spreads to additional tissues but also results in the decline of the hepatic proteasomal function (66–68). Given our findings that CHIP knockdown apparently increases the levels of functionally active P450s, it is tempting to speculate that these stabilized hepatic P450s may partly contribute to such oxidative damage. The CYPs 3A and CYP2E1 are particularly notorious for undergoing futile oxidative cycling and generating H2O2 and other reactive oxygen species (ROS) in the absence of relevant substrates (69–75). Such oxidatively uncoupled P450-dependent metabolism and mitochondrial oxidative activity are proposed to contribute equivalently to the intracellularly generated ROS burden (70). It is not surprising then that in the absence of an effective ERAD/UPD to remove oxidized proteins, cellular proteins including the 26S proteasome, damaged by intracellularly generated ROS, would accumulate. These findings thus suggest that in the face of a compromised hepatic CHIP or gp78 function, the accumulation of functionally active CYPs 3A and CYP2E1, particularly over an extended period (i.e. months), may also be pathophysiologically relevant.
In summary, our findings described above reveal that both gp78 and CHIP Ub-ligases effectively participate in CYP3A ERAD/UPD in vivo, and thus can also regulate hepatic CYP3A content. Abrogation of gp78 and CHIP Ub-ligase function through RNAi results in the stabilization of a functionally active CYP3A species that may be pharmacologically and clinically relevant, given the vast array of clinically prescribed drugs whose metabolism and disposition is dependent on these P450 enzymes. The well recognized proclivity of the CYP3A subfamily to generate ROS through futile catalytic cycling in the absence of relevant substrates, suggests that such a stabilization of functionally active CYP3A enzymes may also be pathophysiologically relevant.
Supplementary Material
Acknowledgments
We are grateful to Chris Her, University of California San Francisco (UCSF) Liver Center Cell and Tissue Biology Core Facility (Dr. J. J. Maher, Director) for hepatocyte isolation. We gratefully acknowledge the US Congress for the NIGMS-ARRA funding, without which this work would not have been possible. We thank Drs. A. M. Weissman (National Institutes of Health, NCI, Frederick, MD), Cam Patterson (University of North Carolina, Chapel Hill), and R. A. DeBose-Boyd (University of Texas Southwestern Medical Center, Dallas) for generously providing some of the plasmids used in these studies. We also thank Dr. Alex Y. So (Keith Yamamoto laboratory, UCSF) for generous advice and training in the use of lentiviral shRNAi technology, Dr. Michelle Flanniken (Raul Andino laboratory, UCSF) for technical advice, and Dr. Allan M. Weissman for valuable discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants GM44037 and DK26506 and the UCSF Liver Center Core on Cell and Tissue Biology funded by the National Institute of Digestive Diseases and Kidney Center Grant P30DK26743. These studies were presented orally at the 2010 International Symposium on Microsomes and Drug Oxidations, Beijing, China (May 18, 2010) and in poster form at the 2010 FASEB Summer Research Conference on Ubiquitin and Cellular Regulation, Saxtons River, VT (June 16, 2010).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
By “native” we refer to the CYP3A/P450 enzyme that has not been structurally and/or functionally inactivated by an exogenous mechanism-based inactivator. Regrettably, the ubiquitination of truly “native state” CYP3A enzymes by UBC7/gp78 or UbcH5a/CHIP complexes cannot be directly evaluated because of the relatively prolonged 90-min incubation periods at 30 °C generally required to detect protein ubiquitination. During such a prolonged incubation, CYP3A proteins unfold and thus can no longer be considered unequivocally native.
We find it interesting that in HepG2 cells, the expression of the soluble C-terminal domain of gp78 (gp78C) rather than the full-length ER membrane-anchored gp78 was more efficient in supporting CYP3A4 degradation. This is consistent with our findings (76) that in the presence of a functional p97 AAA-ATPase complex, the chaperone complex involved in the extraction of ER proteins, a considerable fraction of the parent 55-kDa CYP3A species is found in the cytosol.
Y.-Q. Wang, S. Guan, D. R. Koop, P. Acharya, M. Liao, A. L. Burlingame, and M. A. Correia, manuscript submitted for publication.
- CYP
- cytochrome P450
- P450s
- cytochrome P450 hemoproteins
- BFC
- 7-benzyloxy-4-trifluoromethylcoumarin
- CHIP
- C terminus of Hsp70-interacting protein
- CT
- control
- DDEP
- 3,5-dicarbethoxy-2,6-dimethyl-4-ethyl-1,4-dihydropyridine
- Dex
- dexamethasone
- DDI
- drug-drug interaction
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- HFC
- 7-hydroxy-4-trifluoromethylcoumarin
- HMM
- high molecular mass
- nt
- nucleotide(s)
- qRT
- quantitative real-time
- PCR
- polymerase chain reaction
- ROS
- reactive oxygen species
- Ub
- ubiquitin
- UBC
- genes for Ub-conjugating enzymes
- UPD
- Ub-dependent proteasomal degradation.
REFERENCES
- 1.Guengerich F. P. (2005) in Cytochrome P450: Structure, Mechanism and Biochemistry (Ortiz de Montellano P. ed) pp. 377–530, Kluwer-Academic/Plenum Press, New York [Google Scholar]
- 2.Correia M. A., Decker C., Sugiyama K., Caldera P., Bornheim L., Wrighton S. A., Rettie A. E., Trager W. F. (1987) Arch. Biochem. Biophys. 258, 436–451 [DOI] [PubMed] [Google Scholar]
- 3.Correia M. A., Davoll S. H., Wrighton S. A., Thomas P. E. (1992) Arch. Biochem. Biophys. 297, 228–238 [DOI] [PubMed] [Google Scholar]
- 4.Faouzi S., Medzihradszky K. F., Hefner C., Maher J. J., Correia M. A. (2007) Biochemistry 46, 7793–7803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang H. F., Figueiredo Pereira M. E., Correia M. A. (1999) Arch. Biochem. Biophys. 365, 45–53 [DOI] [PubMed] [Google Scholar]
- 6.Korsmeyer K. K., Davoll S., Figueiredo-Pereira M. E., Correia M. A. (1999) Arch. Biochem. Biophys. 365, 31–44 [DOI] [PubMed] [Google Scholar]
- 7.He K., Bornheim L. M., Falick A. M., Maltby D., Yin H., Correia M. A. (1998) Biochemistry 37, 17448–17457 [DOI] [PubMed] [Google Scholar]
- 8.Wang X., Medzihradszky K. F., Maltby D., Correia M. A. (2001) Biochemistry 40, 11318–11326 [DOI] [PubMed] [Google Scholar]
- 9.Wang Y., Liao M., Hoe N., Acharya P., Deng C., Krutchinsky A. N., Correia M. A. (2009) J. Biol. Chem. 284, 5671–5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pabarcus M. K., Hoe N., Sadeghi S., Patterson C., Wiertz E., Correia M. A. (2009) Arch. Biochem. Biophys. 483, 66–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Correia M. A. (2003) Drug Metab. Rev. 35, 107–143 [DOI] [PubMed] [Google Scholar]
- 12.Correia M. A., Sadeghi S., Mundo-Paredes E. (2005) Annu. Rev. Pharmacol. Toxicol. 45, 439–464 [DOI] [PubMed] [Google Scholar]
- 13.Correia M. A., Liao M. (2007) Expert Opin. Drug Metab. Toxicol. 3, 33–49 [DOI] [PubMed] [Google Scholar]
- 14.Murray B. P., Correia M. A. (2001) Arch. Biochem. Biophys. 393, 106–116 [DOI] [PubMed] [Google Scholar]
- 15.Murray B. P., Zgoda V. G., Correia M. A. (2002) Mol. Pharmacol. 61, 1146–1153 [DOI] [PubMed] [Google Scholar]
- 16.Liao M., Zgoda V. G., Murray B. P., Correia M. A. (2005) Mol. Pharmacol. 67, 1460–1469 [DOI] [PubMed] [Google Scholar]
- 17.Liao M., Faouzi S., Karyakin A., Correia M. A. (2006) Mol. Pharmacol. 69, 1897–1904 [DOI] [PubMed] [Google Scholar]
- 18.Roberts B. J. (1997) J. Biol. Chem. 272, 9771–9778 [DOI] [PubMed] [Google Scholar]
- 19.Raasi S., Wolf D. H. (2007) Semin. Cell Dev. Biol. 18, 780–791 [DOI] [PubMed] [Google Scholar]
- 20.Vembar S. S., Brodsky J. L. (2008) Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakatsukasa K., Huyer G., Michaelis S., Brodsky J. L. (2008) Cell 132, 101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hampton R. Y., Garza R. M. (2009) Chem. Rev. 109, 1561–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bays N. W., Gardner R. G., Seelig L. P., Joazeiro C. A., Hampton R. Y. (2001) Nat. Cell Biol. 3, 24–29 [DOI] [PubMed] [Google Scholar]
- 24.Bordallo J., Plemper R. K., Finger A., Wolf D. H. (1998) Mol. Biol. Cell 9, 209–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swanson R., Locher M., Hochstrasser M. (2001) Genes Dev. 15, 2660–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kreft S. G., Wang L., Hochstrasser M. (2006) J. Biol. Chem. 281, 4646–4653 [DOI] [PubMed] [Google Scholar]
- 27.Haynes C. M., Caldwell S., Cooper A. A. (2002) J. Cell Biol. 158, 91–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kikkert M., Doolman R., Dai M., Avner R., Hassink G., van Voorden S., Thanedar S., Roitelman J., Chau V., Wiertz E. (2004) J. Biol. Chem. 279, 3525–3534 [DOI] [PubMed] [Google Scholar]
- 29.Doolman R., Leichner G. S., Avner R., Roitelman J. (2004) J. Biol. Chem. 279, 38184–38193 [DOI] [PubMed] [Google Scholar]
- 30.Hassink G., Kikkert M., van Voorden S., Lee S. J., Spaapen R., van Laar T., Coleman C. S., Bartee E., Fruh K., Chau V., Wiertz E. (2005) Biochem. J. 388, 647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flierman D., Coleman C. S., Pickart C. M., Rapoport T. A., Chau V. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 11589–11594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tiwari S., Weissman A. M. (2001) J. Biol. Chem. 276, 16193–16200 [DOI] [PubMed] [Google Scholar]
- 33.Fang S., Weissman A. M. (2004) Cell. Mol. Life Sci. 61, 1546–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song B. L., Sever N., DeBose-Boyd R. A. (2005) Mol. Cell 19, 829–840 [DOI] [PubMed] [Google Scholar]
- 35.Fang S., Ferrone M., Yang C., Jensen J. P., Tiwari S., Weissman A. M. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen B., Mariano J., Tsai Y. C., Chan A. H., Cohen M., Weissman A. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong X., Shen Y., Ballar P., Apostolou A., Agami R., Fang S. (2004) J. Biol. Chem. 279, 45676–45684 [DOI] [PubMed] [Google Scholar]
- 38.Kostova Z., Tsai Y. C., Weissman A. M. (2007) Semin. Cell Dev. Biol. 18, 770–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li W., Tu D., Brunger A. T., Ye Y. (2007) Nature 446, 333–337 [DOI] [PubMed] [Google Scholar]
- 40.Ballinger C. A., Connell P., Wu Y., Hu Z., Thompson L. J., Yin L. Y., Patterson C. (1999) Mol. Cell. Biol. 19, 4535–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murata S., Minami Y., Minami M., Chiba T., Tanaka K. (2001) EMBO Rep. 2, 1133–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Connell P., Ballinger C. A., Jiang J., Wu Y., Thompson L. J., Hohfeld J., Patterson C. (2001) Nat. Cell Biol. 3, 93–96 [DOI] [PubMed] [Google Scholar]
- 43.Jiang J., Ballinger C. A., Wu Y., Dai Q., Cyr D. M., Hohfeld J., Patterson C. (2001) J. Biol. Chem. 276, 42938–42944 [DOI] [PubMed] [Google Scholar]
- 44.McDonough H., Patterson C. (2003) Cell Stress Chaperones 8, 303–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qian S. B., McDonough H., Boellmann F., Cyr D. M., Patterson C. (2006) Nature 440, 551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang J., Cyr D., Babbitt R. W., Sessa W. C., Patterson C. (2003) J. Biol. Chem. 278, 49332–49341 [DOI] [PubMed] [Google Scholar]
- 47.Younger J. M., Chen L., Ren H. Y., Rosser M. F., Turnbull E. L., Fan C. Y., Patterson C., Cyr D. M. (2006) Cell 126, 571–582 [DOI] [PubMed] [Google Scholar]
- 48.Peng H. M., Morishima Y., Jenkins G. J., Dunbar A. Y., Lau M., Patterson C., Pratt W. B., Osawa Y. (2004) J. Biol. Chem. 279, 52970–52977 [DOI] [PubMed] [Google Scholar]
- 49.Morishima Y., Peng H. M., Lin H. L., Hollenberg P. F., Sunahara R. K., Osawa Y., Pratt W. B. (2005) Biochemistry 44, 16333–16340 [DOI] [PubMed] [Google Scholar]
- 50.Pratt W. B., Morishima Y., Peng H. M., Osawa Y. (2010) Exp. Biol. Med. (Maywood) 235, 278–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peng H. M., Morishima Y., Clapp K. M., Lau M., Pratt W. B., Osawa Y. (2009) Biochemistry 48, 8483–8490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Acharya P., Engel J. C., Correia M. A. (2009) Mol. Pharmacol. 76, 503–515 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Acharya P., Chen J. J., Correia M. A. (2010) Mol. Pharmacol. 77, 575–592 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Han X. M., Lee G., Hefner C., Maher J. J., Correia M. A. (2005) J. Pharmacol. Exp. Ther. 314, 128–138 [DOI] [PubMed] [Google Scholar]
- 55.LeCluyse E., Bullock P., Madan A., Carroll K., Parkinson A. (1999) Drug Metab. Dispos. 27, 909–915 [PubMed] [Google Scholar]
- 56.Donato M. T., Jimenez N., Castell J. V., Gomez-Lechon M. J. (2004) Drug Metab. Dispos. 32, 699–706 [DOI] [PubMed] [Google Scholar]
- 57.Das R., Mariano J., Tsai Y. C., Kalathur R. C., Kostova Z., Li J., Tarasov S. G., McFeeters R. L., Altieri A. S., Ji X., Byrd R. A., Weissman A. M. (2009) Mol. Cell 34, 674–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shmueli A., Tsai Y. C., Yang M., Braun M. A., Weissman A. M. (2009) Biochem. Biophys. Res. Commun. 390, 758–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meacham G. C., Patterson C., Zhang W., Younger J. M., Cyr D. M. (2001) Nat. Cell Biol. 3, 100–105 [DOI] [PubMed] [Google Scholar]
- 60.Chiu C. G., St-Pierre P., Nabi I. R., Wiseman S. M. (2008) Expert Rev. Anticancer Ther. 8, 207–217 [DOI] [PubMed] [Google Scholar]
- 61.Sjoblom T., Jones S., Wood L. D., Parsons D. W., Lin J., Barber T. D., Mandelker D., Leary R. J., Ptak J., Silliman N., Szabo S., Buckhaults P., Farrell C., Meeh P., Markowitz S. D., Willis J., Dawson D., Willson J. K., Gazdar A. F., Hartigan J., Wu L., Liu C., Parmigiani G., Park B. H., Bachman K. E., Papadopoulos N., Vogelstein B., Kinzler K. W., Velculescu V. E. (2006) Science 314, 268–274 [DOI] [PubMed] [Google Scholar]
- 62.Tsai Y. C., Mendoza A., Mariano J. M., Zhou M., Kostova Z., Chen B., Veenstra T., Hewitt S. M., Helman L. J., Khanna C., Weissman A. M. (2007) Nat. Med. 13, 1504–1509 [DOI] [PubMed] [Google Scholar]
- 63.Joshi B., Li L., Nabi I. R. (2010) J. Biol. Chem. 285, 8830–8839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin D. N., Boersma B. J., Yi M., Reimers M., Howe T. M., Yfantis H. G., Tsai Y. C., Williams E. H., Lee D. H., Stephens R. M., Weissman A. M., Ambs S. (2009) PLoS One 4, e4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morito D., Hirao K., Oda Y., Hosokawa N., Tokunaga F., Cyr D. M., Tanaka K., Iwai K., Nagata A. K. (2008) Mol. Biol. Cell 19, 1328–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Min J. N., Whaley R. A., Sharpless N. E., Lockyer P., Portbury A. L., Patterson C. (2008) Mol. Cell. Biol. 28, 4018–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang C., Xu Z., He X. R., Michael L. H., Patterson C. (2005) Am. J. Physiol. Heart Circ. Physiol. 288, H2836–H2842 [DOI] [PubMed] [Google Scholar]
- 68.Dai Q., Zhang C., Wu Y., McDonough H., Whaley R. A., Godfrey V., Li H. H., Madamanchi N., Xu W., Neckers L., Cyr D., Patterson C. (2003) EMBO J. 22, 5446–5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guengerich F. P., Johnson W. W. (1997) Biochemistry 36, 14741–14750 [DOI] [PubMed] [Google Scholar]
- 70.Zangar R. C., Davydov D. R., Verma S. (2004) Toxicol. Appl. Pharmacol. 199, 316–331 [DOI] [PubMed] [Google Scholar]
- 71.Denisov I. G., Grinkova Y. V., McLean M. A., Sligar S. G. (2007) J. Biol. Chem. 282, 26865–26873 [DOI] [PubMed] [Google Scholar]
- 72.Gorsky L. D., Koop D. R., Coon M. J. (1984) J. Biol. Chem. 259, 6812–6817 [PubMed] [Google Scholar]
- 73.Ekstrom G., Ingelman-Sundberg M. (1989) Biochem. Pharmacol. 38, 1313–1319 [DOI] [PubMed] [Google Scholar]
- 74.Zhukov A., Ingelman-Sundberg M. (1999) Biochem. J. 340, 453–458 [PMC free article] [PubMed] [Google Scholar]
- 75.Goasduff T., Cederbaum A. I. (1999) Arch. Biochem. Biophys. 370, 258–270 [DOI] [PubMed] [Google Scholar]
- 76.Acharya P. A., Liao M., Correia M. A. (2009) Drug Metab. Rev. 41, 53 [Google Scholar]
- 77.Janiesch P. C., Kim J., Mouysset J., Barikbin R., Lochmuller H., Cassata G., Krause S., Hoppe T. (2007) Nat. Cell Biol. 9, 379–390 [DOI] [PubMed] [Google Scholar]
- 78.Custer S. K., Neumann M., Lu H., Wright A. C., Taylor J. P. (2010) Hum. Mol. Genet. 19, 1741–1755 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.