

Abstract
Binding of lymphocyte function-associated antigen-1 (LFA-1) to intercellular adhesion molecule-1 (ICAM-1) mediates leukocyte adhesion under force. Using a biomembrane force probe capable of measuring single bond interactions, we showed ICAM-1 binding to LFA-1 at different conformations, including the bent conformation with the lowest affinity. We quantify how force and conformations of LFA-1 regulate its kinetics with ICAM-1. At zero-force, on-rates were substantially changed by conditions that differentially favor a bent or extended LFA-1 with a closed or open headpiece; but off-rates were identical. With increasing force, LFA-1/ICAM-1 bond lifetimes (reciprocal off-rates) first increased (catch bonds) and then decreased (slip bonds). Three states with distinct off-rates were identified from lifetime distributions. Force shifted the associated fractions from the short- to intermediate- and long-lived states, producing catch bonds at low forces, but increased their off-rates exponentially, converting catch to slip bonds at high forces. An internal ligand antagonist that blocks pulling of the α7-helix suppressed the intermediate-/long-lived states and eliminated catch bonds, revealing an internal catch bond between the αA and βA domains. These results elucidate an allosteric mechanism for the mechanochemistry of LFA-1/ICAM-1 binding.
Keywords: Cell Adhesion, Integrin, Kinetics, Protein Conformation, Single Molecule Biophysics, Affinity Regulation, Allostery, Catch Bond, Conformational Change, Mechanochemistry
Introduction
Integrins are membrane molecules broadly expressed on a wide variety of cells as αβ heterodimers that bind ligands on another cell or the extracellular matrix (1, 2). Integrin/ligand interactions are thought to be capable of not only transmitting forces but also transducing signals bi-directionally across the cell membrane, thereby playing a key role in mechanosensing and mechanotransduction (3, 4).
Integrins can assume distinct conformations with different ligand binding affinities (1, 2). Several types of conformational changes have been described based on structural (5–8) and functional (9–14) studies (see below Fig. 1B for various domains and Fig. 5 for different conformations): coalescent to separated legs (15), bent to extended ectodomains (7, 8), closed-in to swing-out hybrid domain (6–8), and closed to intermediate and open αA (or αI) and βA (or βI) domains (5). A bell rope model has been suggested to relate different ligand binding affinities to distinct conformations of the αA domain, such that pulling down the α7-helix at the bottom opens the αA domain by rearranging the metal ion-dependent adhesion site (MIDAS)2 on the top to change from the low to intermediate and high affinity states (5). The downward movement of the αA domain α7-helix may result from binding of an intrinsic ligand on the α7-helix to the βA domain MIDAS, thereby connecting the conformational changes of the αA domain to those of the βA and other downstream domains of the integrin. Recently published crystal structures of αxβ2 integrin ectodomains reveal unexpected flexible αA domain (16). It is not clear how this flexibility affects binding of external ligand on the MIDAS and the intrinsic ligand on the α7-helix of the αA domain.
FIGURE 1.

BFP experiment. A, BFP photomicrograph. A micropipette-aspirated RBC (left, with a bead attached to the apex to serve as a force probe with 1-pN resolution) was aligned with a Jurkat cell aspirated by another pipette (right), which was driven to contact the bead. The left pipette was held stationary but the position of the bead was tracked to produce the data in C. B, BFP functionalization. LFA-1 was expressed on the Jurkat cell. Either ICAM-1-Fc or anti-LFA-1 mAb (KIM127 or TS1/22) was coated on a streptavidin-decorated bead; their binding sites on LFA-1 are indicated. The XVA143 binding site at the interface between the αA and βA domains is also indicated. C, superposition of force-traces of two contact cycles without and with adhesion. The Jurkat cell was driven to approach the bead (blue), contact for 0.1 s (green), retract (gray), and hold. Jurkat cell retraction resulted in a tensile force in the red trace, indicating adhesion, but not in the purple trace, indicating no adhesion. The Jurkat cell stopped when the force reached a preset value. The lifetime at that force was measured from that point on until bond rupture, signified by a force drop to zero. See also supplemental video S1. D, binding specificity. Adhesion frequencies between LFA-1-expressing Jurkat cells and beads bearing indicated densities of ICAM-1 in 1 mm Ca2+ plus 1 mm of Mg2+ (Ca2+/Mg2+), and conditions known to alter LFA-1 conformations and ligand binding affinities (9, 10) (open bars): Ca2+/Mg2+ plus 10 nm CXCL12 (Ca2+/Mg2+/CXCL12), 2 mm Mg2+ plus 2 mm of EGTA (Mg2+/EGTA), 1 mm Mn2+ (Mn2+), and any of these conditions plus 1 μm XVA143 (closed bars), or in Ca2+/Mg2+ plus a mAb against ICAM-1 or integrin subunit αL, β2, or αM. To ensure bond lifetimes were measured at the single-bond level, different ICAM-1 densities on the beads were selected for different conditions to achieve 15–20% adhesion frequencies: 578 μm−2 for Ca2+/Mg2+, 15 μm−2 for Mg2+/EGTA or Mn2+, or 1078 μm−2 in any cation condition in the presence of 1 μm XVA143, as indicated on the bars. Data are presented as mean ± S.E. of 3–5 pairs of cells and beads repeatedly contacting 50 times with 0.1 s each contact. p values from Welch's t test are indicated. E, plot of horizontal position x of the right edge of the bead versus time t. F, plot of sliding standard deviation σ of 70 consecutive points of the position data versus time t. Horizontal lines represent thresholds to identify bond association (solid line) and dissociation (dashed line) events. Two representative lifetime periods are indicated. See also supplemental video S2.
FIGURE 5.

Model for LFA-1 activation. A, inactive, bent LFA-1 with a closed headpiece. The hybrid domain is closed. The αA domain is in the inactive conformation with the α7-helix in the up position and the MIDAS in the closed conformation. B and C, priming by inside-out signaling or metal ions extends LFA-1 with legs that may or may not separate, a hybrid domain that may or may not swing out, and the ligation between the αA and βA domain may or may not be stable. The αA domain flexibility is depicted by the dotted light yellow ellipsoids moving about the solid yellow αA domain in B, which disappears in C when the ligation between the αA and βA domains is enhanced by the hybrid domain swing-out. The on-rate for ICAM-1 is increased by different amounts depending on this ligation. Without force, however, the αA domain remains in a conformation that yields the same off-rate for ICAM-1 dissociation. D, force applied by ICAM-1 is transmitted from the αA domain MIDAS to the α7-helix (indicated by a curved arrow inside the αA domain) to elicit catch bonds between the intrinsic ligand and the βA domain, which pulls the α7-helix down (indicated) to induce the intermediate- and long-lived states, resulting in LFA-1/ICAM-1 catch bonds. E, regardless of priming, binding of XVA143 blocks the αA/βA ligation, makes the αA domain flexible, and pushes the βA domain α7-helix further downward, inducing conformational changes that propagate further downstream, including hybrid domain swing-out and integrin extension. On-rate for ICAM-1 is reduced but zero-force off-rate is the same. F, blocking the internal ligation shifts a portion of the force originally transmitted via the αA/βA ligation to other contacts between the αA and β-propeller domains, which reduces the force borne by the α7-helix (indicated by a smaller arrow). A much larger external force applied by ICAM-1 is needed to generate a similar internal force to pull down the αA domain α7-helix to induce the intermediate- and long-lived states; but the mechanism of forcing acceleration of dissociation dominates at such high forces, converting the LFA-1/ICAM-1 catch-slip bonds to that of slip-only.
Integrin/ligand bonds are often subjected to forces externally applied to the cell, e.g. during leukocyte adhesion to vascular surfaces, or internally generated by the cell, e.g. during migration. Mechanical forces have been suggested to regulate integrin binding affinity by inducing conformational changes. For example, applying a shear flow to cells has been shown to enhance integrin/ligand binding (12, 17, 18). Atomic force microscopy single-bond experiments have demonstrated that α5β1, an αA domain-lacking integrin, forms catch bonds with fibronectin (FN) in which force prolongs bond lifetimes in the 10–30 pN range (19). Steered molecular dynamics simulations have suggested how force might activate integrin αA domains (20) and the headpiece of integrin αVβ3 (21–24). However, many mechanistic details about the integrin mechanochemistry are still missing.
Using force clamp (25) and thermal fluctuation (26) experiments to measure single bond interactions by a biomembrane force probe (BFP), here we show that lymphocyte function-associated antigen-1 (LFA-1), an αA domain-containing integrin αLβ2, forms catch-slip bonds with intercellular adhesion molecule-1 (ICAM-1) in three cation conditions and in the presence of a chemokine that triggers inside-out signaling, which favor different LFA-1 conformations. Such a force dependence can be explained by two competing mechanisms: 1) at low forces, the dominating mechanism is forcing switch of LFA-1 from short-lived to intermediate- and long-lived states, which generates catch bonds; 2) at high forces, the dominating mechanism is forcing acceleration of dissociation, which results in slip bonds. An internal ligand antagonist molecule XVA143 (27, 28) eliminates the catch bonds by suppressing the former mechanism. These results extend our recent demonstration of α5β1/FN catch bond by elucidating an allosteric regulatory mechanism of LFA-1/ICAM-1 dissociation by force that generates catch bonds.
EXPERIMENTAL PROCEDURES
Cells, Proteins, and Chemicals
Jurkat cells purchased from ATCC (Manassas, VA) were cultured in RPMI 1640 medium with 10% fetal calf serum plus l-glutamine (4 mm) and penicillin/streptomycin (0.1 mg/ml). Human polymorphonuclear leukocytes (PMNs) were isolated from blood using a described protocol (29) approved by the Georgia Institute of Technology Institutional Review Board. Human ICAM-1-Fc chimera, soluble ICAM-1, and CXCL12 were from R&D Systems (Minneapolis, MN). Anti-ICAM-1 blocking mAb (RR1/1) was from Bender Medsystems (San Diego, CA). Anti-αL (TS1/22) and -β2 (TS1/18) blocking mAbs were from Thermo Scientific (Williston, VT). Anti-LFA-1 extension reporter mAb KIM127 was a generous gift of M. Robinson (Celltech, Slough, UK). PE-conjugated anti-LFA-1 closed αA domain MIDAS reporter (HI 111) and open hybrid domain reporter (MEM-148) mAbs were from Santa Cruz Biotechnology (Santa Cruz, CA). PE-conjugated anti-Mac-1 blocking mAb (2LPM19c) was from Dako. PE-conjugated anti-CXCR4 mAb was from eBioscience (San Diego, CA). PE-conjugated goat-anti-mouse secondary antibody F(ab′)2 fragment was from Abcam (Cambridge, MA). The small antagonist molecule XVA143 was kindly provided by P. Gillespie (Roche, NJ).
RBC and Glass Bead Preparations
RBCs were biotinylated as described previously (26). Briefly, fresh human RBCs were isolated from a blood drop via finger prick using a protocol approved by the Georgia Institute of Technology Institutional Review Board. RBCs were resuspended in carbonate/bicarbonate buffer (pH 8.4), and incubated with biotin-PEG3500-SGA (JenKem USA, Allen, TX) for 30 min at room temperature (25). Borosilicate glass beads (Duke Scientific, Palo Alto, CA) were cleaned with a mixture of ammonium hydroxide, hydrogen peroxide and water at boiling temperature. Beads were covalently coupled with mercapto-propyl-trimethoxy silane (United Chemical Technologies, Bristol, PA), and then covalently linked with a mixture of streptavidin-maleimide and maleimide-PEG3500-SGA (JenKem USA) that had been previously covalently coupled with ICAM-1-Fc chimeras or anti-LFA-1 mAbs through its SGA group.
BFP Measurements of Adhesion Frequency and Lifetime under Constant Force
Our BFP apparatus has been described (25, 26). Briefly, an ultra-sensitive force probe was assembled by attaching a streptavidinylated bead bearing biotinylated ICAM-1 or anti-LFA-1 mAb to the apex of a biotinylated RBC that was aspirated by a micropipette (Fig. 1, A and B, left). The aspiration pressure was set by a manometer with micrometer precision. A Jurkat cell aspirated by another micropipette was driven by a piezoelectric translator with sub-nanometer precision via a capacitive sensor feedback control (Fig. 1A, right). The bead and the cell were aligned and observed under an inverted microscope through two cameras. One had a standard video rate of 30 frames per second to provide real-time images (Fig. 1A and supplemental videos S1 and S2). The other had a high speed (1,600 frames per second) when the images were limited to a 24-line strip across the bead, which allowed a custom image analysis software to track bead position with a spatial precision of 5 nm (30). The BFP spring constant was determined from the suction pressure and the radii of the pipette, the spherical portion of the RBC, and the contact area between the probe and the RBC (30, 31). The spring constant was set at 0.3 pN/nm except for the thermal fluctuation experiments, which was <0.1 pN/nm.
Jurkat cells were washed once using Hanks balanced salt solution without Ca2+ and Mg2+ (HBSS-, 150 mOsm, pH 7.4) with 5 mm EDTA to remove any divalent cations bound to LFA-1 during cell culture and washed again using HBSS- to remove EDTA. Depending on the experiments, cells were incubated in one of the four binding buffers (HBSS- containing 0.5% human serum albumin and 1 mm Ca2+ and 1 mm Mg2+ (Ca2+/Mg2+), 1 mm Ca2+ and 1 mm Mg2+ plus 10 nm chemokine (CXC motif) ligand 12 (Ca2+/Mg2+/CXCL12), 2 mm Mg2+ plus 2 mm EGTA (Mg2+/EGTA), or 1 mm Mn2+ (Mn2+)) with or without 1 μm XVA143 or 100 μg/ml soluble ICAM-1 at room temperature for 10 min before adding to the sample chamber that contained the same solution.
In the force-clamp assay (25), the Jurkat cell was driven repeatedly to approach and contact the bead with a 20-pN compressive force for 0.1 s to allow bond formation, retracted at a 3-μm/s speed for adhesion detection, held at a desired force to wait for bond dissociation, and returned to the original position to wait for the next cycle (Fig. 1C and supplemental video S1). Adhesion was detected by a tensile force signal caused by cell retraction. Lifetime was measured from the instant when the force reached the desired level to the instant of bond dissociation. To avoid force drift over long time, a 10-s cutoff was set to rupture bonds with lifetimes >10 s to start the next cycle.
Adhesion frequency (Pa) was determined by dividing the number of resulting adhesions by the total number of contacts repeatedly made at a given duration. Two contact times were used: 1) 0.1 s for specificity controls (Fig. 1D) as this time was used for measuring lifetimes and 2) 2 s for measuring two-dimensional binding affinities (Ka) as this time is sufficiently long for binding kinetics to achieve equilibrium. Ka is lumped with the contact area Ac (a few percents of 3 μm2) and calculated using a published model (32) in Equation 1,
where mr and ml are the respective densities of LFA-1 and ICAM-1 measured by flow cytometry. The effective on-rate is calculated from Ackon = AcKa × koff where the off-rate is from the negative slope of the corresponding zero-force lifetime distribution (Fig. 2E).
FIGURE 2.

Lifetime and adhesion frequency measurements. A and B, plots of short (<10 s) lifetime average (± S.E.) (A) and long (>10 s) lifetime fraction (B) versus force of LFA-1/ICAM-1 bonds in Ca2+/Mg2+ (□), Ca2+/Mg2+/CXCL12 (♢), Mg2+/EGTA ( ), or Mn2+ (○) or of nonspecific adhesions with 10 μg/ml anti-β2 blocking mAb TS1/18 in Ca2+/Mg2+ (▴). Lifetimes at zero- and at peak-forces are significantly different (p < 0.01, Welch's t test). C, plot of short (<10 s) lifetime average (± S.E.) and long (>10 s) lifetime fraction (inset) versus force of bonds of LFA-1 with TS1/22 in Ca2+/Mg2+ (▿) or with KIM127 in Mn2+ plus 100 μg/ml soluble ICAM-1 (to expose the KIM127 binding site on LFA-1) (○). D, adhesion frequencies at 0.1-s contact of LFA-1 expressing Jurkat cells to beads bearing TS1/22 or KIM127 in Ca2+/Mg2+ with or without 1 μm XVA143. p values from Welch's t test are indicated. E, distributions of unstressed lifetimes for LFA-1/ICAM-1 bonds in Ca2+/Mg2+, Ca2+/Mg2+/CXCL12, Mg2+/EGTA, or Mn2+ without or with 1 μm XVA143. Analysis by the Tukey-Kramer method showed no statistical differences among slopes (− off-rates) of different conditions (smallest p > 0.48). F, plots of lifetime (mean ± S.E.) versus force of LFA-1/ICAM-1 bonds in Ca2+/Mg2+, Ca2+/Mg2+/CXCL12, Mg2+/EGTA, or Mn2+ in the presence of 1 μm XVA143. G and H, mean ± S.E. of effective binding affinities (G) and on-rates (H) of LFA-1/ICAM-1 interactions under indicated conditions.
), or Mn2+ (○) or of nonspecific adhesions with 10 μg/ml anti-β2 blocking mAb TS1/18 in Ca2+/Mg2+ (▴). Lifetimes at zero- and at peak-forces are significantly different (p < 0.01, Welch's t test). C, plot of short (<10 s) lifetime average (± S.E.) and long (>10 s) lifetime fraction (inset) versus force of bonds of LFA-1 with TS1/22 in Ca2+/Mg2+ (▿) or with KIM127 in Mn2+ plus 100 μg/ml soluble ICAM-1 (to expose the KIM127 binding site on LFA-1) (○). D, adhesion frequencies at 0.1-s contact of LFA-1 expressing Jurkat cells to beads bearing TS1/22 or KIM127 in Ca2+/Mg2+ with or without 1 μm XVA143. p values from Welch's t test are indicated. E, distributions of unstressed lifetimes for LFA-1/ICAM-1 bonds in Ca2+/Mg2+, Ca2+/Mg2+/CXCL12, Mg2+/EGTA, or Mn2+ without or with 1 μm XVA143. Analysis by the Tukey-Kramer method showed no statistical differences among slopes (− off-rates) of different conditions (smallest p > 0.48). F, plots of lifetime (mean ± S.E.) versus force of LFA-1/ICAM-1 bonds in Ca2+/Mg2+, Ca2+/Mg2+/CXCL12, Mg2+/EGTA, or Mn2+ in the presence of 1 μm XVA143. G and H, mean ± S.E. of effective binding affinities (G) and on-rates (H) of LFA-1/ICAM-1 interactions under indicated conditions.
Zero-force lifetimes were measured using the thermal fluctuation assay (26). After touching the bead, the Jurkat cell was retracted to the null position to allow contact with the bead via thermal motions without compression or tension. Bond association and dissociation under zero-force was manifested as sudden changes in the thermal fluctuations of the bead (Fig. 1E and supplemental video S2). Thermal fluctuations were quantified by 70-point sliding standard deviation σ (Fig. 1F) of the position (Fig. 1E) over time, which had a mean of 8 or 5 nm in the presence or absence of a bond. Lifetime was measured from the instant when σ dropped below 7.0 nm to the instant when σ rose above 7.5 nm. Lifetimes were measured from the instant of bond formation to that of bond dissociation. At each force bin, the survival frequency as a function of time was calculated as the fraction of events with a lifetime >t, which included both short (<10 s) and long (>10 s) lifetime events.
Flow Cytometric Analysis
To characterize the conformations of Jurkat cell LFA-1 in different conditions, cells were washed once using HBSS- with 5 mm EDTA, washed again using HBSS- without EDTA, incubated in one of the four binding buffers with or without 1 μm XVA143 at 37 °C for 10 min, and incubated in the same buffer plus 10 μg/ml PE-conjugated HI 111, PE-conjugated MEM-148, KIM127, or TS1/22 mAb at 37 °C for 30 min. For the last two cases where unconjugated mAbs were used, cells were further incubated with 10 μg/ml PE-conjugated Fab fragment of a goat-anti-mouse antibody at room temperature for an additional 30 min. Cells were then washed and resuspended in the same buffer at 4 °C for immediate analysis by a BD LSR flow cytometer (BD Sciences).
To determine the Mac-1 expression, Jurkat cells or PMNs washed as described in the preceding paragraph were incubated with 10 μg/ml PE-conjugated anti-αM mAb at 37 °C for 30 min in binding butter containing Ca2+/Mg2+ or Ca2+/Mg2+/CXCL12, washed and re-suspended in the same buffer at 4 °C for immediate flow cytometric analysis.
To determine the CXC chemokine receptor 4 (CXCR4) expression, Jurkat cells washed as previously described in the preceding paragraph were incubated in binding butter containing Ca2+/Mg2+ or Ca2+/Mg2+/CXCL12 at room temperature for 1 or 2 h, then incubated with 10 μg/ml PE-conjugated anti-CXCR4 mAb at room temperature for 30 min, washed and re-suspended in the same buffer for immediate flow cytometric analysis.
To measure the LFA-1 site density on Jurkat cells and ICAM-1 site densities on the beads, Jurkat cells stained with PE-conjugated anti-LFA-1 mAb and ICAM-1 beads stained with PE-conjugated anti-ICAM-1 mAb were, respectively, analyzed by flow cytometry. Standard beads (BD Sciences) were used to generate a calibration curve to relate the florescence intensity with the number of fluorescent molecules. The site densities were determined by dividing the number of fluorescent molecules by the respective apparent areas of a Jurkat cell and a bead (32).
Fitting Lifetime Distributions by Multistate Models
The survival frequency and the rupture probability data for each force bin were simultaneously fitted by a model that assumes superposition of exponential decays in Equation 2,
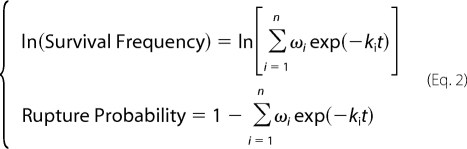 |
where t is time, ki and ωi are, respectively, the off-rate and fraction of the ith state. The sum of fractions equals to 1. Both two- (n = 2) and three-state (n = 3) models were tested.
The Bell equation (33) below was fit to the force-dependent off-rate of each state that was returned from the curve-fitting of lifetime distributions with the three-state model in Equation 3,
where f is force and ki0 and ai are, respectively, zero-force off-rate and reactive compliance of the ith state, kB is Boltzmann constant, and T is absolute temperature.
The average lifetimes that include both the short (<10 s) and long (>10 s) fractions (Fig. 4J) were calculated from the three-state model parameters as <t> = ω1/k1 + ω2/k2 + ω3/k3.
FIGURE 4.

Analysis of lifetimes by a three-state model. Plots of reciprocal off-rates (A–D) or fraction of bonds (F–I and K–N) of the short (□), intermediate (○), and long ()-lived states evaluated from simultaneously fitting the three-state model to the survival frequency (supplemental Fig. S2, upper rows) and rupture probability (supplemental Fig. S2, lower rows) versus lifetime data measured in Ca2+/Mg2+ (A, F, K), Ca2+/Mg2+/CXCL12 (B, G, L), Mg2+/EGTA (C, H, M), or Mn2+ (D, I, M) in the absence (F–I) and presence (K–N) of XVA143. The reciprocal off-rates in A–D were required to fit both data in the absence and presence of XVA143, which were superimposed in E and fitted by the Bell model (Equation 3, lines) with parameters listed in Table 1. J, reciprocal off-rates and associated fractions of the three states were used to calculate the force-dependent average lifetimes that include short (<10 s) as well as long (>10 s) subpopulations.
Statistical Analysis
Experimental results at different conditions were analyzed by the Welch's t test (34). The zero-force off-rates at various conditions were compared with the Tukey-Kramer multiple comparison methods (35). The goodness-of-fit of the three-state and two-state models were assessed by the reduced chi squares (χν2) and compared with the F-test (36). p < 0.01 is defined as significant difference. The details of the Welch's t test, Tukey-Kramer method, and F-test follow.
Welch's t Test
Welch's t test (34) can be viewed as a modified Student's t test to compare two samples with unequal variances. If mi, si, and ni are the respective mean, variance, and sample size of the ith sample, then the t statistic can be calculated in Equation 4,
and the degrees of freedom ν can be approximated in Equation 5,
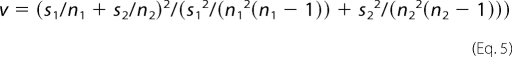 |
t and ν can be used with the t-distribution to test the null hypothesis that two populations have equal means, which will generate a p value for the level of significance of the difference between the two means. The Welch's t test was used to compare adhesion frequencies measured at different conditions. It was also used to compare the maximum value and the zero-force value in the catch-slip bond average lifetime versus force curves.
Tukey-Kramer Method
The Tukey-Kramer method (35) is a one-way layout multiple comparison statistical test generally used with an ANOVA to determine if the means are significantly different with each other among several data sets. Based on a Studentized range distribution, the Tukey-Kramer method calculates a minimum significant difference between each data pair. The difference of the two data sets is not significant if this minimum significant difference is larger than the actual difference of the “means” of the two pairs. The Tukey-Kramer multiple-comparison method was used to compare the differences of zero-force off-rates at various conditions. The zero-force off-rates were obtained from the negative slopes of the linear fits to the ln(survival frequency) versus lifetime data measured by the thermal fluctuation assay (see Fig. 2E). The slope was treated as the “mean” for a data set. The differences between the means and the experimental values of the survival frequency at each lifetime are treated as the “variances” of the data set.
F-test
F-test (36) is generally used to determine which model can better fit the data when the population has an F-distribution under null hypothesis. We used a two-state model with three parameters (p2 = 3) and a three-state model with five parameters (p3 = 5) to fit the lifetime distributions. At first glance, the three-state model should be able to fit the data better than the two-state model because the latter model can be viewed as a special case of the former mode. However, this was not always the case in our problem because we constrained the three-state model fitting by requiring the off-rate of each state to obey the Bell equation but did not constrain the two-state model fitting. For a total number of n data points, if χ2 and χ3 are the respective residual sums of squares of the two- and three-state models, the F statistic can be calculated as Equation 6,
If the three-state model is not significantly better than the two-state model, the null hypothesis predicts that F follows an F-distribution with the degrees of freedom (p3 − p2, n − p3). If the calculated F statistic is greater than the critical value of the F-distribution for a desired false-rejection probability, then the null hypothesis is rejected. Based on the degrees of the freedom and the F statistic, a p value can be calculated. p < 0.01 means that the three-states model is significantly better than the two-state model.
RESULTS
Using a BFP (Fig. 1A and supplemental video S1), we measured frequencies of adhesions and lifetimes of bonds between LFA-1 on a Jurkat cell (Fig. 1, A and B, right) and recombinant ICAM-1 or anti-LFA-1 monoclonal antibodies (mAbs) coated on a glass bead attached to the apex of a RBC (Fig. 1, A and B, left). The absence or presence of an adhesion was detected from the force signal upon cell retraction (Fig. 1C), allowing calculation of adhesion frequency from repeated tests (Fig. 1D). The adhesion lifetimes were measured under constant forces (Fig. 1C). To avoid force errors due to drift over long time, only were the fractions of adhesion events with lifetimes >10 s at the set forces measured instead of the precise lifetimes and the forces at rupture. To ensure single-bond events in most adhesions, the ICAM-1 densities (indicated in Fig. 1D) were so adjusted that specific adhesion frequencies were 15–20% for all conditions tested.
To verify binding specificity, we measured adhesion frequencies in the presence of blocking mAbs. Blocking ICAM-1 or integrin αL or β2 subunit reduced adhesion frequencies to that of Jurkat cell adhesion to beads not coated with ICAM-1 (<5%) whereas blocking the integrin αM subunit had no effect. This suggests that adhesions were mediated by LFA-1/ICAM-1 binding. To further confirm that Mac-1 (integrin αMβ2) did not contribute to ICAM-1 binding, we measured the expression of αM subunit on Jurkat cells by flow cytometry and did not detect any (supplemental Fig. S1, A and B), consistent with published data (37).
The single bond sensitivity of the BFP (30) enabled us to measure specific ICAM-1 binding to LFA-1 in Ca2+/Mg2+, a physiological condition under which LFA-1 has low affinity not measurable by most assays. Priming integrin with Mg2+/EGTA or Mn2+ (9, 10) induced much higher binding affinities, as only one-fortieth of ICAM-1 density was required to produce similar adhesion frequencies in these cation conditions (Fig. 1D). However, using soluble CXCL12 (Fig. 1D) that binds CXCR4 on Jurkat cells (supplemental Fig. S1C) to induce inside-out signaling did not significantly increase ligand binding affinity, consistent with published data (10). Interestingly, XVA143, a small molecule that blocks binding of the αA domain α7-helix to the βA domain MIDAS (27, 28) greatly reduced ligand binding affinity and its dependence on Mg2+/EGTA and Mn2+, as >70-fold higher ICAM-1 density was required to generate similar adhesion frequencies in the presence of XVA143 (Fig. 1D). The thermal fluctuation assay (26) was used to measure lifetimes at zero-force (Fig. 1, E and F and supplemental video S2).
Observation of LFA-1/ICAM-1 Catch Bonds
The averages of short lifetimes (<10 s) and the fractions of long lifetimes (>10 s) of the LFA-1/ICAM-1 bonds are plotted versus force in Fig. 2, A and B, respectively. As force increased from 0 to 10–15 pN, so did the short-lifetime averages and the long-lifetime fractions, revealing the counterintuitive catch bonds. After reaching their respective maxima, both lifetime metrics decreased with further force increase, showing slip bonds. The catch-slip bond biphasic curves were qualitatively similar for all four conditions tested. Adding CXCL12 right- and up-shifted both curves. Changing the divalent cations from Ca2+/Mg2+ to Mn2+ also shifted the maximum of the short-lifetime average curve from 10 to 15 pN and lifted the long-lifetime fraction curve upward (Fig. 2, A and B). By comparison, all lifetimes of nonspecific adhesions (measured with specific adhesions blocked by the anti-β2 mAb TS1/18) were <10 s (Fig. 2B) and their average decreased with increasing force, i.e. slip bonds (Fig. 2A).
To further confirm that the catch bond behavior was specific to the LFA-1/ICAM-1 interaction, we measured the force-dependent dissociation of LFA-1 from two mAbs. For bonds between LFA-1 and TS1/22, both curves of the short-lifetime averages and the long-lifetime fractions decreased monotonically with force (Fig. 2C). For bonds between LFA-1 and KIM127, all lifetimes were <10 s the average of which also decreased monotonically with force (Fig. 2C). These slip bond characteristics are similar to those of the antigen/antibody interactions previously studied (19, 38–40). Unlike TS1/22 whose bond lifetimes with LFA-1 were measured in Ca2+/Mg2+, the LFA-1/KIM127 bond lifetimes were measured in Mn2+ plus 100 μg/ml soluble ICAM-1 because the adhesion frequency in Ca2+/Mg2+ without soluble ICAM-1 was at the background level unless XVA143 was added (Fig. 2D). This is consistent with the reports (9, 15) that ligand or XVA143 binding extends LFA-1 to expose the KIM127 epitope at the β subunit genu region (Fig. 1B). Adding XVA143 had no effects on the TS1/22 binding frequency to LFA-1 (Fig. 2D), as expected for this conformation-independent mAb (9, 41).
Identification of Multiple LFA-1/ICAM-1 Bond States
Although no LFA-1/ICAM-1 bond lifetime was >10 s at 0 and >30 pN, 20–30% of total lifetimes were >10 s in 10–15 pN (Fig. 2B). Yet the peaks of short-lifetime averages were just 0.64–1.6 s, only moderately higher than the corresponding values at zero-force and at the highest force tested (Fig. 2A). This suggests that lifetimes of events in the >10 s fraction have far longer than those of events in the <10 s fraction. In other words, LFA-1/ICAM-1 bonds exist in at least two states that dissociate with short and long timescales. To test this hypothesis, we analyzed force-dependent distributions of total lifetimes that included both short (<10 s) and long (>10 s) fractions. Lifetimes at zero-force follow single-exponential distributions, as evidenced by the linear appearance of the semi-log survival frequency versus lifetime plots (supplemental Fig. S2, A–D, leftmost column, top panels). The negative slopes of the lines equal off-rates, or reciprocal timescales, of LFA-1/ICAM-1 bonds dissociating at zero-force. Bond lifetimes become multi-exponentially distributed as force increases in the catch bond regime but tend to return to single-exponentially distributed as force increases further into the slip bond regime (supplemental Fig. S2, A–D, from second column rightward, top panels). The curvy appearance of the semi-log plots reveals contributions from multiple subpopulations of bonds with distinct timescales to the pooled lifetimes. Such multi-exponentially distributed lifetimes are similar to those of the α5β1/FN catch bonds (19) but distinct from those of the selectin/ligand catch-slip bonds which are distributed as single exponentials (38, 39). The control lifetimes of LFA-1/KIM127 bonds follow single exponential distributions at all forces as they line up in the semi-log plots (supplemental Fig. S3).
Internal Force between αA and βA Domains Is Required for LFA-1/ICAM-1 Catch Bonds
Our observation of multi-state LFA-1/ICAM-1 bonds is consistent with previous structural (5, 7, 42, 43) and functional (9–14) studies suggesting multiple affinity states of LFA-1 that correspond to different conformations of the MIDAS allosterically regulated by multiple positions of the αA domain α7-helix (5, 20). XVA143 has been reported to inhibit LFA-1/ICAM-1 binding by blocking the internal ligation of the αA domain α7-helix to the βA domain MIDAS, thereby impairing the downward movement of the α7-helix (28). Therefore, we tested the effect of XVA143 on the force-dependent lifetimes of LFA-1/ICAM-1 bonds. Thermal fluctuation experiments observed indistinguishable zero-force lifetimes in the absence or presence of 1 μm XVA143 regardless of the divalent cations and chemokine used (Fig. 2E). Their specificity was confirmed by the significantly (p < 10−6, Student's t test) decreased average lifetime of nonspecific adhesions at zero-force (Fig. 2A). Remarkably, force-clamp experiments showed that XVA143 completely eliminated the long-lifetime (>10 s) fractions, converting the cation-dependent catch-slip bonds to cation-independent slip bonds in the entire force range tested (Fig. 2F). In the semi-log survival frequency versus lifetime plots, lifetime distributions with XVA143 (supplemental Fig. S2, E–H) were much less curved than those without XVA143 (supplemental Fig. S2, A–D), confirming that XVA143 suppressed the bond states with long timescales. These data suggest that both the applied force and the αA/βA ligation are required for the LFA-1/ICAM-1 catch bonds. Because this ligation affects lifetimes only when LFA-1 is pulled by ICAM-1, it must bear some internal force within LFA-1, which was generated by such pulling. In other words, the external force must be transmitted, at least partly, from the ICAM-1 binding site (αA domain MIDAS) to the α7-helix and further downstream through its internal ligation with the βA domain. It is this internal force that induces the LFA-1/ICAM-1 bond states with long timescales.
Affinity Regulation without Force Alters LFA-1/ICAM-1 On-rate but Not Off-rate
The lack of dependence of zero-force lifetimes on cations, chemokine, and XVA143 (Fig. 2E) is remarkable because these agents are known to regulate LFA-1/ICAM-1 binding affinity. However, bond lifetimes only reflect off-rates, i.e. how fast LFA-1/ICAM-1 bonds dissociate. We thus measured adhesion frequencies at a sufficiently long contact time to allow the LFA-1/ICAM-1 binding kinetics to reach equilibrium (similar to Fig. 1D but measured at 2 s) before the molecular interaction could be further altered by cellular regulations. These were then used to calculate the effective two-dimensional binding affinities (Fig. 2G) and on-rates (Fig. 2H) (Equation 1, see “Experimental Procedures”) (26, 32). Although LFA-1/ICAM-1 interaction in Ca2+/Mg2+ had a low affinity, the value was still much higher than those of Fcγ receptor III binding to IgG (32). Changing cations to Mg2+/EGTA or Mn2+ increased affinity and on-rate by two orders of magnitude. However, adding CXCL12 only slightly increased affinity and on-rate. XVA143 reduced binding affinities and on-rates in all conditions to similar levels slightly lower than that in Ca2+/Mg2+ without XVA143, consistent with previous studies (28, 44).
Forcing Extension and Hybrid Domain Swing-out Are Not Required for LFA-1/ICAM-1 Catch Bonds
We next used flow cytometry to define the conformations of LFA-1 in the conditions where its on-rate, off-rate, and affinity for ICAM-1 were measured. Staining of the extension reporter mAb KIM127 was low in Ca2+/Mg2+ (Fig. 3A). Adding CXCL12 or changing the cations to Mg2+/EDTA or Mn2+ progressively increased KIM127 staining. Adding XVA143 further increased KIM127 staining. These results are consistent with previous reports that most LFA-1 are bent under physiological conditions, that more LFA-1 assume an extended conformation when stimulated by chemokines or in Mg2+/EDTA or Mn2+, and that XVA143 induces LFA-1 extension (9, 10). These data show that, unlike on-rate that controls how fast LFA-1 forms bonds with ICAM-1, the force-dependent off-rate that defines the type of bonds (i.e. catch or slip) is independent of the prior bent or extended conformation of the integrin as manipulated by the eight treatments tested, because 1) the same LFA-1/ICAM-1 off-rates at zero-force and their biphasic force dependence (i.e. catch-slip bonds) were observed in all conditions without XVA143 and 2) XVA143 converted the catch-slip bonds to slip-only bonds without changing the zero-force off-rates regardless of the different conditions. A corollary of this result is that force-induced extension is not required for LFA-1 to form catch bonds with ICAM-1, consistent with our recent finding that force-induced extension is not required for integrin α5β1 to form catch bonds with FN (19).
FIGURE 3.

Characterization of LFA-1 conformations. Fluorescence intensity hisotograms of Jurkat cells stained by isotype-matched irrelevant IgG (control), KIM127 plus a PE-conjugated secondary goat-anti-mouse antibody (A), PE-conjugated MEM-148 (B), PE-conjugated HI 111 (C), or TS1/22 plus a PE-conjugated secondary goat-anti-mouse antibody (D) in Ca2+/Mg2+, Ca2+/Mg2+/CXCL12, Mg2+/EGTA, or Mn2+ without or with 1 μm XVA143. Data are representative of three experiments.
Staining of MEM-148, the reporter mAb for the hybrid domain swing-out (16, 45), showed a similar pattern, such that most LFA-1 kept the hybrid domain closed in Ca2+/Mg2+, adding CXCL12 slightly increased the fraction of LFA-1 with a swing-out hybrid domain, changing the cations to Mg2+/EGTA or Mn2+ and adding XVA143 induced more LFA-1 to swing-out the hybrid domain (Fig. 3B). Using the same reasoning as the preceding paragraph, these data show that, unlike the on-rate, the off-rate at zero-force and whether LFA-1 forms catch-slip bonds or slip-only bonds with ICAM-1 under tensile force are independent of the prior conformation of its hybrid domain as manipulated by the eight treatments tested. Again, a corollary of this result is that force-induced hybrid domain swing-out is not required for LFA-1 to form catch bonds with ICAM-1.
As controls, staining of HI 111, a reporter mAb for the closed conformation of the LFA-1 αA domain MIDAS (41), and TS1/22, a conformation-independent mAb (9, 27), were similar (Fig. 3, C and D). These data corroborate with the zero-force off-rate data, indicating that, in any of the conditions tested, the αA domain MIDAS conformation is in the closed conformation before and after ICAM-1 binding at zero-force. In other words, without applied force, short-time engagement of ICAM-1 alone does not induce any conformational change in the αA domain MIDAS that affects dissociation kinetics.
Characterizing the Short-, Intermediate-, and Long-lived States and the Switches Among Them by Force
Our data suggest that an external force applied via an engaged ICAM to induce an internal force pulling the αA domain α7-helix is necessary and sufficient for catch bond formation, regardless of the conformations of domains downstream to the αA domain MIDAS regulated by cations, chemokine, and XVA143. Catch bonds are generated by mixing multiple subpopulations of bond states with different timescales whose fractions change with force. To obtain further corroborations, we characterized the force-induced switch from short- to long-lived states in different conditions by fitting the lifetime data with a three-state model. The model assumes superposition of three subpopulations of bonds each of which follows first-order dissociation kinetics with a respective short, intermediate, or long timescale to govern its exponential distribution of lifetimes. Both the semi-log survival frequency (supplemental Fig. S2, A–D, upper row blue circles) and the rupture probability (supplemental Fig. S2, A–D, lower row blue circles) data were well-fitted by the model (Equation 2, “Experimental Procedures”; supplemental Fig. S2, A–D, solid black curves); the simultaneously fitting was designed to avoid overweighting of the rarely observed exceedingly long lifetimes. The best-fit results are plotted versus force in Fig. 4 for the four conditions studied: Ca2+/Mg2+ (first column), Ca2+/Mg2+/CXCL12 (second column), Mg2+/EGTA (third column), and Mn2+ (fourth column). The top row plots reciprocal off-rates (timescales) versus force for LFA-1/ICAM-1 bonds dissociating from the three states. Remarkably, the force-dependent timescales of the three states are indistinguishable (Fig. 4. A–D) despite that they were separately obtained by fitting four sets of survival frequency and rupture probability data independently measured in four different conditions (supplemental Fig. S2, A–D). Indeed, remarkable agreement is observed when these data are superimposed (Fig. 4E). The excellent linear fits in the semi-log plot show that all three reciprocal off-rates decrease exponentially with increasing force (Fig. 4, A–E). This agrees with the Bell model (33) (Equation 3, see “Experimental Procedures”), the parameters (k0 and a) of which are summarized in Table 1.
TABLE 1.
Bell model parameters of the three-state model under different conditions
| Off-rate states | Ca2+/Mg2+ |
Ca2+/Mg2+/CXCL12 |
Mg2+/EGTA |
Mn2+ |
All |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| k0 | a | k0 | a | k0 | a | k0 | a | k0 | a | |
| s−1 | nm | s−1 | nm | s−1 | nm | s−1 | nm | s−1 | nm | |
| Short-lived | 2.66 | 0.47 | 3.70 | 0.33 | 2.60 | 0.41 | 2.65 | 0.45 | 2.79 | 0.43 |
| Intermediate-lived | 0.20 | 0.75 | 0.13 | 0.71 | 0.36 | 0.70 | 0.28 | 0.53 | 0.22 | 0.69 |
| Long-lived | 0.001 | 1.28 | 0.00097 | 1.24 | 0.0011 | 1.33 | 0.0011 | 1.10 | 0.00091 | 1.29 |
We next analyzed the XVA143 data. Because XVA143 binds the MIDAS of βA rather than αA domain, it is assumed that XVA143 changes how force regulates the fractions associated with the three states but does not alter the three force-dependent off-rates of the LFA-1/ICAM-1 bonds. Therefore, the off-rates were calculated from the corresponding Bell model parameters in Table 1; only the fractions of the three states were allowed to change to fit the data with XVA143 (supplemental Fig. S2, E–H, solid black curves).
How force regulates LFA-1/ICAM-1 dissociation from each of the three states is characterized by their Bell model parameters (Table 1). The short-lived state has a stress-free off-rate (k0) an order of magnitude faster (smaller 1/k0 in Fig. 4A-E) than the intermediate-lived state value, which is ∼240-fold faster than the long-lived state value. However, force induces a faster decline in the reciprocal off-rate of the long-lived state than the intermediate-lived state, which in turn declines faster with force than the short-lived state (steeper slopes of the linear fits in Fig. 4E and larger a values in Table 1). The rapid declines of the reciprocal off-rates of all three states with increasing force explain the eventual slip bonds beyond an “optimal” force where lifetimes reach maximal.
The middle (Fig. 4, F–I) and bottom (Fig. 4, K–N) rows of Fig. 4 show how force regulates the fractions associated with the three states in the absence and presence of XVA143, respectively. Without XVA143, increasing force causes the fractions of intermediate- and long-lived bonds to grow at the expense of the short-lived bond fraction (Fig. 4, F–I). In all conditions, the short-lived state fractions decrease monotonically. By comparison, the intermediate-lived state fractions increase monotonically in Ca2+/Mg2+ and Ca2+/Mg2+/CXCL12, but are biphasic in Mg2+/EGTA and Mn2+, which increase initially, reach a maximum, and decline with further force increase. The increases in the long-lived state fractions occur at a larger force and less rapidly, which are biphasic in Ca2+/Mg2+ and Ca2+/Mg2+/CXCL12 but monotonic in Mg2+/EDTA and Mn2+ in the force ranges tested. These data indicate that the short-, intermediate-, and long-lived states co-exist in a dynamic equilibrium. Force progressively tilts this equilibrium toward more intermediate- and long-lived state fractions, thus explaining the catch bonds. Indeed, the timescales (Fig. 4, A–D) and associated fractions (Fig. 4, F–I) allow us to predict the force-dependent average lifetimes that include measurements >10 s, which show strong catch bonds for all conditions (Fig. 4J).
With XVA143, the decline of the short-lived state fractions and the growth of the intermediate-lived state fractions occur at higher forces and slower paces compared with the corresponding conditions without XVA143 (Fig. 4, K–N). The growth of the long-lived state fractions is eliminated in Ca2+/Mg2+ and Ca2+/Mg2+/CXCL12 and further right-shifted in Mg2+/EGTA and Mn2+ to forces so high that even the timescale of the long-lived state becomes very short. These results indicate that the external force applied by a bound ICAM-1 elicits LFA-1 catch bonds via an internal force that pulls on the αA domain α7-helix. Blocking the internal ligation between the αA and βA domains reduces the α7-helix-shared portion of the forces internally distributed within the stretched LFA-1; therefore, requires compensation by a higher external force to generate comparable pulling of the α7-helix via its C-terminal connection to the β-propeller domain.
As a comparison, we fitted a two-state model to the data without XVA143 (supplemental Fig. S2, A–D, dashed red curves) and with XVA143 (supplemental Fig. S2, E–H, dashed red curves) using the off-rates shown in supplemental Fig. S4, A–D and associated fractions shown in supplemental Fig. S4, E–H and supplemental Fig. S4, I–L for data without and with XVA143, respectively. However, the best-fit χv2 values of the two-state model are significantly higher (p < 0.001) than those of the three-state model in all conditions (supplemental Fig. S5), indicating that the three-state model is significantly better than the two-state model.
Fitting the zero-force data by either model (supplemental Fig. S2, leftmost column) returned a short timescale of ∼0.37 s with 100% bonds in the short-lived state regardless of XVA143. This indicates that no force pulled on the intrinsic ligand at αA domain α7-helix even in the absence of XVA143, regardless of the downstream domain conformations regulated by cations and chemokine.
Together, our results explain the LFA-1/ICAM-1 catch-slip bonds and their elimination by XVA143 in terms of two competing force-induced mechanisms: switching LFA-1 from short-lived to intermediate- and long-lived states and accelerating dissociation. At low forces, the former mechanism dominates, resulting in catch bonds. At high forces, the latter mechanism takes over, resulting in slip bonds. XVA143 greatly impairs the former mechanism, converting the catch-slip bond behavior to that of slip-only. Without force, ICAM-1 engagement alone does not induce state transitions, suggesting that the α7-helix remains in the “up” position despite that the βA domain may have already been activated by the integrin extension (Fig. 3A) and by the hybrid domain swing-out (Fig. 3B) via inside-out signaling or treatment with Mg2+/EGTA or Mn2+. Thus, our data also suggest a structural mechanism for force to allosterically regulate bond states.
DISCUSSION
Following our recent demonstration of a catch bond between FN and α5β1, an αA domain-lacking integrin (19), we have now shown a catch bond between ICAM-1 and LFA-1 (Fig. 2, A and B), an αA domain-containing integrin. These findings elucidate a molecular-level mechanical regulatory mechanism for force to enhance adhesive functions of integrins (3, 9, 11, 12, 17, 18). More importantly, here we provide a structurally-based explanation for the LFA-1/ICAM-1 catch bond as forcing switch from the short- to intermediate- and long-lived states by pulling the αA domain α7-helix with ligation to the βA domain, which represents a major conceptual advance beyond the previous work.
Like the α5β1/FN case, LFA-1/ICAM-1 catch bonds also were not observed in a previous dynamic force spectroscopy study (46). This study measured rupture forces by constant rate pulling and assumed dissociation along a single pathway with off-rates that increased exponentially with force as modeled by Bell (33), which precluded catch bonds. In a more recent study (47, 48), LFA-1/ICAM-1 off-rates of the Bell model type were derived from rupture forces >15 pN, beyond the catch bond regime observed here. Interestingly, our off-rates of the three states also obey the Bell equation (Fig. 4, A–E) with model parameters (Table 1) for the intermediate- and long-lived states comparable to those of Kinoshita et al. (48).
Because changes in adhesive functions (3, 9, 11, 12, 14, 17, 18) and ligand binding affinities/kinetics (5, 49–51) of integrins are associated with their conformational changes, it has been proposed that forcing integrin conformational changes would allosterically result in catch bonds (52, 53). We show that the LFA-1/ICAM-1 catch bond results from forcing downward movement of the αA domain α7-helix. However, catch bond formation does not require forced unbending of LFA-1 and/or hybrid domain swing-out. This is because more LFA-1 molecules are in an extended conformation with an out-swung hybrid domain in Mg2+/EGTA or Mn2+ than in Ca2+/Mg2+ and Ca2+/Mg2+/CXCL12 (Fig. 3, A and B) (7, 9, 10), yet similar catch bonds were observed in all of these conditions (Fig. 2, A and B). Our finding is consistent with recent publications showing that unbending of integrins is not necessary for the high affinity integrin-ligand interactions under certain conditions (54, 55).
Regulation of integrin affinity by conformation is also manifested as the substantial increases in LFA-1/ICAM-1 on-rate resulted from cation changes from Ca2+/Mg2+ to Mg2+/EGTA or Mn2+ (Fig. 2H), conditions that also induced LFA-1 unbending and hybrid domain swing-out (Fig. 3, A and B). It has been reported that hybrid domain swing-out activates the βA domain, enhancing its binding to the intrinsic ligand on the αA domain α7-helix (9). This may stabilize the flexible αA domain (16) to better orient it for more rapid ICAM-1 association. The key role of αA/βA ligation in on-rate regulation is supported by the XVA143 data that blocking this ligation suppressed LFA-1/ICAM-1 on-rate in all cation conditions tested (Fig. 2H) regardless of the extended conformation of LFA-1 with a swing-out hybrid domain (Fig. 3, A and B).
Our data also suggest an internal catch bond between the αA and βA domains. When the external force applied via ICAM-1 to the αA domain MIDAS increases, the internal force applied via the α7-helix to the βA domain MIDAS also increases. The external force prolongs the LFA-1/ICAM-1 bond lifetime. But this lifetime cannot be longer than the αA/βA bond lifetime, for dissociation of the αA/βA bond would release the internal force required to maintain the LFA-1/ICAM-1 bond. Therefore, the αA/βA bond lifetime must also increase with force, a hallmark of catch bonds. This result is remarkable but not surprising, as the ability of the βA domain to form catch bonds with its external ligand has been demonstrated for an αA domain-lacking integrin (19).
It is intriguing that LFA-1/ICAM-1 off-rates at zero-force were indistinguishable regardless of the integrin conformations as manipulated by the conditions tested (Fig. 2E). This implies that, without force, the αA domain α7-helix remains in the up position regardless of its ligation to the βA domain MIDAS and transient ICAM-1 engagement to the αA domain MIDAS. Upon hybrid domain swing-out, the αA domain may tilt about the β-propeller to allow the internal ligand on the α7-helix to better dock onto the βA domain MIDAS, but this may not cause movement of the α7-helix relative to the αA domain. This view is supported by the indistinguishable staining of mAb HI 111 (Fig. 3C), suggesting that the αA domain MIDAS remains closed (41) regardless of other downstream domain conformational changes induced by the cation conditions.
Our results extend the model for activation of αA domain-containing integrins (Fig. 5). Published work suggests that the integrin is inactive with a bent ectodomains and a closed headpiece in Ca2+/Mg2+ (Fig. 5A) (2, 3, 7, 9, 16). The hybrid domain is bent toward the β-propeller domain of the α subunit. The αA domain is flexible (depicted by the dotted light yellow ellipsoids moving from both sides of the solid yellow αA domain in Fig. 5, A and B) (16) with a “closed” MIDAS and “up” α7-helix. Priming by Mg2+/EGTA, Mn2+, or chemokines extends the integrin. Depending on the type and degree of priming, the hybrid domain may (Fig. 5C) or may not (Fig. 5B) swing out to activate the βA domain MIDAS to bind the intrinsic ligand on the αA domain α7-helix, resulting in differential enhancements of ligand binding affinity (9, 10, 16).
Our modification and extension includes identifying separate contributions to affinity from on- and off-rates, highlighting the role of force, demonstrating two catch bonds working in series, and elucidating their mechanism of action. We propose that stabilization and better orientation of the flexible αA domain through its ligation to the βA domain substantially increases on-rate; however, without tensile force applied via a bound ligand, the αA domain α7-helix is not pulled down and the αA domain MIDAS remains in the short-lived state regardless of any downstream conformational changes (Fig. 5, A–C). Comparing to the internal ligand, binding of XVA143 is much stronger and longer lasting, as it can block the αA/βA ligation to destabilize the αA domain orientation and can induce conformational changes that propagate from the βA domain further downstream (Fig. 5E).
An upward force applied via ICAM-1 would pull the αA domain α7-helix up unless it is anchored to the βA domain. The ligation between the αA and βA domains allows the applied force to be transmitted along the α7-helix (indicated by an arrow), which pulls it down (relative to the rest of the αA domain that is being pulled up) to induce the intermediate- and long-lived states in the αA domain MIDAS to form a catch bond with the ligand, regardless of the conformations of the downstream domains (Fig. 5, A–D). The prolonged lifetimes of the LFA-1/ICAM-1 bond at increased forces require continued down-pulling of the α7-helix, implying prolonged lifetimes of its bond with the βA domain MIDAS at increased forces, i.e. an internal catch bond between the αA and βA domains (Fig. 5D).
XVA143 blocks the αA/βA internal ligation, preventing force transmission from the αA to the βA domains (Fig. 5F). Other connections between the αA and β-propeller domains have to bear the load, resulting in a reduced force in the α7-helix (indicated by a smaller arrow). Because of this load shift, a much larger external force is required to generate a similar internal force to pull down the α7-helix, which is now anchored to the β-propeller domain via its C-terminal linker (Fig. 5F).
The above model highlights the important role of force in outside-in signaling. Without force, the interactions between the αA and βA domains are weak and transient. Transient ligand engagement per se cannot change the αA domain MIDAS conformation (as assessed by the unchanged off-rate for ligand dissociation), cannot move its α7-helix downward, and cannot induce conformational changes downstream. In other words, force applied via a bound ligand is required to induce conformational changes in the αA domain MIDAS and α7-helix, to strengthen the engagement of the βA domain MIDAS with the internal ligand, and to propagate the conformational changes further downstream to induce signaling. Catch bonds are key elements of this allosteric model of the integrin mechanochemistry.
Supplementary Material
Acknowledgments
We thank M. Robinson for providing KIM127, P. Gillespie for providing XVA143, R. McEver and F. Kong for critical reading of the manuscript, and Larissa Doudy for technical support.
This work was supported, in whole or in part, by National Institutes of Health Grant AI44902 (to C. Z.), an American Heart Association Scientist Development Grant 0735224N and the “Hundred Talents Program” of the Chinese Academy of Sciences (to J. L.), and an American Heart Association Predoctoral Fellowship (to W. C.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5 and videos S1 and S2.
- MIDAS
- metal ion-dependent adhesion site
- FN
- fibronectin
- BFP
- biomembrane force probe
- RBC
- red blood cell.
REFERENCES
- 1.Hynes R. O. (2002) Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- 2.Luo B. H., Carman C. V., Springer T. A. (2007) Annu. Rev. Immunol. 25, 619–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alon R., Dustin M. L. (2007) Immunity. 26, 17–27 [DOI] [PubMed] [Google Scholar]
- 4.Schwartz M. A., DeSimone D. W. (2008) Curr. Opin. Cell Biol. 20, 551–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimaoka M., Xiao T., Liu J. H., Yang Y., Dong Y., Jun C. D., McCormack A., Zhang R., Joachimiak A., Takagi J., Wang J. H., Springer T. A. (2003) Cell. 112, 99–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao T., Takagi J., Coller B. S., Wang J. H., Springer T. A. (2004) Nature. 432, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishida N., Xie C., Shimaoka M., Cheng Y., Walz T., Springer T. A. (2006) Immunity. 25, 583–594 [DOI] [PubMed] [Google Scholar]
- 8.Takagi J., Petre B. M., Walz T., Springer T. A. (2002) Cell. 110, 599–611 [DOI] [PubMed] [Google Scholar]
- 9.Salas A., Shimaoka M., Kogan A. N., Harwood C., von Andrian U. H., Springer T. A. (2004) Immunity. 20, 393–406 [DOI] [PubMed] [Google Scholar]
- 10.Shamri R., Grabovsky V., Gauguet J. M., Feigelson S., Manevich E., Kolanus W., Robinson M. K., Staunton D. E., von Andrian U. H., Alon R. (2005) Nat. Immunol. 6, 497–506 [DOI] [PubMed] [Google Scholar]
- 11.Salas A., Shimaoka M., Phan U., Kim M., Springer T. A. (2006) J. Biol. Chem. 281, 10876–10882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woolf E., Grigorova I., Sagiv A., Grabovsky V., Feigelson S. W., Shulman Z., Hartmann T., Sixt M., Cyster J. G., Alon R. (2007) Nat. Immunol. 8, 1076–1085 [DOI] [PubMed] [Google Scholar]
- 13.Shulman Z., Shinder V., Klein E., Grabovsky V., Yeger O., Geron E., Montresor A., Bolomini-Vittori M., Feigelson S. W., Kirchhausen T., Laudanna C., Shakhar G., Alon R. (2009) Immunity 30, 384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanley P., Smith A., McDowall A., Nicol A., Zicha D., Hogg N. (2008) EMBO J. 27, 62–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim M., Carman C. V., Springer T. A. (2003) Science 301, 1720–1725 [DOI] [PubMed] [Google Scholar]
- 16.Xie C., Zhu J., Chen X., Mi L., Nishida N., Springer T. A. (2009) EMBO J. 666–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Astrof N. S., Salas A., Shimaoka M., Chen J., Springer T. A. (2006) Biochemistry 45, 15020–15028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedland J. C., Lee M. H., Boettiger D. (2009) Science 323, 642–644 [DOI] [PubMed] [Google Scholar]
- 19.Kong F., García A. J., Mould A. P., Humphries M. J., Zhu C. (2009) J. Cell Biol. 185, 1275–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin M., Andricioaei I., Springer T. A. (2004) Structure 12, 2137–2147 [DOI] [PubMed] [Google Scholar]
- 21.Puklin-Faucher E., Vogel V. (2009) J. Biol. Chem. 284, 36557–36568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puklin-Faucher E., Gao M., Schulten K., Vogel V. (2006) J. Cell Biol. 175, 349–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaillard T., Dejaegere A., Stote R. H. (2009) Proteins 76, 977–994 [DOI] [PubMed] [Google Scholar]
- 24.Provasi D., Murcia M., Coller B. S., Filizola M. (2009) Proteins 77, 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lou J., Yago T., Klopochi A. G., Metha P., Chen W., Zarnitsyna V. I., Bovin N. V., Zhu C., McEver R. P. (2006) J. Cell Biol. 174, 1107–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen W., Evans E. A., McEver R. P., Zhu C. (2008) Biophys. J. 94, 694–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welzenbach K., Hommel U., Weitz-Schmidt G. (2002) J. Biol. Chem. 277, 10590–10598 [DOI] [PubMed] [Google Scholar]
- 28.Shimaoka M., Salas A., Yang W., Weitz-Schmidt G., Springer T. A. (2003) Immunity 19, 391–402 [DOI] [PubMed] [Google Scholar]
- 29.Yago T., Wu J., Wey C. D., Klopocki A. G., Zhu C., McEver R. P. (2004) J. Cell Biol. 166, 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen W., Zarnitsyna V. I., Sarangapani K. K., Huang J., Zhu C. (2008) Cell. Mol. Bioeng. 1, 276–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans E., Heinrich V., Leung A., Kinoshita K. (2005) Biophys. J. 88, 2288–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chesla S. E., Selvaraj P., Zhu C. (1998) Biophys. J. 75, 1553–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell G. I. (1978) Science 200, 618–627 [DOI] [PubMed] [Google Scholar]
- 34.Welch B. L. (1947) Biometrika 34, 28–35 [DOI] [PubMed] [Google Scholar]
- 35.Wu C. F., Hamada M. S. (2009) Experiments: Planning, Analysis, and Optimization, 2nd Ed., pp. 53–57, John Wiley & Sons, Inc., New York [Google Scholar]
- 36.Lomax R. G., Hahs-Vaughn D. L. (2007) Statistical Concepts: A Second Course, p. 10, Routledge Academic, New York [Google Scholar]
- 37.Miller L. J., Schwarting R., Springer T. A. (1986) J. Immunol. 137, 2891–2900 [PubMed] [Google Scholar]
- 38.Marshall B. T., Long M., Piper J. W., Yago T., McEver R. P., Zhu C. (2003) Nature. 423, 190–193 [DOI] [PubMed] [Google Scholar]
- 39.Sarangapani K. K., Yago T., Klopochi A. G., Lawrence M. B., Fieger C. B., Rosen S. D., McEver R. P., Zhu C. (2004) J. Biol. Chem. 279, 2291–2298 [DOI] [PubMed] [Google Scholar]
- 40.Wu T., Lin J., Cruz M. A., Dong J. F., Zhu C. (2010) Blood 115, 370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma Q., Shimaoka M., Lu C., Jing H., Carman C. V., Springer T. A. (2002) J. Biol. Chem. 277, 10638–10641 [DOI] [PubMed] [Google Scholar]
- 42.Lee J. O., Bankston L. A., Arnaout M. A., Liddington R. C. (1995) Structure 3, 1333–1340 [DOI] [PubMed] [Google Scholar]
- 43.Lee J. O., Rieu P., Arnaout M. A., Liddington R. (1995) Cell 80, 631–638 [DOI] [PubMed] [Google Scholar]
- 44.Zhang F. (2007) Department of Biomedical Engineering, Georgia Institute of Technology, Atlanta, Georgia [Google Scholar]
- 45.Drbal K., Angelisová P., Cerný J., Hilgert I., Horejsí V. (2001) Immunobiology 203, 687–698 [DOI] [PubMed] [Google Scholar]
- 46.Zhang X., Wojcikiewicz E., Moy V. T. (2002) Biophys. J. 83, 2270–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans E., Kinoshita K., Simon S., Leung A. (2010) Biophys. J. 98, 1458–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kinoshita K., Leung A., Simon S., Evans E. (2010) Biophys. J. 98, 1467–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimaoka M., Lu C., Palframan R. T., von Andrian U. H., McCormack A., Takagi J., Springer T. A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 6009–6014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Labadia M. E., Jeanfavre D. D., Caviness G. O., Morelock M. M. (1998) J. Immunol. 161, 836–842 [PubMed] [Google Scholar]
- 51.Zhang F., Marcus W. D., Goyal N. H., Selvaraj P., Springer T. A., Zhu C. (2005) J. Biol. Chem. 280, 42207–42218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu C., Lou J., McEver R. P. (2005) Biorheology 42, 443–462 [PubMed] [Google Scholar]
- 53.Zhu J., Luo B. H., Xiao T., Zhang C., Nishida N., Springer T. A. (2008) Mol. Cell. 32, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blue R., Li J., Steinberger J., Murcia M., Filizola M., Coller B. S. (2010) J. Biol. Chem. 285, 17604–17613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuwano Y., Spelten O., Zhang H., Ley K., Zarbock A. (2010) Blood 116, 617–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.