

Abstract
Past studies of bone extracellular matrix phosphoproteins such as osteopontin and bone sialoprotein have yielded important biological information regarding their role in calcification and the regulation of cellular activity. Most of these studies have been limited to proteins extracted from mammalian and avian vertebrates and nonvertebrates. The present work describes the isolation and purification of two major highly glycosylated and phosphorylated extracellular matrix proteins of 70 and 22 kDa from herring fish bones. The 70-kDa phosphoprotein has some characteristics of osteopontin with respect to amino acid composition and susceptibility to thrombin cleavage. Unlike osteopontin, however, it was found to contain high levels of sialic acid similar to bone sialoprotein. The 22-kDa protein has very different properties such as very high content of phosphoserine (∼270 Ser(P) residues/1000 amino acid residues), Ala, and Asx residues. The N-terminal amino acid sequence analysis of both the 70-kDa (NPIMA(M)ETTS(M)DSKVNPLL) and the 22-kDa (NQDMAMEASSDPEAA) fish phosphoproteins indicate that these unique amino acid sequences are unlike any published in protein databases. An enzyme-linked immunosorbent assay revealed that the 70-kDa phosphoprotein was present principally in bone and in calcified scales, whereas the 22-kDa phosphoprotein was detected only in bone. Immunohistological analysis revealed diffusely positive immunostaining for both the 70- and 22-kDa phosphoproteins throughout the matrix of the bone. Overall, this work adds additional support to the concept that the mechanism of biological calcification has common evolutionary and fundamental bases throughout vertebrate species.
Keywords: Bone, Evolution, Extracellular Matrix Proteins, Glycosylation, Protein Phosphorylation, Fish Bone
Introduction
The noncollagenous phosphoproteins of bone extracellular matrix (ECM)2 have been of major scientific interest for more than three decades and continue to be the subject of a number of studies. This is due to observations and experimental evidence that they are involved in a number of biological events such as regulation of biomineralization and cellular activity/behavior in normally mineralizing tissues including bone (1–7), cartilage (8), and dentin (9). ECM phosphoproteins have been also found in the pathologically mineralizing tissues such as atherosclerotic plaque (10), kidney stones (11), dental calculus (12), and breast tumors (13). Two most abundant and well known are bone sialoprotein (BSP) and osteopontin (OPN), which are synthesized by osteoblasts during bone formation (1–7, 14, 15). In addition to their involvement in biomineralization and its regulation (6, 7, 16–19), bone ECM phosphoproteins are implicated in modulating cellular function and the behavior of bone cells such as osteoblasts (20–22), osteoclasts (23–27), and other cell types such as tumor and immune (28, 29) where they promote cell adhesion, motility, and transmembrane signaling. The involvement of both BSP and OPN in such biological functions has been predominantly linked to the presence of an integrin receptor-binding tripeptide Arg-Gly-Asp sequence in these proteins that interacts with cell surface integrins αvβ3 and αvβ1 (26, 30–32). Other non-Arg-Gly-Asp amino acid sequences of these proteins have also been found to participate in cell binding with additional integrins such as α9β1 and α4β1 (22, 33, 34). Although the covalently bound phosphate groups play a direct role in the nucleation of calcium phosphate crystals during biomineralization (1), the organic phosphate groups also participate in cell attachment properties of these phosphoproteins (35). In terms of the effects on cellular activity and behavior, BSP stimulates osteoblast differentiation and bone formation in vitro, and in vivo these effects are abolished by the addition of anti-BSP antibody (36–39). In contrast, OPN enhances bone resorption and the absence of OPN suppresses parathyroid hormone-induced bone resorption (40, 41). Although BSP is also shown to promote bone resorption in vitro (40, 42), immunocytochemistry of bone tissue revealed no accumulation of BSP on bone surfaces facing osteoclasts; on the contrary, an enhanced concentration of osteopontin was found in bone opposite to the clear zone of osteoclasts (43). These observations suggested the involvement of BSP and OPN in overall bone remodeling. More recent studies using BSP knock-out mice showed impaired bone growth and mineralization with resultant dramatic reduction in overall bone formation (44).
Analysis of the OPN and BSP genes has suggested that they belong to a genetically related family of small integrin-binding ligand N-linked glycoproteins and are in fact the result of duplication and subsequent divergent evolution of a single ancient gene (45). To date, the studies related to ECM phosphoproteins of mineralizing tissues have been carried out in mammalian and avian vertebrates (1–15) as well as invertebrate species such as sea urchin (46, 47). However, it is of major interest to study components of bone ECM in other vertebrate species such as fish. This has 2-fold implications: one for the evolutionary point of view regarding the mechanism of biomineralization/bone formation and the other concerning whether fundamentally basic common physicochemical steps are conserved during bone formation throughout vertebrate species. Furthermore, because intramuscular bones of fish such as herring and pickerel have quite different bone architecture, the relationship between the collagen fibrils and mineral is highly simplified. As such, this model provided the advantage of a clear determination of the three-dimensional spatial relationship between these components (48, 49). The present study reports on the isolation and characterization of two novel fish bone glycosylated phosphoproteins of 70 and 22 kDa (70 k and 22 k, respectively) from alewife (Alosa pseudoharengus) and blueback herring (Alosa aestivalis) (Teleostei Clupeidae).
EXPERIMENTAL PROCEDURES
Tissue Extraction
Fresh marine herring (∼5 years old), alewives, and small amounts of closely related blueback herring were provided by the Massachusetts Division of Marine Fisheries (Pocasset, MA). After removal of soft tissues, the vertebrae including intramuscular bone, rib, and spine (∼300 g) were powdered under liquid nitrogen and extracted with 0.1 m HCl containing protease and phosphatase inhibitors (1 mm phenylmethylsulfonyl fluoride, 1 mm α-amino-n-caproic acid, 1 mm p-hydroxymercuribenzoic acid, 1 mm benzamidine hydrochloride, 1 mm levamisole, and 5 mm sodium pyrophosphate) at a constant pH of ∼2.0 for 4 h at 4 °C. The supernatant (HCl extract) was obtained by centrifugation at 15,000 × g for 30 min at 4 °C, dialyzed against 10 liters of H2O for 48 h with one change after 24 h, lyophilized, and stored at −20 °C until use.
In a separate experiment, the herring roe, rib bone, liver, skin, muscle, stomach, and scale each (∼10 g) were collected, lyophilized, and cut into small pieces (<1 mm) using scissors on dry ice, whereas the rib bone (∼10 g) was powdered as above. These tissues were first extracted with 4 m guanidine HCl, containing protease inhibitors, 50 mm Tris-HCl, pH 7.4, at 4 °C for 5 h. The residues were separated from the supernatants (4 m guanidine HCl extracts) by centrifugation and further extracted with the same buffer containing 0.5 m EDTA for 3 days. The extracts each were clarified by centrifugation, pooled together with the respective 4 m guanidine HCl extracts, dialyzed against 10 liters of H2O with one change after 24 h, lyophilized, and stored at −20 °C.
Purification
The HCl extract was dissolved in 7 m urea, 50 mm Tris-HCl, pH 7.4, and applied to a DE-52 column (2.5 × 7.0 cm; Fisher) in the same buffer at 4 °C, and the bound proteins were eluted by using a linear gradient of 0–0.5 m NaCl in 7 m urea, 50 mm Tris-HCl, pH 7.4, at a flow rate of 3 ml/min. The separated fractions containing the proteins of interest were concentrated by ultrafiltration using an Amicon cell concentrator ultrafilter membrane (Mr 5000 cutoff; Millipore Co., Billerica, MA), brought to 7 m urea, 10 mm Tris-HCl, 50 mm sodium acetate, pH 4.0, and chromatographed on another DE-52 column (0.9 × 5.2 cm) using stepwise gradients of 0.05, 0.1, 0.15, 0.2, and 0.5 m NaCl in 7 m urea, 10 mm Tris-HCl, 50 mm sodium acetate, pH 4.0, at a flow rate of 1 ml/min. The fractions containing the 70- and 22-kDa proteins were concentrated using another membrane YM 10 (Mr 10,000 cutoff; Millipore). After 10-fold dilution with 7 m urea, 0.4 m NaCl, 50 mm KH2PO4, pH 6.8, the samples were concentrated. This procedure was repeated 10 times to well separate the two proteins. The concentrated samples containing the 70-kDa protein were treated by the addition of 20 mm DTT at 4 °C for 15 h, purified by chromatography on a TSK gel G3000SW (TSK-3000) HPLC column (0.75 × 60 cm; TosoHaas Co., Montgomeryville, PA) in 7 m urea, 0.4 m NaCl, 50 mm KH2PO4, pH 6.8, at a flow rate of 0.6 ml/min and rechromatographed on the same column, whereas the filtrates containing the 22-kDa protein were concentrated with the Mr 5000 cutoff membrane, treated with DTT, and further purified on the TSK-3000 column followed by reverse phase (RP)-HPLC on a Delta-Pak C4 column (0.8 × 10 cm; Waters Co., Milford, MA) using a linear gradient from 20% (v/v) acetonitrile, 0.3% (v/v) TFA to 60% acetonitrile, 0.3% TFA in 40 min at a flow rate of 1 ml/min. For comparative purposes, OPN and BSP were purified from chicken and bovine bones using a previously described protocol (5).
Electrophoresis
SDS-PAGE was performed using 10 or 15% mini-gels (1.5 × 55 × 80 mm) by the Laemmli method (50). All of the samples were reduced with 1% β-mercaptoethanol unless indicated otherwise. In another set of experiments, polyacrylamide gel and electrode buffer were prepared as those used for SDS-PAGE but without SDS, and the samples were not treated with SDS and β-mercaptoethanol (nondenaturing PAGE). The gels were stained with Coomassie Brilliant Blue R-250 (CBB) or Stains-all (Sigma).
Chemical Analyses
The proteins of interest were hydrolyzed with 4 and 6 n HCl for 6 and 24 h for phosphoamino acid and total amino acid analyses, respectively. The hydrolysates were analyzed with a Beckman model 121-M amino acid analyzer. Protein, neutral sugar, and sialic acid contents were determined as described previously (51).
Internal Peptide Generation and Peptide Isolation
∼50 μg of the purified 70k-I-2 phosphoprotein was subjected to a 10% SDS-PAGE, stained with CBB, destained with acetic acid/methanol, and washed with H2O. The 70-kDa band was excised, cut into 1-mm pieces, and in-gel digested with 2.5 μg/25 μl of Promega modified sequencing grade porcine trypsin in 50 mm NH4HCO3 for 20 h at 37 °C. The tryptic peptides were extracted by alternative use of 50 mm NH4HCO3 and 50% acetonitrile in 50 mm NH4HCO3, and the extracts were pooled. This was repeated twice, the pooled samples containing the peptides were dried using a SpeedVac and resuspended in buffer A (0.06% TFA/H2O), and the peptides were separated by RP-HPLC on a Zorbax C18 column (1 × 150 mm). The peptides were eluted by using sequential linear gradients from buffer A to 67% buffer A + 33% buffer B (0.04% TFA, acetonitrile) in 130 min followed by 45% buffer A + 55% buffer B in 30 min at a flow rate of 0.3 ml/min.
Approximately ∼50 μg of the purified 22-kDa protein, 22k-I, was subjected to a 15% SDS-PAGE, and the band of interest was excised, reduced with 10 mm DTT, 0.02% EDTA, in 100 mm NH4HCO3 at 37 °C for 30 min, and incubated with 55 mm iodoacetamide in 100 mm NH4HCO3 at room temperature for 30 min. The protein was then digested with 0.5 μg/5 μl of thermolysin (Sigma) in 50 mm NH4HCO3 at 37 °C for 15 min. The resulting peptides were extracted from the gel as described above and separated by the C18 column chromatography using sequential gradients from 100% buffer A to 67.5% A + 32.5% buffer B for 130 min followed by 44.5% buffer A + 55.5% buffer B over 29 min and washed at 5% buffer A + 95% buffer B.
N-terminal Sequence Analysis of the Phosphoproteins and Their Internal Peptides
The purified proteins and their internal peptides were sequenced using PE/ABD Procise 494 HT protein sequencing system (Applied Biosystems Co., Bedford, MA) at the Microchemistry Facility of Harvard University. Approximately 40 μg of the purified 70k-I-2 was subjected to the nondenaturing PAGE (10% gel), and the major band was cut and further separated by SDS-PAGE (10% gels). The protein bands were then transferred onto an Immobilon-P membrane (Millipore Co.), visualized with CBB, and the excised bands were N-terminally sequenced directly on the Immobilon-P membrane. The purified 70k-I-1 was sequenced directly without running SDS-PAGE. The 22k-I (∼20 μg) was electrophoresed (10% SDS-PAGE gels), transferred onto an Immobilon-P membrane, and N-terminally sequenced directly on the Immobilon-P membrane. The major internal peptide fractions were screened by matrix-assisted laser desorption time-of-flight mass spectrometry performed using a Perseptive Biosystems Voyager-DE STR (Framingham, MA), and the fractions containing single peptide were selected for N-terminal sequence analysis.
Nano-flow Liquid Chromatography and Electrospray Ionization Tandem Mass Spectrometry (LC-ESI-MS/MS) Analysis of Fish Bone Phosphoproteins
10 μg of each of the purified 70- and 22-kDa phosphoproteins and 300 μg of crude HCL bone extract were digested by trypsin, 2% (w/w) at 37 °C overnight as described previously (6). The samples were dried and suspended in 97.4% H2O, 2.5% CH3CN, 0.1% formic acid. LC-ESI-MS/MS analyses were carried out using LTQ-linear ion trap mass spectrometer (Thermo Electron, San Jose, CA) and an on-line autosampler (Micro AS, ThermoFinnigan, CA) with auto-injections of 3 μl onto an in-line fused silica microcapillary column (75 μm × 10 cm), packed in-house with C18 resin (Micron Bioresource, Inc.,+ Auburn, CA), and developed at a flow rate of 250 nl/min. The peptides were separated by a 55-min elution comprising multi-step linear gradient using solvent A (H2O, 2.5% CH3CN, 0.1% formic acid) and solvent B (CH3CN, 0.1% formic acid). The gradient steps were from 100% solvent A to 8% solvent B in 5 min, to 15% solvent B in 10 min, to 25% solvent B in 10 min, to 50% solvent B in 20 min, and to 95% solvent B in 10 min using a Surveyor MS Pump Plus (ThermoFinnigan, CA). The eluted peptides were directly nano-electrosprayed, and the MS/MS data were generated using data-dependent acquisition with a MS survey scan range between 390 and 2000 m/z.
Database Search and Phosphoprotein/Protein Identification
All of the MS/MS spectra from LC-ESI-MS/MS were searched against the zebrafish database and general fish databases: Uniprot (Universal Protein Resource, version 9.0), which combines the data from Swiss-Prot (version 51), TreMBL (version 34), and PIR using Bioworks 3.3.1 software and SEQUEST search engine. The mass spectral file containing MS data for scans during SEQUEST analysis generation was with precursor ion tolerance of 1.5 atomic mass units, fragment ion tolerance of 1.0 atomic mass unit, and automated calculated charged states +1, +2, and +3 which also included 5-point smoothing. The searches were performed with parameters: partial trypsin, two miscleavages, and modifications of serine and threonine residues by phosphate (+80 Da) and run as a dynamic modification that would identify both phosphopeptides and nonphosphopeptides. The database search results were filtered using the criteria: ΔCn (sequence coverage) ≥ 0.1; probability ≤ 0.1; for full tryptic peptides, XCorr (cross-correlation) ≥ 1.7, 2.0, 3.5 for Z = +1, +2, +3; and for half-tryptic peptides, XCorr ≥ 1.9, 2.2, 3.75 for Z = +1, +2, +3.
Thrombin Digestion
The purified proteins were incubated at 37 °C in the absence and presence of human thrombin (Sigma) at a ratio of 1 unit of thrombin/8 μg of protein in 0.1 m NH4HCO3 for 2 and 4 h. The resulting samples were analyzed by SDS-PAGE.
Antibody Production
∼150 μg of purified 70k-I-1 was treated with thrombin for 2 h as described above and then subjected to SDS-PAGE. The 32-, 38-, and 45-kDa fragments (see Fig. 3B, lane 9) were cut out for antibody production in rabbit as described previously (51). ∼100 μg of the purified 22k-II was also used to raise polyclonal antibodies following SDS-PAGE.
FIGURE 3.

SDS-PAGE of the purified phosphoproteins. The purified proteins were separated using 10% SDS-PAGE gels and visualized with CBB (A) or Stains-all (B) or using 15% SDS-PAGE gels and visualized with Stains-all (C). A, lanes 1, 70k-I-1 (1 μg); lane 2, 70k-I-2 (1 μg); lane 3, 70k-II (1 μg). B, lane 1, 70k-I-2 (1 μg) under nonreducing conditions; lanes 2–7, 70k-I-1, 70k-I-2, and 70k-II (1 μg each) incubated without thrombin (lanes 2, 4, and 6, respectively) or with thrombin (lanes 3, 5, and 7, respectively) for 2 h; lanes 8–10, 70k-I-1 (3 μg) incubated without thrombin for 4 h (lane 8) or with thrombin for 2 h (lane 9) and 4 h (lane 10). C, lane 1, 22k-I (1 μg); lane 2, 22k-II (1 μg); lane 3, 22k-I (1 μg) under nonreducing conditions.
Enzyme-linked Immunosorbent Assay
The EDTA/guanidine HCl extracts of herring roe, liver, skin, muscle, stomach, scale, and bone were analyzed by enzyme-linked immunosorbent assay (52), with some modifications. Briefly, each extract was dissolved in 7 m urea, 50 mm Tris-HCl, pH 7.4, and aliquots of the extracts were added to 1% gelatin, 0.05% Tween 20, 0.15 m NaCl, 10 mm Tris-HCl, pH 8.0 (TBS), containing antiserum against the purified protein at a dilution of 1:4000. After incubation for 15 h at room temperature and centrifugation at 14,000 × g for 20 min, the supernatants were added to a 96-well Nunc-Immuno plate (VWR Scientific Products Co., Bridgeport, NJ) precoated with the purified protein and blocked with 2% gelatin in TBS containing 0.05% Tween 20. The bound antibodies were detected at 405 nm after incubation with alkaline phosphatase-conjugated goat anti-rabbit IgG and p-nitrophenylphosphate as the substrate (Sigma).
Immunohistochemical Procedures
Intramuscular bone of fresh alewife herring (∼5 years old) was dissected, fixed for 20 h at 4 °C with 2% paraformaldehyde, 0.1 m sodium cacodylate, pH 7.4, embedded in low melting point paraffin Tissue Prep (Fisher) via xylene after dehydration in ethanol, and cut at a thickness of 5 μm. After deparaffinization and rehydration, the sections were treated with 2.5% glutaraldehyde, 0.1 m sodium cacodylate, pH 7.4, for 15 min and decalcified with 7% EDTA, 2.5% glutaraldehyde, 0.1 m sodium cacodylate, pH 7.4, for 20 min. They were then washed with TBS, blocked with 15% goat serum in TBS for 40 min, and incubated with antiserum to the purified proteins or preimmune rabbit sera (negative control) at a dilution of 1:50–100 in TBS containing 5% goat serum for 1 h at room temperature. After several washes, the bound antibodies were reacted with biotinated anti-rabbit IgG at a dilution of 1:200 followed by incubation with colloidal gold-labeled streptavidin and silver enhancer (Sigma-Aldrich). For counterstaining, the sections were further stained with 0.01% methyl green for ∼3 s. In other sets of experiments, the decalcified and undecalcified sections were stained with hematoxylin/eosin and AgNO3 for phosphate determination (Sigma), respectively. For hematoxylin/eosin staining, the sections were stained with 0.4% hematoxylin, 3.52% aluminum sulfate, 0.04% sodium iodate for ∼3 s and washed with H2O followed by incubation with 1% eosin Y in 95% ethanol for ∼2 s. The undecalcified sections were used for von Kossa stain (53) and counterstained with 0.01% methyl green.
RESULTS
Purification
Two major herring bone phosphoproteins of 70 and 22 kDa were extracted by HCl demineralization. The ion exchange chromatography of the HCl extract on DE-52 column at pH 7.4 led to separation of the 70-kDa protein into two fractions designated as 70k-I and 70k-II (Fig. 1A). Most of the 22 k protein was co-eluted with the 70k-I. The 70k-I was further separated into 70k-I-1 and 70k-I-2 forms when rechromatographed on DE-52 column at pH 4.0. Similarly the 22 k phosphoprotein was fractionated into 22k-I and 22k-II forms (Fig. 1B). The 70k-II phosphoprotein was also rechromatographed using this column at pH 4.0 (Fig. 1C). These isolated proteins were each then subjected to gel filtration chromatography using a HPLC TSK-3000 column (Fig. 2, A–C, G, and H). The final purification of the 70k-I-1, 70k-I-2, and 70k-II (Fig. 2, D–F) was achieved by repeating this gel filtration step. Quite distinct from this, the final purification of the 22k-I and 22k-II was attained by RP-HPLC using C4 column (Fig. 2, I and J). SDS-PAGE of both purified 70k-I-1 (Fig. 3, A, lane 1, and B, lane 2) and 70k-II (Fig. 3, A, lane 3, and B, lane 6) revealed a single band of 70 kDa. The purified 70k-I-2 phosphoprotein (Fig. 3, A, lane 2, and B, lane 4) on SDS-PAGE showed a major band at 70 kDa with small amounts of lower (55 kDa ∼ 69 kDa) and higher (>70 kDa) molecular mass bands. These lower and higher molecular mass bands as well as the major 70-kDa band of 70k-I-2 phosphoprotein were all susceptible to thrombin cleavage as described below and were reactive to the antibody against the 70k-I-1 (data not shown), indicating that they were fragments and aggregates of the major 70-kDa protein identical to 70k-I-1. SDS-PAGE of the 22k-I and 22k-II showed a single band as defined by Stains-all stained gels (Fig. 3C, lanes 1 and 2), whereas no bands were observed when CBB used for staining (data not shown). Under reducing conditions, the 22k-I and 22k-II migrated as 22 kDa on the 10% (Fig. 1A, inset) but as 14 kDa on the 15% SDS-PAGE gels (Fig. 3C, lanes 1 and 2). The addition of mercaptoethanol did not change the migration of the 70k-I-2 (Fig. 3B, lanes 2 and 4), whereas the 22k-I moved more slowly (∼15 kDa) under nonreducing conditions relative to reducing conditions (∼14 kDa) (Fig. 3C, lanes 1 and 3). Interestingly, on the nondenaturing PAGE gels, the 70k-I-1, 70k-I-2, and 70k-II proteins migrated at a position similar to BSP, and the purified 70k-II protein behaved like OPN (Fig. 4).
FIGURE 1.

DE-52 anion exchange chromatography of herring bone phosphoproteins. Bone HCl extract was applied to a DE-52 column (2.5 × 7.0 cm) in 7 m urea, 50 mm Tris-HCl, pH 7.4, as described in the text (A). An aliquot of each fraction was subjected to PAGE and visualized with Stains-all (inset). The fractions 70k-I/22k and 70k-II were concentrated by ultrafiltration over a Mr 5,000 cutoff membrane, brought to 7 m urea, 10 mm Tris-HCl, 50 mm sodium acetate, pH 4.0, and rechromatographed on another DE-52 column (0.9 × 5.2 cm) equilibrated with the same buffer (B and C).
FIGURE 2.

HPLC of the partially purified phosphoproteins. The fractions 70k-I-1/22k-I, 70k-I-2/22k-II, and 70k-II (Fig. 1, B and C) were concentrated by ultrafiltration over a Mr 10,000 cutoff membrane. The concentrated samples were treated with DTT and chromatographed on a TSK-3000 column as described in the text (A–C) followed by rechromatography on the same column (D–F). The filtrates were concentrated using a Mr 5,000 cutoff membrane, treated with DTT, and chromatographed on the TSK-3000 column (G and H) followed by RP-HPLC on a Delta-Pak C4 column (I and J).
FIGURE 4.
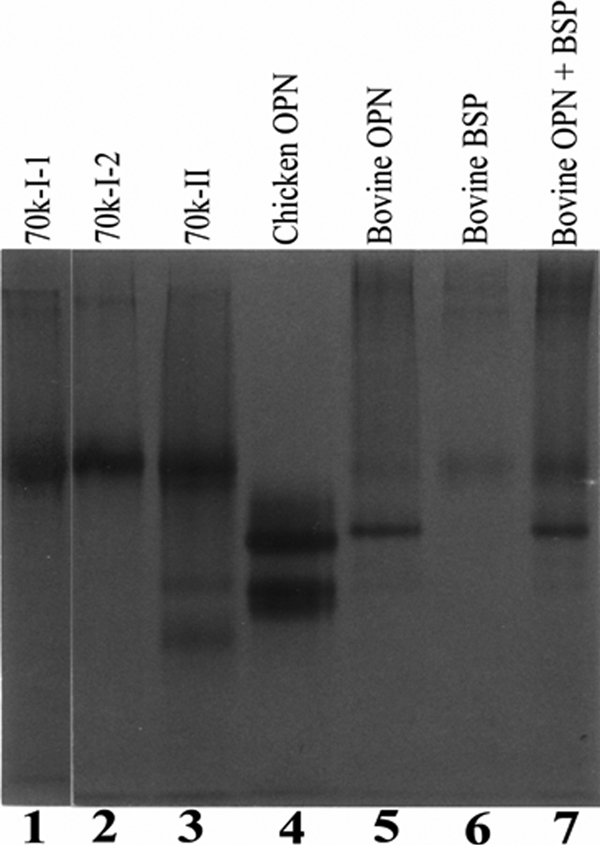
Nondenaturing PAGE of the purified phosphoproteins. The proteins were incubated in TBS at 37 °C for 1 h, subjected to nondenaturing PAGE using 10% gels, and visualized with Stains-all as described in the text. Lane 1, 70k-I-1 (1 μg); lane 2, 70k-I-2 (1 μg); lane 3, 70k-II (1 μg); lane 4, chicken OPN (2 μg); lane 5, bovine OPN (1 μg); lane 6, bovine BSP (1 μg); lane 7, bovine OPN (1 μg) and BSP (1 μg). Note that 70k-I-1, 70k-I-2, and 70k-II migrated like BSP, whereas two additional minor bands were observed in the 70K-II, acting like OPN.
Amino Acid Compositions and N-terminal Amino Acid Sequence Analyses
The amino acid compositions of the purified proteins are shown in Table 1. The 70-kDa phosphoprotein forms 70k-I-1, 70k-I-2, and 70k-II all have almost identical amino acid composition and show similarities to those of OPN and bone acidic glycoprotein 75 (another bone phosphoprotein of 75 kDa) (54). The amino acid compositions of the 22k-I and 22k-II are almost the same, with strikingly high levels of Ala (∼250 residues/1000 residues) and Ser (∼380 residues/1000 residues). Importantly and surprisingly, most of the Ser residues of this fish phosphoprotein were phosphorylated (∼270 residues/1000 residues). In contrast, 70k-I-1, 70k-I-2, and 70k-II contained ∼35 Ser(P)/1000 residues, almost equal to that of OPN, one of the most highly phosphorylated proteins in bone (Table 2). Both neutral sugars and sialic acid were detected in the 22- and 70-kDa phosphoproteins (Table 2).
TABLE 1.
Amino acid compositions of the purified proteins
| 70k-I-1 | 70k-I-2 | 70k-II | OPNa | BAG-75b | 22k-I | 22k-II | PPc | |
|---|---|---|---|---|---|---|---|---|
| Asxd | 118g | 110 | 120 | 180 | 117 | 153 | 156 | 447 |
| Thr | 77 | 68 | 74 | 42 | 47 | 35 | 32 | 6 |
| Ser | 175 | 168 | 185 | 131 | 112 | 388 | 387 | 484 |
| Glxe | 166 | 149 | 171 | 183 | 175 | 80 | 74 | 14 |
| Pro | 57 | 70 | 55 | 55 | 49 | 36 | 47 | 6 |
| Gly | 73 | 107 | 79 | 40 | 103 | 5 | 4 | 24 |
| Ala | 116 | 96 | 104 | 67 | 79 | 246 | 245 | 6 |
| Cysf | 3 | 4 | 4 | 1 | 0 | 0 | Trace | |
| Val | 47 | 45 | 46 | 44 | 32 | 25 | 24 | 3 |
| Met | 9 | 18 | 7 | 9 | 9 | 18 | 17 | Trace |
| Ile | 15 | 14 | 15 | 20 | 13 | 4 | 4 | 3 |
| Leu | 44 | 46 | 48 | 55 | 40 | 3 | 3 | 4 |
| Tyrf | 7 | 11 | 1 | 20 | 29 | Trace | Trace | 2 |
| Phe | 16 | 16 | 16 | 29 | 23 | Trace | Trace | 2 |
| His | 15 | 18 | 20 | 43 | 29 | Trace | Trace | 4 |
| Lys | 41 | 34 | 33 | 49 | 40 | 2 | 1 | 40 |
| Arg | 22 | 27 | 23 | 32 | 42 | 7 | 6 | 3 |
TABLE 2.
Contents of phosphate and carbohydrates in the purified proteins
The N-terminal sequences of the 70 k-I-1 and the 70k-I-2 phosphoproteins (Table 3) were identical: NPIMA(M)ETTS(M)DSKVNPLL. When the tryptic internal peptides of the 70k-I-2 protein were separated, the numbered major ones were screened, and several N-terminal were sequenced (Fig. 5 and Table 3). No sequences similar to these N-terminal and internal sequences were found in the nonredundant GenBankTM CDS translations + Protein Data Bank + SwissProt + Spupdate + PIR database when the computation was performed at the National Center for Biotechnology Information using the Blast network service. When the FASTA program in the Genetics Computer Group's Wisconsin Sequence Analysis Package was performed, some low homology sequences were defined toward a single internal peptide in Swiss-Prot database; however, no combination of the internal peptides and the N-terminal sequences could be matched to a known protein. The second amino acid residue within the N-terminal sequence of the 70k-I-1 and the 70k-I-2 proteins was identical to that of OPN proline. This is a residue that is conserved for OPN across all known animal species from which OPN has been isolated, including chicken, mouse, rat, cow, pig, and human. Similarly, several internal peptides of the 22k-I were obtained and sequenced (Fig. 5 and Table 3). These sequences and the N-terminal sequence of the intact 22k-I were overlapped, and the assembly of the sequences led to a 15-residue N-terminal sequence. The sequence analysis indicated no similar sequence in the databanks for this smaller fish phosphoprotein.
TABLE 3.
N-terminal sequences of the purified proteins and their tryptic internal peptides
| Sequence | |
|---|---|
| 70k-I-1 | |
| N-terminal | XP(A)XMA(Y)ETT(A) |
| 70k-I-2 | |
| N-terminal | NPIMA(M)ETTS(M)DSKVNPLL(S) |
| Peptide 31 | D(S)D(S)D(S)RGGKENLR |
| Peptide 41 | VYAVK |
| FH(C)G(P)ERGP | |
| Peptide 53 | MGYXDYK |
| Peptide 54 | S(K)A(G)P(A)TFDDGK(A)NNVEK |
| Peptide 91 | FNLVETGVTR |
| 22k-I | |
| N-terminal | NQDMAMEASS |
| Peptide 21 | MEASSDPEAA |
| NQDMAMEASSDPE | |
| Peptide 33 | MAMEASSDPEA |
| EDMXMEASSDPE | |
| Assembly | NQDMAMEASSDPEAA |
FIGURE 5.

RP-HPLC of the tryptic peptides of the purified proteins. The purified 70k-I-2 (A) and 22k-I (B) were subjected to PAGE, the major bands were in-gel trypsin-digested, and the peptides were extracted followed by RP-HPLC on a C18 column as described in the text.
Mass Spectrometric Analysis of Purified 70- and 22-kDa Phosphoproteins and Crude HCL Bone Extract Using LC-ESI-MS/MS Approach
MS analysis of the tryptic samples of purified 70- and 22-kDa phosphoproteins did not match to any protein in the zebrafish database or general fish databases. Similar analysis of the crude HCL herring bone extract, on the other hand, identified 15 proteins with more than two peptides in the zebrafish database. Interestingly within these identified proteins there were six different collagens when the database search included hydroxylproline as an additional peptide modification. As expected the dominant collagen was collagen type I with 12 different identified peptides for the alpha 1 chain and six different peptides for the alpha 2 chain.
Thrombin Cleavage
Unlike other bone ECM proteins such as osteocalcin, matrix Gla protein, osteonectin, decorin, biglycan, etc., OPN can be cleaved by thrombin. When 70k-I-1, 70k-I-2, and 70k-II were incubated with thrombin, they were all cleaved into five fragments: 32, 34, 38, 45, and 55 kDa (Fig. 3B, lanes 3, 5, and 7). The lower (<70 kDa) and higher (>70 kDa) Mr contaminants in the purified 70k-I-2 fraction disappeared (Fig. 3B, lanes 4 and 5). Long time digestion of 70k-I-1 led to decreasing levels of the 55-kDa fragment and the disappearance of the 45 kDa (Fig. 3B, lanes 8–10).
Tissue Distribution
The tissue distribution and concentrations of the 70-kDa phosphoprotein was determined by enzyme-linked immunosorbent assay and indicated this protein to be predominantly present in rib/back bone (88 μg/100 mg of total extracted proteins) and calcified scale (60 μg/100 mg of total extracted proteins). Other tissues, however, such as brain, roe, liver, muscle, skin, and stomach were shown to contain very small amounts (1–4 μg/100 mg of total extracted proteins). In contrast, the 22-kDa protein was detected in bone at the comparatively high level of 26 μg/100 mg of total extracted proteins only in bone tissue.
Immunolocalization
There was diffusely positive immunostaining for both the 70- and 22-kDa phosphoproteins throughout the sparsely cellular matrix of intramuscular bone, whereas the control was negative (Fig. 6). There was no immunoreactivity for the 70- or 22-kDa protein in the tip of intramuscular bone, a nonmineralized region.
FIGURE 6.

Immunolocalization of the 70 k phosphoprotein in intramuscular bone matrix. Immunolocalization of the 70 k and 22 k phosphoproteins in herring intramuscular bone is shown. A, hematoxylin/eosin staining. B, von Kossa/methyl green staining. C, immunostaining with preimmune serum (control). D, immunostaining for the 70 k phosphoprotein. E, immunostaining for the 22 k phosphoprotein. Longitudinal sections are ×200. oc, osteocyte.
DISCUSSION
Two major glycosylated phosphoproteins of 70 and 22 kDa were purified from herring bone by acid extraction followed by a series of chromatographic procedures comparable with those of well known avian and mammalian major bone phosphoproteins such as OPN and BSP (4–7). The 70-kDa fish phosphoprotein had many characteristics similar to mammalian and avian bone ECM phosphoproteins in terms of amino acid composition. This protein possessed high levels of both acidic residues such as Asp and Glu and hydroxyl-containing amino acids, i.e. Ser and Thr residues. Similarly, it contained high levels of sugars and sialic acid and high levels of covalently bound phosphates (∼35 residues/1000 residues). However, the presence of cysteine residues sets this protein apart from other known phosphoproteins such as OPN and BSP. The 22-kDa fish phosphoprotein, on the other hand, was found to be of quite distinct composition in its inclusion of uncharacteristically high levels of Ser and Ser(P) residues (∼270 Ser(P) residues/1000 residues). This remarkable number of Ser(P) residues determined by partial acid hydrolysis is not corrected for possible loss of phosphate groups by its cleavage from the serine residue during such analysis. In essence the actual Ser(P) residues/1000 residues of this protein is likely to be even higher than 270, but unfortunately any correction for the loss of Ser(P) during partial acid hydrolysis would still be only an estimate because there are no ideal standards that can be used to establish a perfect correction factor. Another unique component of this protein is the presence of very high levels of Ala residues. This low molecular mass 22-kDa protein was associated with the 70-kDa protein during purification by chromatography on DE-52 column (Fig. 1). These two proteins were separated by the use of ultrafilter membrane with an Mr 10,000 cutoff, and the filtrates contained the 22-kDa protein, indicating its actual molecular mass being <10,000 Da. The apparent anomalous mobility on SDS-PAGE is related to its post-translational modifications. Similar to other known glycosylated phosphoproteins of bone, the fish phosphoprotein stained blue with Stains-all and showed no staining with CBB. The difference in the migration on SDS-PAGE gels under reducing and nonreducing conditions indicates that the molecule contains intramolecular disulfide bond(s) (Fig. 3C).
No significant sequence similarities were observed by directly blasting the N-terminal and internal sequences of the 70- or 22-kDa phosphoprotein. The presence of a family of genes of the 22-kDa protein is suggested by sequence analysis of the putative cDNAs obtained by reverse transcription of herring rib bone total RNA using an oligo(dT) adapter. This was carried out by polymerase chain reaction using a degenerate primer based on the N-terminal sequence of the 22-kDa protein and a primer corresponding to the adapter sequence, and phagemid cloning, although the cDNA sequence whose 5′ end was identical to that derived from the N-terminal sequence has not yet been determined (data not shown). The susceptibility of the 70-kDa fish phosphoprotein to specific thrombin cleavage suggests its OPN type characteristic, because the thrombin cleavage site of OPN YGLRS is found to be conserved between species (YGLKSRS for bovine and YGFRA for chicken). A comparison of the OPN amino acid sequences for the mouse, rat, cow, pig, and human species reveals that ∼40% of their positions are identical (55). When the OPN sequence from a lower species such as chicken is added to the analysis, the degree of identity is only ∼19%. However, it is noteworthy that the N-terminal second and third amino acids, Pro-Val, of OPN are conserved within the six animal species above. Consistent with this, the herring fish 70-kDa protein and OPN from fish species such as trout, zebrafish, and channel catfish also contain Pro residue as a second amino acid. Recently, comparison of OPN sequences for trout versus tetrapod and zebrafish showed low amino acid sequence homologies, ∼20 and ∼35%, respectively (56, 57). The data generated by MS analysis of the purified herring fish phosphoproteins and the use of the available zebrafish database for protein identification did not provide positive identification. This was most likely due to a lack of full genomic protein sequence database for the herring fish and low protein sequence homologies among fish species. Importantly, a similar analysis of the crude herring bone HCL extract by MS analysis using the zebrafish database did identify several proteins including dominant collagen type I. This suggests that although noncollagenous proteins among the fish species have low homologies, collagens appear to retain significant sequence homologies.
The high phosphoserine content of the 22-kDa fish phosphoprotein is reminiscent of phosphophoryn, a cleavage product of dentin sialophosphoprotein belonging to the same small integrin binding ligand N-linked glycoprotein family of OPN and BSP, in which phosphorylation is important for the deposition of calcium phosphate crystals and that the acidic carboxylate groups alone are not sufficient (46, 58). The generation of dentin sialophosphoprotein null mice resulted in tooth defects similar to human dentinogenesis imperfecta III (59). The post-translational modifications of bone and dentin ECM proteins have been found to impact their biological functions. These range from effects on hydroxyapatite nucleation and accumulation of calcium phosphate crystals (1, 7, 16–19, 60–62) to cell attachment and behavior (36, 42, 63). Other studies have indicated that post-translational modifications influence in vitro osteoclast activity and bone resorption (40, 42, 64) and osteoblast differentiation (37).
Tissue distribution of the 70-kDa fish phosphoprotein showed its high levels in herring bone and scale with small amounts detected in soft tissues such as roe, brain, liver, skin, muscle, and stomach. This is somewhat similar to OPN in avian and mammalian species, where most of the known bone noncollagenous ECM proteins such as osteonectin, matrix Gla protein, decorin, biglycan, and fibronectin are also found in other connective tissues. These latter proteins may play functional roles in bone that are different from those in other tissues. This may, with some proteins, relate to different post-translational modifications leading to alterations in their observed biological functions. The 70-kDa protein was estimated at 50 μg/g of wet weight of bone, a concentration different from those of OPN and BSP at 0.3 and 1.5 mg/g, respectively, in bovine and rat bones (3, 66), suggesting lower remodeling of the fish bone. Immunohistological analysis led to localization of the 70-kDa phosphoprotein as well as the 22-kDa phosphoprotein to calcified bone matrix (Fig. 6). As expected, the 70-kDa phosphoprotein was detected in the calcified acellular scale and at low levels in soft tissues. In contrast to the 70-kDa protein, the 22-kDa protein was only detected in bone and not in the other tissues examined. However, this low Mr fish protein with very high levels of phosphorylation was absent in the calcified scale, unlike the bone/calcified tissue-specific osteocalcin, which is identified in fish scale as well as in fish bone (67).
Overall, the two major glycosylated phosphoproteins of fish bone represent proteins analogous to those already defined for mammalian and avian vertebrates and those for invertebrates. To date, the evidence accumulating on the biological functions of ECM phosphoproteins have led to the proposal that these proteins may represent evolutionary conservation with similar biological functions (45, 68, 69). The fish bone ECM phosphoproteins defined in this study provide additional evidence in support of the common fundamental mechanistic bases for biomineralization throughout vertebrate species and evolution. Indeed, collagen has remained as one of the major components of all vertebrate bone throughout species. The identification of collagen fragments, albeit short peptides, that survived in ∼200,000-year-old mastodon (Mammut americanum) bone fossils and perhaps even in ∼70 million-year-old dinosaur bone fossils from Tyrannosaurus rex (65) suggests that phosphoproteins along with collagen may have served the same biological and mechanistic function in vertebrate biomineralization processes in both ancient and contemporary times.
Acknowledgments
We thank Thelma Gows, Dee Adams, and Thelma Young for bone powder preparation; Marie Torres for amino acid composition analysis; and Phillips Brady et al. at the Massachusetts Division of Marine Fisheries for fish collection. We also acknowledge tryptic internal peptide isolation and amino acid sequencing work provided by Dr. W. S. Lane et al. at the Microchemistry Facility of Harvard University.
This work was supported, in whole or in part, by National Institutes of Health Grants AG17969-0H (to E. S.), R01 AG14701-16, and 2R01 AG14701-17A2. This work was also supported by a grant from the Peabody Foundation, Inc. (to M. J. G.).
- ECM
- extracellular matrix
- OPN
- osteopontin
- BSP
- bone sialoprotein
- RP
- reverse phase
- CBB
- Coomassie Brilliant Blue R-250
- LC-ESI-MS/MS
- liquid chromatography and electrospray ionization tandem mass spectrometry.
REFERENCES
- 1.Glimcher M. J. (1989) Anat. Rec. 224, 139–153 [DOI] [PubMed] [Google Scholar]
- 2.Fisher L. W., Whitson S. W., Avioli L. V., Termine J. D. (1983) J. Biol. Chem. 258, 12723–12727 [PubMed] [Google Scholar]
- 3.Franzén A., Heinegård D. (1985) Biochem. J. 232, 715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salih E., Ashkar S., Gerstenfeld L. C., Glimcher M. J. (1996) J. Bone Miner. Res. 11, 1461–1473 [DOI] [PubMed] [Google Scholar]
- 5.Salih E., Zhou H. Y., Glimcher M. J. (1996) J. Biol. Chem. 271, 16897–16905 [DOI] [PubMed] [Google Scholar]
- 6.Salih E., Ashkar S., Gerstenfeld L. C., Glimcher M. J. (1997) J. Biol. Chem. 272, 13966–13973 [DOI] [PubMed] [Google Scholar]
- 7.Salih E., Wang J., Mah J., Fluckiger R. (2002) Biochem. J. 364, 465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glimcher M. J., Kossiva D., Rouosse A. (1979) Calcif. Tissue Int. 27, 187–191 [DOI] [PubMed] [Google Scholar]
- 9.Veis A. (1985) in The Chemistry and Biology of Mineralized Tissues, (Butler W. T. ed) pp. 170–176, Ebsco Media, Inc., Birmingham, AL [Google Scholar]
- 10.Giachelli C. M., Bae N., Almeida M., Denhardt D. T., Alpers C. E., Schwartz S. M. (1993) J. Clin. Invest. 92, 1686–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okada A., Nomura S., Saeki Y., Higashibata Y., Hamamoto S., Hirose M., Itoh Y., Yasui T., Tozawa K., Kohri K. (2008) J. Bone Miner. Res. 23, 1629–1637 [DOI] [PubMed] [Google Scholar]
- 12.Kido J., Kasahara C., Ohishi K., Nishikawa S., Ishida H., Yamashita K., Kitamura S., Kohri K., Nagata T. (1995) Arch. Oral Biol. 40, 967–972 [DOI] [PubMed] [Google Scholar]
- 13.Bellahcène A., Castronovo V. (1997) Bull. Cancer 84, 17–24 [PubMed] [Google Scholar]
- 14.Midura K. J., McQuilan D. J., Benham K. J., Fisher L. W., Hascall V. C. (1990) J. Biol. Chem. 265, 5285–5291 [PubMed] [Google Scholar]
- 15.Wang J., Glimcher M. J., Mah J., Zhou H. Y., Salih E. (1998) Bone 22, 621–628 [DOI] [PubMed] [Google Scholar]
- 16.Endo A., Glimcher M. J. (1989) Connect. Tissue Res. 21, 179–196 [DOI] [PubMed] [Google Scholar]
- 17.Hunter G. K., Goldberg H. A. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 8562–8565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boskey A. L., Maresca M., Ullrich W., Doty S. B., Butler W. T., Prince C. W. (1993) Bone Miner. 22, 147–159 [DOI] [PubMed] [Google Scholar]
- 19.Gericke A., Qin C., Spevak L., Fujimoto Y., Butler W. T., Sørensen E. S., Boskey A. L. (2005) Calcif. Tissue Int. 77, 45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gordon J. A., Tye C. E., Sampaio A. V., Underhill T. M., Hunter G. K., Goldberg H. A. (2007) Bone 41, 462–473 [DOI] [PubMed] [Google Scholar]
- 21.Stubbs J. T., 3rd, Mintz K. P., Eanes E. D., Torchia D. A., Fisher L. W. (1997) J. Bone Miner. Res. 12, 1210–1222 [DOI] [PubMed] [Google Scholar]
- 22.Buttler W. T., Ridall A. L., McKee M. D. (1996) In Osteopontin: Principles of Bone Biology (Bilezikian J. P., Raisz L. G., Rodan G. A. eds) pp. 167–181, Academic Press, San Diego, CA [Google Scholar]
- 23.Denhardt D. T., Noda M. (1998) J. Cell. Biochem. Suppl. 30–31, 92–102 [PubMed] [Google Scholar]
- 24.Chambers T. J., Fuller K., Darby J. A., Pringle J. A., Horton M. A. (1986) Bone Miner. 1, 127–135 [PubMed] [Google Scholar]
- 25.Davies J., Warwick J., Totty N., Philp R., Helfrich M., Horton M. A. (1989) J. Cell Biol. 109, 1817–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reinholt F. P., Hultenby K., Oldberg A., Heinegård D. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 4473–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ross F. P., Chappel J., Alvarez J. I., Sander D., Butler W. T., Farach-Carson M. C., Mintz K. A., Robey P. G., Teitelbaum S. L., Cheresh D. A. (1993) J. Biol. Chem. 268, 9901–9907 [PubMed] [Google Scholar]
- 28.Senger D. R., Perruzzi C. A. (1996) Biochim. Biophys. Acta 1314, 13–24 [DOI] [PubMed] [Google Scholar]
- 29.O'Regan A. W., Chupp G. L., Lowry J. A., Goetschkes M., Mulligan N., Berman J. S. (1999) J. Immunol. 162, 1024–1031 [PubMed] [Google Scholar]
- 30.Helfrich M. H., Nesbitt S. A., Dorey E. L., Horton M. A. (1992) J. Bone Miner. Res. 7, 335–343 [DOI] [PubMed] [Google Scholar]
- 31.Horton M. A., Nesbit M. A., Helfrich M. H. (1995) Ann. N.Y. Acad. Sci. 760, 190–200 [DOI] [PubMed] [Google Scholar]
- 32.Väänänen H. K., Horton M. A. (1995) J. Cell Sci. 108, 2729–2732 [DOI] [PubMed] [Google Scholar]
- 33.Bayless K. J., Meininger G. A., Scholtz J. M., Davis G. E. (1998) J. Cell Sci. 111, 1165–1174 [DOI] [PubMed] [Google Scholar]
- 34.Yokosaki Y., Matsuura N., Sasaki T., Murakami I., Schneider H., Higashiyama S., Saitoh Y., Yamakido M., Taooka Y., Sheppard D. (1999) J. Biol. Chem. 274, 36328–36334 [DOI] [PubMed] [Google Scholar]
- 35.Ek-Rylander B., Flores M., Wendel M., Heinegård D., Andersson G. (1994) J. Biol. Chem. 269, 14853–14856 [PubMed] [Google Scholar]
- 36.Wang J., Zhou H. Y., Salih E., Xu L., Wunderlich L., Gu X., Hofstaetter J. G., Torres M., Glimcher M. J. (2006) Calcif. Tissue Int. 79, 179–189 [DOI] [PubMed] [Google Scholar]
- 37.Zhou H. Y., Takita H., Fujisawa R., Mizuno M., Kuboki Y. (1995) Calcif. Tissue Int. 56, 403–407 [DOI] [PubMed] [Google Scholar]
- 38.Cooper L. F., Yliheikkilä P. K., Felton D. A., Whitson S. W. (1998) J. Bone Miner. Res. 13, 620–632 [DOI] [PubMed] [Google Scholar]
- 39.Mizuno M., Imai T., Fujisawa R., Tani H., Kuboki Y. (2000) Calcif. Tissue Int. 66, 388–396 [DOI] [PubMed] [Google Scholar]
- 40.Razzouk S., Brunn J. C., Qin C., Tye C. E., Goldberg H. A., Butler W. T. (2002) Bone 30, 40–47 [DOI] [PubMed] [Google Scholar]
- 41.Ihara H., Denhardt D. T., Furuya K., Yamashita T., Muguruma Y., Tsuji K., Hruska K. A., Higashio K., Enomoto S., Nifuji A., Rittling S. R., Noda M. (2001) J. Biol. Chem. 276, 13065–13071 [DOI] [PubMed] [Google Scholar]
- 42.Raynal C., Delmas P. D., Chenu C. (1996) Endocrinology 137, 2347–2354 [DOI] [PubMed] [Google Scholar]
- 43.Flores M. E., Norgård M., Heinegård D., Reinholt F. P., Andersson G. (1992) Exp. Cell Res. 201, 526–530 [DOI] [PubMed] [Google Scholar]
- 44.Malaval L., Wade-Guéye N. M., Boudiffa M., Fei J., Zirngibl R., Chen F., Laroche N., Roux J. P., Burt-Pichat B., Duboeuf F., Boivin G., Jurdic P., Lafage-Proust M. H., Amédée J., Vico L., Rossant J., Aubin J. E. (2008) J. Exp. Med. 205, 1145–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fisher L. W., Torchia D. A., Fohr B., Young M. F., Fedarko N. S. (2001) Biochem. Biophys. Res. Commun. 280, 460–465 [DOI] [PubMed] [Google Scholar]
- 46.Alvares K., Dixit S. N., Lux E., Veis A. (2009) J. Biol. Chem. 284, 26149–26160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mann K., Poustka A. J., Mann M. (2010) Proteome Science 8, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glimcher M. J. (1959) Rev. Mod. Phys. 31, 359–393 [Google Scholar]
- 49.Lee D. D., Glimcher M. J. (1991) J. Mol. Biol. 217, 487–501 [DOI] [PubMed] [Google Scholar]
- 50.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 51.Zhou H. Y., Salih E., Glimcher M. J. (1998) Biochem. J. 330, 1423–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rennard S. I., Berg R., Martin G. R., Foidart J. M., Robey P. G. (1980) Anal. Biochem. 104, 205–214 [DOI] [PubMed] [Google Scholar]
- 53.Von Kossa J. (1901) Beitr. Path. Anat. 29, 163 [Google Scholar]
- 54.Gorski J. P., Shimizu K. (1988) J. Biol. Chem. 263, 15938–15945 [PubMed] [Google Scholar]
- 55.Fisher L. W. (1992) in Chemistry and Biology of Mineralized Tissues (Slavkin H., Price P. eds) pp. 177–186, Elsevier, New York [Google Scholar]
- 56.Bobe J., Goetz F. W. (2001) FEBS Lett. 489, 119–124 [DOI] [PubMed] [Google Scholar]
- 57.Kawasaki K., Suzuki T., Weiss K. M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 11356–11361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saito T., Arsenault A. L., Yamauchi M., Kuboki Y., Crenshaw M. A. (1997) Bone 21, 305–311 [DOI] [PubMed] [Google Scholar]
- 59.Sreenath T., Thyagarajan T., Hall B., Longenecker G., D'Souza R., Hong S., Wright J. T., MacDougall M., Sauk J., Kulkarni A. B. (2003) J. Biol. Chem. 278, 24874–24880 [DOI] [PubMed] [Google Scholar]
- 60.Hunter G. K., Goldberg H. A. (1994) Biochem. J. 302, 175–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hunter G. K., Kyle C. L., Goldberg H. A. (1994) Biochem. J. 300, 723–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tye C. E., Rattray K. R., Warner K. J., Gordon J. A., Sodek J., Hunter G. K., Goldberg H. A. (2003) J. Biol. Chem. 278, 7949–7955 [DOI] [PubMed] [Google Scholar]
- 63.Shanmugam V., Chackalaparampil I., Kundu G. C., Mukherjee A. B., Mukherjee B. B. (1997) Biochemistry 36, 5729–5738 [DOI] [PubMed] [Google Scholar]
- 64.Curtin P., McHugh K. P., Zhou H. Y., Flückiger R., Goldhaber P., Oppenheim F. G., Salih E. (2009) Biochemistry 48, 6876–6886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Asara J. M., Schweitzer M. H., Freimark L. M., Phillips M., Cantley L. C. (2007) Science 316, 280–285 [DOI] [PubMed] [Google Scholar]
- 66.Fujisawa R., Butler W. T., Brunn J. C., Zhou H. Y., Kuboki Y. (1993) J. Dent. Res. 72, 1222–1226 [DOI] [PubMed] [Google Scholar]
- 67.Nishimoto S. K., Araki N., Robinson F. D., Waite J. H. (1992) J. Biol. Chem. 267, 11600–11605 [PubMed] [Google Scholar]
- 68.Simkiss K., Wilbur K. (1989) Cell and Mineral Deposition, pp. 1–337, Academic Press, San Diego [Google Scholar]
- 69.Lowenstam H. A., Weiner S. (1989) On Biomineralization, pp. 1–324, Oxford University Press, New York [Google Scholar]