

Abstract
A total synthesis of the oxo-polyene macrolide (+)-roxaticin is achieved in 20 steps from 1,3-propanediol. In this approach, nine of ten C-C bonds formed in the longest linear sequence are made via metal catalysis, including 7 C-C bonds formed via iridium catalyzed alcohol C-C coupling. Notably, the present synthesis, which represents the most concise preparation of any oxo-polyene macrolide reported to date, is achieved in the absence of chiral reagents, chiral auxiliaries with minimal use of premetallated C-nucleophiles.
Polyketide natural products represent a broad class of secondary metabolites used extensively in human medicine.1,2 Approximately 20% of the top-selling small molecule drugs are polyketides,2,3 and it is estimated that polyketides are five times more likely to possess drug activity compared to other natural product families.3 As investigations into polyketide biosynthesis4 and chemical biology continue to uncover important biochemical pathways and novel therapeutic targets, the design of synthetic5 and bio-engineered6 methods for polyketide construction remains at the forefront of research.
The complex issues of stereoselectivity posed by polyketides are most often addressed through the use of chiral auxiliaries, chiral reagents and premetallated nucleophiles.5 Although such methods are highly effective, their use often mandates multi-stage preactivation and excessive byproduct generation. Inspired by the atom-economy of catalytic hydrogenation, we have developed a broad family of enantioselective “C-C bond forming hydrogenations,”7 which provide an alternative to stoichiometric organometallic reagents in certain carbonyl and imine additions. Here, we showcase the utility of this methodology in a total synthesis of the oxo-polyene macrolide (+)-roxaticin.8,9d–h,10
Our approach takes advantage of carbonyl allylation7f,11a–d and crotylation7f,11e protocols recently developed in our laboratory, wherein primary alcohol dehydrogenation concurrently triggers aldehyde formation and reductive generation of allyliridium nucleophiles, enabling asymmetric carbonyl allylation and crotylation directly from the alcohol oxidation level. Iterative two-directional carbonyl allylation of 1,3-diols11c,d,12 was envisioned to deliver the requisite C2-symmetric 1,3-polyol substructure spanning C14–C28. Elaboration of the protected 1,3-polyol 3 to (+)-roxaticin requires differential elongation of the homotopic diol termini, which is potentially achieved via sequential dehydration-cross-metathesis at C28 and direct carbonyl crotylation from the alcohol oxidation level at C14. Installation of the oxo-pentaene fragment takes advantage of a sequence involving cross-metathesis-Horner-Wadsworth-Emmons olefination. Subsequent macrocyclization and global deprotection would deliver (+)-roxaticin (Scheme 1).
Scheme 1.

Representative oxo-polyene macrolides and retrosynthetic analysis of the (+) roxaticin.a
aSee supporting information for a graphical summary of prior syntheses.
Double C-allylation of 1,3-propanediol was accomplished as previously described using the ortho-cyclometallated iridium C,O-benzoate generated in situ from [Ir(cod)Cl]2, (R)-Cl,MeO-BIPHEP, 4-chloro-3-nitrobenzoic acid and allyl acetate (eqn. 1).11c,d The C2-symmetric product of double allylation 1 is obtained in 70% yield and > 99% ee, as the minor enantiomer of the mono-adduct is converted to the meso-diastereomer of 1.13,14 On larger scale (20 mmol), to mitigate cost, double allylation reactions were conducted using the corresponding (R)-BINAP complex, which provides 1 in 51% yield with equivalent levels of absolute stereocontrol.
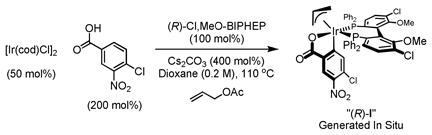 |
(eqn. 1) |
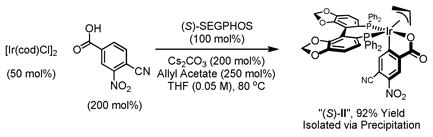 |
(eqn. 2) |
Conversion of 1 to the acetonide followed by ozonolysis of the olefinic termini provides the homologous diol 2, constituting “one iteration” of two-directional carbonyl allylation from the alcohol oxidation level. In principle, subsequent iterations of two-directional carbonyl allylation could be performed from the diol or dialdehyde oxidation level. However, the β-alkoxy aldehydes were found to be quite unstable, whereas diol intermediates, such as 2, are highly tractable and may be stored for long periods of time under ambient conditions without decomposition. Therefore, in subsequent iterations of two-directional carbonyl allylation, ozonolysis reactions were quenched with NaBH4 to furnish the homologous diols.15 In the following two double allylation reactions, complete levels of catalyst-directed diastereoselectivity were observed, providing rapid access to the acetonide-protected C2-symmetric 1,3-polyol 3 as a single diastereomer. Thus, tris-acetonide 3, which possesses 6 stereocenters, is prepared in only 9 steps from 1,3-propanediol (Scheme 2).
Scheme 2.

Total synthesis of the oxo-polyene macrolide (+)-roxaticin via C-C bond forming transfer hydrogenation.a
aYields are of material isolated by silica gel chromatography and represent the average of two runs. Enantiomeric excess of 1 was determined by chiral stationary phase HPLC analysis. See Supporting Information for preparation of homoallylic ether A and other experimental details.
Differential elongation of the diol termini of tris-acetonide 3 begins with Grieco’s two-step method for primary alcohol dehydration.16 Because the diol termini of 3 are homotopic, the product appears as a component of a statistical mixture. The resulting allylic ether was exposed to the previously prepared homoallylic ether A17 in the presence of the second generation Hoveyda-Grubbs catalyst to furnish the product of cross-metathesis 5 in 53% yield with complete (E)-selectivity, as determined by 1H NMR.18 Direct carbonyl crotylation from the alcohol oxidation level at C14 using the preformed iridium C,O-benzoate complex (S)-II (eqn. 2) delivers the desired adduct 6a in 85% yield with 14:1 anti-diastereoselectivity.
With homoallylic alcohol 6a in hand, installation of the polyene substructure was undertaken. The unprotected homoallylic alcohol 6a was subjected to cross-metathesis with acrolein to deliver the corresponding enal 7a in 68% yield with complete (E)-selectivity, as determined by 1H NMR.19 Subsequent protection of the C14 alcohol to provide the triethylsilyl (TES) ether 7b was followed by oxidative removal of the para-methoxybenzyl ether to furnish hydroxy enal 7c. This specific sequence of functional group interconversions was essential as the TES-ether 6b does not participate in cross-metathesis, presumably due to the sterically demanding environment at the homoallylic olefin. Additionally, selective protection of the C14 and C30 diol 6c was not possible under a variety of conditions. Finally, attempted macrolactonization of 7d, which lacks a C14 hydroxyl protecting group, was unsuccessful.
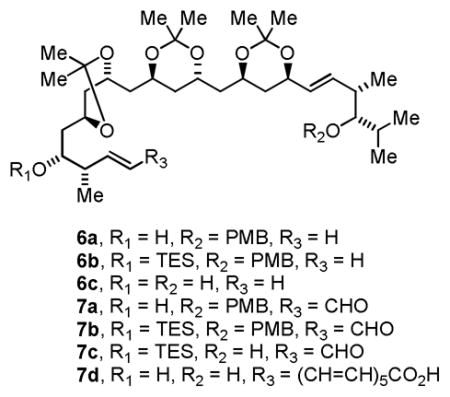
Elaboration of hydroxy enal 7c to (+)-roxaticin was accomplished as follows. Exposure of 7c to the previously reported Horner-Wadsworth-Emmons reagent, EtO2C(CH=CH)3CH2PO(OEt)2,20 delivered the polyunsaturated ester in 61% yield. This transformation, which involves LHMDS mediated lithium enolate generation, represents the sole use of a premetallated C-nucleophile in the longest linear sequence. Ester hydrolysis followed by Yamaguchi lactonization21 and global deprotection, as practiced in prior syntheses by Mori9e–g and Evans,9h delivers (+)-roxaticin in 31% yield over the three-step sequence. The last step of the longest linear sequence, which involves hydrolysis of three acetonide moieties and a TES-ether, was particularly daunting, as in the Evan’s synthesis9h acetonide protecting groups could not be removed and were abandoned for cyclopentylidene ketals, which are hydrolytically more labile. Hence, in the present macrolactonization-global deprotection sequence, it is possible that even higher efficiencies would be observed were cyclopentylidene ketals employed as protecting groups.
In summary, in this initial application of our “C-C bond forming hydrogenation” methodology, (+)-roxaticin is prepared from 1,3-propane diol in 20 steps (longest linear sequence), with a total number of 29 manipulations. Notably, 9 of 10 C-C bonds formed in the longest linear sequence are made via metal catalysis (7 hydrogen-mediated C-C couplings, 2 cross-metatheses) in processes that exploit “native functionality” (alcohols, olefins). This approach circumvents additional manipulations associated with the use of non-native functional groups or substructures to mediate bond construction, as evident in the use of chiral auxiliaries, and minimizes (re)functionalizations, especially redox manipulations. Indeed, as demonstrated in carbonyl additions from the alcohol oxidation level, as in the conversion of 5 to 6a, the union of oxidation-construction events allows to one to bypass discrete alcohol oxidation while gaining access to more tractable synthetic intermediates in the form of alcohols. These features are in line with longstanding ideals of synthetic efficiency.22,23
Supplementary Material
Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM069445), the American Chemical Society Green Chemistry Institute Pharmaceutical Roundtable and the University of Texas at Austin, Center for Green Chemistry and Catalysis. Dr. Yasunori Ino, Dr. Wataru Kuriyama and Dr. Taichiro Touge of Takasago are thanked for the generous donation of SEGPHOS. The Higher Education Commission of Pakistan is acknowledged for graduate student fellowship support (AH). The Korea Research Foundation (KRF-2007-356-E00037) is acknowledged for postdoctoral fellowship support (ISK).
Footnotes
Supporting Information Available: Detailed experimental procedures, spectral data for new compounds, including scanned images of 1H and 13C NMR spectra. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews on polyketide natural products, see: O’Hagan D. The Polyketide Metabolites. Ellis Horwood; Chichester, U.K: 1991. Rimando AM, Baerson SR, editors. ACS Symposium Series 955. American Chemical Society; Washington, DC: 2007. Polyketides Biosynthesis, Biological Activity, and Genetic Engineering.
- 2.(a) Newman DJ, Cragg GM. J Nat Prod. 2007;70:461. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]; (b) Newman DJ, Grothaus PG, Cragg GM. Chem Rev. 2009;109:3012. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 3.Rohr J. Angew Chem, Int Ed. 2000;39:2847. doi: 10.1002/1521-3773(20000818)39:16<2847::aid-anie2847>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 4.For selected reviews on polyketide biosynthesis, see: Birch AJ. Science. 1967;156:202. doi: 10.1126/science.156.3772.202.Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Nat Prod Rep. 2007;24:162. doi: 10.1039/b507395m.Van Lanen SG, Shen B. Curr Opin Drug Discovery Dev. 2008;11:186.Das A, Khosla C. Acc Chem Res. 2009;42:631. doi: 10.1021/ar8002249.Gallimore AR. Nat Prod Rep. 2009;26:266. doi: 10.1039/b807902c.
- 5.For selected reviews on synthetic methods for polyketide construction, see: Paterson I, Doughty VA, Florence G, Gerlach K, McLeod MD, Scott JP, Trieselmann T. ACS Symp Ser. 2001;783:195.Koskinen AMP, Karisalmi K. Chem Soc Rev. 2005;34:677. doi: 10.1039/b417466f.Yeung KS, Paterson I. Chem Rev. 2005;105:4237. doi: 10.1021/cr040614c.Schetter B, Mahrwald R. Angew Chem, Int Ed. 2006;45:7506. doi: 10.1002/anie.200602780.Morris JC, Nicholas GM, Phillips AJ. Nat Prod Rep. 2007;24:87. doi: 10.1039/b602832m.Paterson I. Total Synthesis of Polyketides using Asymmetric Aldol Reactions. In: Christmann M, Bräse S, editors. Asymmetric Synthesis. 2. Wiley-VCH Verlag GmbH & Co; Weinheim, Germany: 2008. pp. 293–298.Paterson I, Findlay AD. Aust J Chem. 2009;62:624.
- 6.For selected reviews on bio-engineered polyketide construction, see: Katz L. Chem Rev. 1997;97:2557. doi: 10.1021/cr960025+.Khosla C. Chem Rev. 1997;97:2577. doi: 10.1021/cr960027u.Khosla C, Keasling JD. Nat Rev Drug Discovery. 2003;2:1019. doi: 10.1038/nrd1256.McDaniel R, Welch M, Hutchinson CR. Chem Rev. 2005;105:543. doi: 10.1021/cr0301189.Abe I. Chem Pharm Bull. 2008;56:1505. doi: 10.1248/cpb.56.1505.Horinouchi S. Curr Opin Chem Biol. 2009;13:197. doi: 10.1016/j.cbpa.2009.02.004.Lu D, Williams PG, Wang G. Biocatal Pharm Ind. 2009:247.
- 7.For selected reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Ngai MY, Kong JR, Krische MJ. J Org Chem. 2007;72:1063. doi: 10.1021/jo061895m.Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394. doi: 10.1021/ar7001123.Patman RL, Bower JF, Kim IS, Krische MJ. Aldrichim Acta. 2008;41:95.Shibahara F, Krische MJ. Chem Lett. 2008;37:1102. doi: 10.1246/cl.2008.1102.Bower JF, Kim IS, Patman RL, Krische MJ. Angew Chem, Int Ed. 2009;48:34. doi: 10.1002/anie.200802938.Han SB, Kim IS, Krische MJ. Chem Commun. 2009:7278. doi: 10.1039/b917243m.
- 8.For reviews on oxo-polyene macrolide natural products, see: Rychnovsky SD. Chem Rev. 1995;95:2021.Thirsk C, Whiting AJ. Chem Soc, Perkin Trans. 2002;1:999.Dougherty J, Zhou M, O’Doherty GA. Chemtracts. 2005;18:349.Cereghetti DM, Carreira EM. Synthesis. 2006:914.
- 9.For formal and total syntheses of oxo-polyene macrolides, see: Amphotericin B: Nicolaou KC, Daines RA, Uenishi J, Li WS, Papahatjis DP, Chakraborty TK. J Am Chem Soc. 1988;110:4672.Mycoticin A: Poss CS, Schreiber SL. J Am Chem Soc. 1993;115:3360.Dreher SD, Leighton JL. J Am Chem Soc. 2001;123:341. doi: 10.1021/ja0035102.Roxaticin: Rychnovsky SD, Hoye RC. J Am Chem Soc. 1994;116:1753.Mori Y, Asai M, Kawade J-i, Okumura A, Furukawa H. Tetrahedron Lett. 1994;35:6503.Mori Y, Asai M, Okumura A, Furukawa H. Tetrahedron. 1995;51:5299.Mori Y, Asai M, Kawade J-i, Furukawa H. Tetrahedron. 1995;51:5315.Evans DA, Connell BT. J Am Chem Soc. 2003;125:10899. doi: 10.1021/ja027638q.Filipin III: Richardson TI, Rychnovsky SD. J Am Chem Soc. 1997;119:12360.Roflamycoin: Rychnovsky SD, Khire UR, Yang G. J Am Chem Soc. 1997;119:2058.Dermostatin: Sinz CJ, Rychnovsky SD. Angew Chem, Int Ed. 2001;40:3224. doi: 10.1002/1521-3773(20010903)40:17<3224::AID-ANIE3224>3.0.CO;2-D.RK-397: Burova SA, McDonald FE. J Am Chem Soc. 2004;126:2495. doi: 10.1021/ja039618+.Denmark SE, Fujimori S. J Am Chem Soc. 2005;127:8971. doi: 10.1021/ja052226d.Mitton-Fry MJ, Cullen AJ, Sammakia T. Angew Chem, Int Ed. 2007;46:1066. doi: 10.1002/anie.200602601.Guo H, Mortensen MS, O’Doherty GA. Org Lett. 2008;10:3149. doi: 10.1021/ol801055b.
- 10.For isolation of (+)-roxaticin, see: Maehr H, Yang R, Hong LN, Liu CM, Hatada MH, Todaro LJ. J Org Chem. 1992;57:4793.
- 11.For enantioselective carbonyl allylation via iridium catalyzed C-C bond forming transfer hydrogenation and related processes, see: Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e.Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ. Angew Chem, Int Ed. 2009;48:5018. doi: 10.1002/anie.200901648.Lu Y, Krische MJ. Org Lett. 2009;11:3108. doi: 10.1021/ol901096d.Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w.Han SB, Kim IS, Han H, Krische MJ. J Am Chem Soc. 2009;131:6916. doi: 10.1021/ja902437k.Itoh J, Han SB, Krische MJ. Angew Chem, Int Ed. 2009;48:6316. doi: 10.1002/anie.200902328.Hassan A, Lu Y, Krische MJ. Org Lett. 2009;11:3112. doi: 10.1021/ol901136w.Han SB, Han H, Krische MJ. J Am Chem Soc. 2010;132:1760. doi: 10.1021/ja9097675.Zhang YJ, Yang JH, Kim SH, Krische MJ. J Am Chem Soc. 2010;132:4562. doi: 10.1021/ja100949e.Han SB, Gao X, Krische MJ. J Am Chem Soc. 2010;132:9153. doi: 10.1021/ja103299f.Zbieg JR, Fukuzumi T. Adv Synth Catal. 2010. In Press.For a recent application in total synthesis, see: Harsh P, O’Doherty GA. Tetrahedron. Vol. 65. 2009. p. 5051.
- 12.For a review on two-directional chain synthesis, see: Poss CS, Schreiber SL. Acc Chem Res. 1994;27:9.and literature cited in reference 11c
- 13.This mechanism for enantiomeric enrichment is documented by Eliel and Midland: Kogure T, Eliel EL. J Org Chem. 1984;49:576.Midland MM, Gabriel J. J Org Chem. 1985;50:1144.Also see, reference 11c
- 14.In the course of a synthetic approach to (+)-phorboxazole A, the mono-TBS (tert-butyldimethylsilyl) derivative of C2-symmetric diol (S,S)-1 was prepared in 7 steps from propylene glycol through successive use of Brown’s reagent for asymmetric carbonyl allylation, Ipc2(n-C3H5)B, and an improved procedure delivers (S,S)-1 in 4 steps from acetylacetone: Smith AB, III, Minbiole KP, Verhoest PR, Schelhaas M. J Am Chem Soc. 2001;123:10942. doi: 10.1021/ja011604l.Rychnovsky SD, Griesgraber G, Zeller S, Skalitsky DJ. J Org Chem. 1991;56:5161.Rychnovsky SD, Griesgraber G, Powers JP. Org Synth. 2000;77:1.
- 15.In large volume ozonolysis reactions, NaBH4 reduction of the ozonide is preferred as this protocol circumvents generation of stoichiometric organic byproducts and, hence, facilitates product purification: Caron S, Dugger RW, Gut Ruggeri S, Ragan JA, Brown Ripin DH. Chem Rev. 2006;106:2943. doi: 10.1021/cr040679f.
- 16.Grieco PA, Gilman S, Nishizawa M. J Org Chem. 1976;41:1487. [Google Scholar]
- 17.For preparation of homoallylic ether A, see reference 9l.
- 18.Although the second generation Grubb’s catalyst is known to promote closely related cross-metathesis reactions of allylic and homoallylic alcohols, the second generation Hoveyda-Grubb’s catalyst was superior in this particular case: Nyavanandi VK, Nadipalli P, Nanduri S, Naidu A, Iqbal J. Tetrahedron Lett. 2007;48:6905.
- 19.For cross-metathesis of an unprotected homoallylic alcohols with acrolein employing the second generation Hoveyda-Grubb’s catalyst, see: Chan KP, Ling YH, Loh TP. Chem Commun. 2007:939. doi: 10.1039/b616558c.
- 20.The Horner-Wadsworth-Emmons reagent EtO2C(CH=CH)3CH2PO(OEt)2 was prepared in 6 steps using a combination of established procedures: Lira R, Roush WR. Org Lett. 2007;9:533. doi: 10.1021/ol0629869.Doyle MP, Wang Y, Ghorbani P, Bappert E. Org Lett. 2005;7:5035. doi: 10.1021/ol0520121.
- 21.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Soc Chem Jpn. 1979;52:1989. [Google Scholar]
- 22.“The ideal synthesis creates a complex skeleton… in a sequence only of successive construction reactions involving no intermediary refunctionalizations, and leading directly to the structure of the target, not only its skeleton but also its correctly placed functionality.” Hendrickson JB. J Am Chem Soc. 1975;97:5784.
- 23.For an outstanding review on the topic of “redox economy” in organic synthesis, see: Baran PS, Hoffmann RW, Burns NZ. Angew Chem Int Ed. 2009;48:2854. doi: 10.1002/anie.200806086.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.