

Abstract
Genome-wide association mapping is a popular method for using natural variation within a species to generate a genotype–phenotype map. Statistical association between an allele at a locus and the trait in question is used as evidence that variation at the locus is responsible for variation of the trait. Indirect association, however, can give rise to statistically significant results at loci unrelated to the trait. We use a haploid, three-locus, binary genetic model to describe the conditions under which these indirect associations become stronger than any of the causative associations in the organism—even to the point of representing the only associations present in the data. These indirect associations are the result of disequilibrium between multiple factors affecting a single trait. Epistasis and population structure can exacerbate the problem but are not required to create it. From a statistical point of view, indirect associations are true associations rather than the result of stochastic noise: they will not be ameliorated by increasing sampling size or marker density and can be reproduced in independent studies.
GENOME-WIDE association mapping is a powerful tool that leverages the natural variation of a trait in a population to identify genetic factors that influence the trait. The theory is that due to the large number of recombination events in the genetic history of the population, only markers in tight linkage disequilibrium with loci responsible for the trait variation will exhibit significant statistical association with the trait. There are two ways in which genome-wide association mapping will fail by identifying loci that are not responsible for the variation in the trait (i.e., false positives): stochastic noise can generate an association in a sample that is not present in the larger population, or patterns of correlation among loci and factors causing trait variation can create indirect associations between markers and traits where no causal relation exists. While the former can be well quantified and managed with traditional sampling theory and replication, genomic control, and properly specified error terms in statistical models, these techniques do little to address the latter. As the association is true and not a statistical aberration, all accurate tests of association will point to the same noncausative loci; increasing sample sizes and marker densities will only heighten the misleading results, and these results can be reproduced in all follow-up studies.
It has long been recognized that population structure can cause these kinds of spurious, nonrandom associations (Li 1969; Lander and Schork 1994), and considerable effort has been devoted to addressing this problem statistically (Devlin and Roeder 1999; Pritchard et al. 2000; Price et al. 2006; Yu et al. 2006). However, attention has almost exclusively focused on the case where a noncausal marker is falsely identified as causal (or closely linked to a causal polymorphism) because both it and the trait are correlated with a single unobserved variable (e.g., geographic origin in a structured population). The effect of including multiple causal loci has not adequately been considered.
That this matters has been demonstrated by two recent articles. Dickson et al. (2010) used simulations to show that the presence of two or more rare causal variants in disequilibrium that can themselves not be detected due to lack of statistical power can produce spurious associations that are only distantly linked to the causal polymorphisms, and Atwell et al. (2010) showed that negative disequilibrium between two causal polymorphisms in the gene FRIGIDA interfered with the ability to find either of them but created strong signals at several distantly linked markers in a genome-wide association study in Arabidopsis thaliana.
To understand these cases we need a model with at least three variables: a noncausal marker and two background, unobserved factors. Here we present the simplest possible model—a haploid model of three binary loci—and use it to illustrate what conditions give rise to misleading genome-wide association mapping results.
MODEL AND RESULTS
The simplest model possible:
Table 1 defines the model: C denotes the causative locus we are trying to identify; L is a latent variable, be it a second locus or an environmental factor, that may also influence the organism's phenotype; and N is a noncausal marker locus. Parameters a, …, h are the population frequencies of all the possible “genotypes” (Table 2). βC and βL represent the additive component of the influence on phenotype of the designated causative allele and state of the latent variable, respectively. βLC is an epistatic term defined as the deviation from additivity of the combined effects of L and C. Without loss of generality, the causative alleles and latent variables are labeled so that βC and βL are both ≥0 and the noncausal marker is labeled so that cov(N, P) ≥ 0. In every case we consider the phenotype, P, to be fully determined by L and C. There is no stochastic noise included in our analyses.
TABLE 1.
Model specification
| Latent variable | Causative polymorphism | Noncausal marker | Phenotype | Genotype frequency |
|---|---|---|---|---|
| 0 | 0 | 0 | 0 | a |
| 0 | 0 | 1 | 0 | b |
| 0 | 1 | 0 | βC | c |
| 0 | 1 | 1 | βC | d |
| 1 | 0 | 0 | βL | e |
| 1 | 0 | 1 | βL | f |
| 1 | 1 | 0 | βC + βL + βLC | g |
| 1 | 1 | 1 | βC + βL + βLC | h |
The model is defined as a “genotype” of three binary factors, L, C, and N. Every combination of these factors perfectly describes a phenotype P and occurs with a frequency indicated by a, … , h. Table 2 defines some useful parameterizations.
TABLE 2.
Parameterization
| Symbol | Description | Definition |
|---|---|---|
| ρL | Frequency of variable L | e + f + g + h |
| ρC | Frequency of allele C | c + d + g + h |
| ρN | Frequency of allele N | b + d + f + h |
| DNC | Disequilibrium between N and C | d + h − ρNρC |
| DNL | Disequilibrium between N and L | f + h − ρNρL |
| DLC | Disequilibrium between L and C | g + h − ρNρC |
| DNLC | Three-locus disequilibrium | h − ρNρLρC |
Reparameterizing the model in terms of the frequencies of individual factors and the disequilibrium between them facilitates biological understanding of what creates associations between factors and phenotypes.
With this model we can describe simple traits with only a single factor influencing the phenotype by setting βL and βLC to 0. A trait governed by purely additive contributions from two factors is modeled by letting βC and βL vary freely but keeping βLC at 0. Varying βLC gives us a wide range of epistatic effects. Positive values of βLC give us synergistic epistasis and negative values are antagonistic.
In association mapping we are looking for nonindependence between alleles and phenotypes. Nonindependence can be quantified in many ways. Our analytical work focuses on covariance between proposed factors and observed phenotypes. A significantly nonzero covariance indicates an association between the trait and the marker being examined. The hope is that this indicates that the associated locus contributes biologically to variation for the trait or is very closely linked to a locus that does. In our model, we want the covariance between the causal polymorphism and the trait, cov(C, P), to be high (or we will not be able to detect the causal association), and we want the covariance between the noncausal marker, cov(N, P), and the trait to be high if and only if the marker is tightly linked to the causal polymorphism. We do not want cov(N, P) > cov(C, P) lest we misidentify the noncausal marker as causal. The covariance between the latent variable and the trait, cov(L, P), finally, is just a nuisance from the point of view of identifying the causal polymorphism. For our model, we have
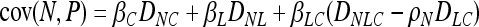 |
(1) |
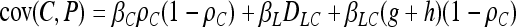 |
(2) |
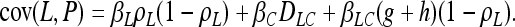 |
(3) |
By looking at these covariance terms in various settings we illustrate when we can expect association mapping to be misleading. For clarity, we focus on expectations and do not consider the stochastic error introduced by finite sample sizes.
Simple traits:
Setting βL = 0 and βLC = 0 we describe a trait that is influenced only by a single causative polymorphism. In this case Equations 1 and 2 reduce to
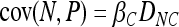 |
and
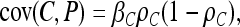 |
respectively. The causative allele will give the most significant results when its effect on the phenotype is large and it is at an intermediate frequency in the sample. The noncausal marker will give significant results when the effect of the causative allele is large and there is disequilibrium between the two loci. In expectation, however, the noncausal marker should not give a more significant result than the causative polymorphism. Indeed,
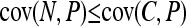 |
(4) |
expands to
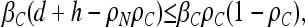 |
which simplifies to
 |
This is always true as c, g, b, f, and ρC are all defined on the interval [0, 1]. While disequilibrium can generate significant results for noncausal markers, with sufficient sample size the most significant results can be expected to be for the causative polymorphism or, if it is not present in the marker panel, the marker in greatest disequilibrium with it.
Thus, while false positives, in the sense of significantly associated but unlinked noncausal markers may exist (especially if population structure induces long-distance linkage disequilibrium across the genome), sufficiently powered association studies should always also locate the causal polymorphism if it exists. However, with traits with more than one contributing factor there is no such guarantee. This is the problem we turn to next. (Association studies can of course always be misleading if no causal polymorphism exists but noncausal markers covary with a nongenetic latent variable: this is readily seen by setting βC = 0 and βLC = 0 in our model).
Complex traits:
When two or more factors contribute to variation in a trait, association studies may be misleading in the sense that noncausal markers can be expected to be more strongly associated than either causal polymorphism. To see this we consider several scenarios beginning with causative factors with only additive effects.
Additive effects, strong latent variable:
In an extreme case where effects are additive (βLC = 0), but βL ≫ βC, Equations 1 and 2 can be approximated by
 |
and
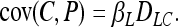 |
Under these conditions the causative polymorphism acts like a noncausal marker and the most significant signals will come from whichever one has the greatest disequilibrium with the latent variable that is responsible for most of the variation in the phenotype. If the latent variable is another genetic locus, this is not a problematic result as we have simply approximated the previously described case of a simple genetic trait. If the latent variable is an exogenous factor, however, we now see that we may erroneously ascribe its effect to a genetic locus that happens to be correlated with it.
Equivalent additive factors:
Less trivially, setting βLC = 0 and βL = βC = β describes a trait controlled equally by two factors and gives us covariance terms
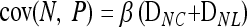 |
(5) |
 |
(6) |
 |
(7) |
In this case, the noncausal marker is expected to have a more significant result than the causative allele whenever
 |
(8) |
which makes it intuitive to see how rare causative alleles can give rise to the kind of “synthetic” association described by Dickson et al. (2010). The term involving ρC on the right becomes small, leaving ample opportunity for the two disequilibrium terms on the left to swamp out the one disequilibrium term on the right. The specific pattern described in that article is one where the latent variable is a second causative genetic variant at a locus. This creates strong negative covariance between the two causative factors and eliminates the opportunity for genetic interactions to play any role. In this case the only haplotypes that occur with appreciable frequencies correspond in our model to a, b, d, and f. Setting all other haplotype frequencies to 0 in Equation 8 and simplifying show us that under these conditions the strongest association will be expected at the noncausal locus whenever ρN < 1 − bd/f. For this scenario to cause problematic results, the noncausal marker cannot be too common or it cannot be in sufficiently strong linkage disequilibrium with the rare causative loci.
Epistasis:
There are limits to the degree of confounding possible when interactions are purely additive. Within the restriction of additivity, even when the strongest signal in an association study is coming from a noncausal locus, we should expect at least one of the truly causative factors to exhibit at least some association. This is because the covariance between the noncausal marker and the phenotype will never be larger than the sum of the covariance between the causative locus and the phenotype and the latent variable and the phenotype. From Equations 1–3,
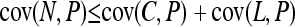 |
expands to
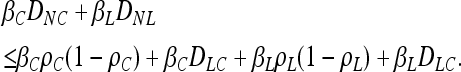 |
From Equation 4 it follows that
 |
which is also true if you replace all the C's with L's. Doing so and substituting lets us cancel and get
 |
which is always true.
A nonzero interaction term does away with this upper bound for cov(N, P), however. Consider, for example, the case where βC = βL = β but βLC = −β (negative epistasis: either causative allele is sufficient for the phenotype), a = b = c = e = h = 0, and d = f = g = 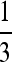 (negative covariance between the two causal factors). In this example, cov(C, P) = cov(L, P) = 0, but cov(N, P) = 2β/3. In other words, the noncausal marker can have an arbitrarily large covariance with the trait even though there is no association for any of the truly causative factors, no matter how powerful the study.
(negative covariance between the two causal factors). In this example, cov(C, P) = cov(L, P) = 0, but cov(N, P) = 2β/3. In other words, the noncausal marker can have an arbitrarily large covariance with the trait even though there is no association for any of the truly causative factors, no matter how powerful the study.
Simulated example:
To illustrate the behavior of our model using real polymorphism data, we use the data of Atwell et al. (2010), who carried out a genome-wide association study using 216,130 single-nucleotide polymorphism (SNP) markers in a set of 199 inbred lines of A. thaliana. The sample is characterized by complex population structure (Platt et al. 2010), which makes it ideal for illustrative purposes. Many traits are strongly correlated with latitude in A. thaliana. This can come about through geographically distributed causative genetic polymorphism of large effect, the combined effect of many causative polymorphisms of small effect, or nongenetic confounding factors. We performed two sets of simulations. A first causative locus is picked at random from the 216,130 SNPs and a random allele is assigned an effect. The second causative factor is then either a SNP or a binary environmental factor where both possibilities for an effect allele are used. This is repeated for 10% of the SNPs in the data set and a new trait is generated, resulting in ∼43,200 nonconstant traits for each of the sets of simulations. For the first set, the traits are correlated with the population structure of the organism, and the second causative variable is a latent indicator variable that identifies each individual as having been collected north of 50° latitude, a line that lies midway between London and Paris, and that divides the sample roughly in half. In the second set of simulations, the second causative variable is another randomly selected SNP.
Phenotypes were calculated for three different trait architectures, letting βC = βL = β with differing degrees of interaction (Table 3). Setting βLC = 0 gives a purely additive model. With βLC = −β we get an “or” model where either causative factor is sufficient to create phenotypic change. When describing two genetic loci, this model can reflect the interaction between loss-of-function mutations in different genes in a common pathway. With an environmental cofactor this represents a canalized trait whose genetic variation is revealed phenotypically only in certain environments. As described above, this kind of negative epistasis can give rise to situations where only the noncausal marker is correlated with the phenotype. Setting βLC = −2β gives us an “xor” model where individuals with zero and two labeled factors share a common phenotype but are different from those with only one (regardless of which one it is). Genetically, this model can reflect the interaction between a compensatory pair of mutations, such as one in a transcription factor and one in a binding site. As an environmental effect this scenario occurs whenever there are trade-offs between responses in different environments. Pathogen resistance is one example. Functional resistance genes can increase seed production where pathogens are present but reduce it where they are not (Korves and Bergelson 2004).
TABLE 3.
Simulated phenotypes
| “Genotype” |
Phenotype |
|||
|---|---|---|---|---|
| Latent variable | Causative polymorphism | Additive: |
or: |
xor: |
| βLC = 0 | βLC = −P | βLC = −2P | ||
| North | 0 | 0 | 0 | 0 |
| South | 0 | β | β | β |
| North | 1 | β | β | β |
| South | 1 | 2β | β | 0 |
Model is shown for generating phenotypes from data with one causative genetic locus and a nongenetic, geographic factor that is treated as a latent variable.
For each simulated phenotype we performed a genome-wide association study using the nonparametric Wilcoxon rank sum test on every marker. For the first set of simulations, where the latent variable is a North–South split, Figure 1, A–C, shows how far down in the list of associated markers one would have to go to find the correct locus. In the purely additive simulations there are few problems (Figure 1A). The correct locus is easily identified as one of the very strongest results in almost all cases, with the vast majority of exceptions being associated with cases where the causative locus has a very low minor allele frequency. The or model exhibits greater confounding (Figure 1B). The locus is perfectly identified less than half of the time and is sometimes missed even when the minor allele frequency is intermediate. The correct locus was essentially never found in the xor model regardless of the minor allele frequency (Figure 1C). Measurements of the distance between the causative locus and the locus with the lowest P-value followed the same pattern. When the causative locus is among the highest ranked SNPs, it is near the locus with the lowest P-value. As its rank falls, it tends to be farther and farther away, and by the time it is not within the top 1000 SNPs it is often on the wrong chromosome.
Figure 1.—






Simulation results for a geographical latent variable, a North–South split. (A–C) Rank of the causative SNP: illustration of how many markers had a stronger association than the causative SNP in a given analysis under (A) the “additive” genetic model, (B) the “or” model, and (C) the “xor” model. Colors indicate the minor allele frequency of the causative SNP. (D–F) Maximum distance to the causative SNP of all SNPs with greater or equal association than the causative SNP under (D) the additive genetic model, (E) the or model, and (F) the “xor” model. Colors indicate whether the causative marker was found to be significant at the Bonferroni threshold. Only results where at least one SNP was found significant were included in the analysis.
Figure 1, D–F, shows the distribution of maximum distances to the causative SNP for all markers with association greater than or equal to that of the causative locus. It is evident that when the causative marker is not the most significant, a very distant marker usually is. This is true even in the simple additive case. In the xor model the causative marker is not significant most of the time.
Turning to the simulations with two randomly chosen causative loci, Figure 2, A–C, shows the P-value rank distribution of the two causative alleles, both the top ranking and the second ranking. A true causative locus is essentially always found in the additive case (Figure 2A), and the more weakly associated locus is often among the most significant ones. For the epistatic or and xor models a true causative locus is missed one time in eight and two times in five, respectively (Figure 2, B and C). The rank of the second-ranking causative locus also becomes lower in the epistatic models. Figure 2, D–F, shows the distribution of maximum distances to the nearest causative SNP for all markers with association greater than that of the second-ranking causative locus. This demonstrates that there are often unlinked loci with greater significance than the second-ranking causative locus, even when both causative loci are significant. This is a particularly serious problem in the epistatic models (see also Table 4).
Figure 2.—






Simulation results for two causative SNPs, where both are chosen at random. (A–C) Rank of the top-ranking causative SNP (blue) and the second-ranking causative SNP (orange) under (A) the “additive” genetic model, (B) the “or” model, and (C) the “xor” model. (D–F) Maximum distance to nearest causative SNP among SNPs with greater association than the more weakly associated causative SNP under (D) the additive genetic model, (E) the or model, and (F) the xor model. Colors indicate whether two, one, or none of the causative SNPs were found significant at the Bonferroni threshold. Only results where at least one SNP was found significant were included in the analysis.
TABLE 4.
Summary of simulation result
| At least one significant?a |
Top-ranking causal?b |
Distant noncausal found?c |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Model | Additive | or | xor | Additive | or | xor | Additive | or | xor |
| Latent North–South variable | 1.00 | 1.00 | 0.86 | 0.70 | 0.49 | 0.00 | 0.23 | 0.43 | 1.00 |
| Two causal loci | 1.00 | 1.00 | 0.94 | 0.96 | 0.80 | 0.86 | 0.25 | 0.76 | 0.81 |
Fraction of results with at least one significant SNP (at a Bonferroni-corrected threshold of 0.05) and that were used for subsequent analysis.
Fraction of results in which the top-ranking association was a causal polymorphism (the causal polymorphism in the case of a latent variable).
Fraction of results in which a SNP more strongly associated with the phenotype than a casual polymorphism (the causal polymorphism in the case of a latent variable) was >50 kb away from the nearest causal polymorphism.
DISCUSSION
Causes of confounding:
We used a very simple three-locus model to clarify the conditions under which genome-wide association studies are expected to be reproducibly misleading. We believe there are three distinct problem sources: correlation between causal factors and (unlinked) noncausal markers, more than a single causal factor (especially if the factors themselves are correlated), and epistasis (i.e., nonlinear interactions between causal factors in the determining the phenotype).
Consider each in turn.
Correlation with unlinked markers:
Correlation between causal factors and unlinked, noncausal markers (note that all noncausal markers are unlinked if the causal factors are nongenetic) violates the basic assumption of genome-wide association studies (GWAS) and causes false positives.
Population structure, by definition, causes genome-wide correlations between alleles (linkage disequilibrium), which can easily lead to genome-wide occurrence of false positives (Rosenberg and Nordborg 2006), a problem that has long been recognized (Li 1969; Lander and Schork 1994) and for which many statistical solutions have been proposed (Devlin and Roeder 1999; Pritchard et al. 2000; Price et al. 2006; Yu et al. 2006). However, it is important to realize that associations at unlinked, noncausal markers can also arise because of pleiotropy. Consider, for example, a scenario in which one polymorphism affects both skin and eye color and another affects just skin color. If skin color variation is locally adaptive, then selection causes correlation (linkage disequilibrium) between the two loci. A GWAS for eye color would detect associations at both loci, even though one of them has nothing to do with this trait. Unlike false positives caused by population structure, these types of false positives would not occur at random throughout the genome: they would occur only at noncausal markers correlated with causal factors through selection on pleiotropic traits. This might make them less common: it would certainly make them more difficult to eliminate through statistical methods.
More than a single causative factor:
Whenever a trait is controlled by more than a single factor, it is possible that the strongest associations in the data are indirect ones. As biologically uninformative as these associations are, they are true associations and will respond as such to statistical tests, gaining significance with increased sampling and reproducing in multiple data sets.
Without any population structure, strong indirect associations can arise at loci that are genetically linked to two or more causative factors, even if the causative factors are in equilibrium with each other. This linkage-only case has been well documented in linkage mapping literature (Haley and Knott 1992; Martinez and Curnow 1992). Here, two genetically linked quantitative trait loci combine to produce a false or “ghost” peak of association between them. In the presence of natural selection it is no longer necessary for the indirectly associated marker to be linked to more than one causative locus (as in the ghost peak version) as correlations will already exist between the causative factors. A marker linked to one is likely to be in disequilibrium with all of them. With population structure or selection and pleiotropy, however, these indirect associations can be far removed from all causative factors.
Epistasis:
When the causative loci interact epistatically, it is possible that the only loci exhibiting any association with the phenotype are noncausal. While it has long been recognized that epistatically interacting loci may be difficult to find due to lack of marginal effect (Eaves 1994), correlated noncausal loci can serve as excellent markers for the joint state of several causative loci working in concert.
Tests for association based on multilocus haplotypes (or that model explicit interaction terms) will improve results but not completely ameliorate the problem. While we have mostly been describing the factors L, C, and N as single loci, they can just as easily represent arbitrarily complex combinations of loci (and external factors). A statistician who perfectly models the trait architecture, and knows that he or she has done so, will have effectively recast the complex trait as a simple trait (albeit with complex inputs). It would be guaranteed that no noncausal marker complex will have a stronger association than the causative factor complex, but there is nothing stopping noncausal marker complexes from having associations just as strong as the causative ones. Even simple noncausal markers may have associations as strong as the causative marker complex, which would mislead any sort of model-selection algorithm.
Conclusions:
Our purpose in writing this article was to clarify the conditions under which GWAS are expected to be reproducibly misleading. As our simulation results demonstrate, severe problems may arise when we attempt to model traits that are really due to multiple, possibly correlated, possibly epistatically interacting factors using single-locus models that assume that unlinked, noncausal markers are not correlated with the causal factors. Not only do we face the well-known problem of false positives across the genome, but also we see that the strongest associations may appear on chromosomes completely devoid of causative loci and that the true positives may be undetectable.
In this light, the common practice of “correcting for population structure” may be misguided. The real goal should be correcting for the confounding effects of multiple causative factors. Some of the techniques currently employed as population structure correction actually do this very well. The mixed-model approach (Yu et al. 2006), for instance, can be interpreted as removing the effect of a large number of unlinked selectively neutral factors, each with an uninterestingly small effect on the studied trait (Kang et al. 2010). Approaches such as structured analysis (Pritchard et al. 2000) and principal components analysis (Price et al. 2006), on the other hand, aid in correcting for the correlations among multiple causative factors only to the extent that clustering on global patterns of genetic variation approximates the distributions of the individual causative factors. Attempting to correct for population structure directly, as opposed to correcting for correlations among multiple causative factors, runs the risk of eliminating the effects of the largest, most interesting loci from the study. This will happen whenever alleles at those loci have a distribution similar to the genomic patterns of correlation. Such factors can easily and accurately be identified as being associated, although they will be in disequilibrium with many noncausal loci, making them difficult to locate with any precision.
This is not to say, however, that the presence of any of these confounding attributes of complex traits dooms a genome-wide association study to failure. All of them, multiple factors, natural selection, epistasis, and population structure, contribute to confounding in quantitative ways and in amounts that will be greatly influenced by their specific details. A carefully constructed human case–control study, for instance, may not suffer from appreciable population structure and would therefore introduce an imprecision only in the location of the cause of the associations. Larger, population-based cohort studies, however, may soon find themselves running into the kinds of large-scale population structure inherent in the human species (Freedman et al. 2004; Novembre et al. 2008). The results may still be mostly accurate if natural selection is weak and the additive effects of the majority of the causative loci are large, but may become questionable when considering highly polygenic traits under strong selection. Genome-wide association studies applied to other organisms, however, may be considerably more problematic. The very worst situation is likely to arise in species that have undergone strong local adaptation or have experienced artificial selection to create numerous different phenotypes. In these cases the correlated effects of population structure and selection may well be expected to swamp any remaining causative associations with rampant and excessive indirect associations spread all across the genome. Organisms like A. thaliana may be intermediate, with confounding ranging from almost nonexistent to extremely problematic depending on the architecture of the trait. In organisms with high levels of confounding, it is necessary to proceed with caution and treat identified associations as hypotheses for follow-up confirmatory studies (Atwell et al. 2010).
It is also worth noting that these indirectly associated sites confound not just the scientist attempting to discover the map between phenotype and genotype, but similarly interfere with the process of natural selection as well. In the example of epistasis described above, in which marginal effects of the causal factors are completely missing, any selection applied to the trait in question would change the allele frequency (producing a partial selective sweep) only at the noncausal, neutral locus, not at any of the loci that actually contribute to the phenotype. Where natural selection has an advantage over the scientist is that the scientist is generally restricted to a snapshot of a population and its patterns of disequilibrium. Natural selection is a process that unfolds over successive generations and may have the opportunity to break apart disadvantageous correlations. Scientists can mimic this process in some cases by performing experimental crosses, genetic transformations, or pedigree- or family-based analyses and thereby disrupting the extant patterns of disequilibrium, although this is often not feasible in clinical studies.
Acknowledgments
We thank David Conti, Sergey Nuzhdin, Paul Marjoram, Juan Pablo Lewinger, Thomas Turner, Quingrun Zhang, and Quan Long for helpful discussions. This work was supported by the National Science Foundation (DEB-0723935), the National Institutes of Health (P50 HG002790), and the Austrian Academy of Sciences.
Available freely online through the author-supported open access option.
References
- Atwell, S., Y. S. Huang, B. J. Vilhjálmsson, G. Willems, M. Horton et al., 2010. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin, B., and K. Roeder, 1999. Genomic control for association studies. Biometrics 55 997–1004. [DOI] [PubMed] [Google Scholar]
- Dickson, S. P., K. Wang, I. Krantz, H. Hakonarson and D. B. Goldstein, 2010. Rare variants create synthetic genome-wide associations. PLoS Biol. 8 e1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaves, L. J., 1994. Effect of genetic architecture on the power of human linkage studies to resolve the contribution of quantitative trait loci. Heredity 72 175–192. [DOI] [PubMed] [Google Scholar]
- Freedman, M. L., D. Reich, K. L. Penney, G. J. McDonald, A. A. Mignault et al., 2004. Assessing the impact of population stratification on genetic association studies. Nat. Genet. 36 388–393. [DOI] [PubMed] [Google Scholar]
- Haley, C. S., and S. A. Knott, 1992. Maximum-likelihood mapping of quantitative trait loci using full-sib families. Genetics 132 1211–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, H. M., J. H. Sul, S. K. Service, N. A. Zaitlen, S.-y. Kong et al., 2010. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korves, T., and J. Bergelson, 2004. A novel cost of r gene resistance in the presence of disease. Am. Nat. 163 489–504. [DOI] [PubMed] [Google Scholar]
- Lander, E. S., and N. J. Schork, 1994. Genetic dissection of complex traits. Science 265 2037–2048. [DOI] [PubMed] [Google Scholar]
- Li, C. C., 1969. Population subdivision with respect to multiple alleles. Ann. Hum. Genet. 33 23–29. [DOI] [PubMed] [Google Scholar]
- Martinez, O., and R. N. Curnow, 1992. Estimating the locations and the sizes of the effects of quantitative trait loci using flanking markers. Theor. Appl. Genet. 85 480–488. [DOI] [PubMed] [Google Scholar]
- Novembre, J., T. Johnson, K. Bryc, Z. Kutalik, A. R. Boyko et al., 2008. Genes mirror geography within Europe. Nature 456 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt, A., M. Horton, Y. S. Huang, Y. Li, A. E. Anastasio et al., 2010. The scale of population structure in Arabidopsis thaliana. PLoS Genet. 6 e1000843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, A. L., N. J. Patterson, R. M. Plenge, M. E. Weinblatt, N. A. Shadick et al., 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38 904–909. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens, N. A. Rosenberg and P. Donnelly, 2000. Association mapping in structured populations. Am. J. Hum. Genet. 67 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, N., and M. Nordborg, 2006. A general population-genetic model for the production by population structure of spurious genotype-phenotype associations in discrete, admixed, or spatially distributed populations. Genetics 173 1665–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J., G. Pressoir, W. Briggs, I. Vroh Bi, M. Yamasaki et al., 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38 203–208. [DOI] [PubMed] [Google Scholar]