

Abstract
The Bayesian LASSO (BL) has been pointed out to be an effective approach to sparse model representation and successfully applied to quantitative trait loci (QTL) mapping and genomic breeding value (GBV) estimation using genome-wide dense sets of markers. However, the BL relies on a single parameter known as the regularization parameter to simultaneously control the overall model sparsity and the shrinkage of individual covariate effects. This may be idealistic when dealing with a large number of predictors whose effect sizes may differ by orders of magnitude. Here we propose the extended Bayesian LASSO (EBL) for QTL mapping and unobserved phenotype prediction, which introduces an additional level to the hierarchical specification of the BL to explicitly separate out these two model features. Compared to the adaptiveness of the BL, the EBL is “doubly adaptive” and thus, more robust to tuning. In simulations, the EBL outperformed the BL in regard to the accuracy of both effect size estimates and phenotypic value predictions, with comparable computational time. Moreover, the EBL proved to be less sensitive to tuning than the related Bayesian adaptive LASSO (BAL), which introduces locus-specific regularization parameters as well, but involves no mechanism for distinguishing between model sparsity and parameter shrinkage. Consequently, the EBL seems to point to a new direction for QTL mapping, phenotype prediction, and GBV estimation.
REGULARIZATION or shrinkage methods are gaining increasing recognition as a valuable alternative to variable selection techniques in dealing with oversaturated or otherwise ill-defined regression problems in both the classical and Bayesian frameworks (e.g., O'hara and Sillanpää 2009). Many studies (e.g., Xu 2003; Wang et al. 2005; Zhang and Xu 2005; De los Campos et al. 2009; Usai et al. 2009; Wu et al. 2009; Xu et al. 2009) have documented the potential of shrinkage methods for quantitative trait locus (QTL) mapping and genomic breeding value (GBV) estimation using genome-wide dense sets of markers. Lee et al. (2008) make a clear connection between phenotype prediction and GBV estimation, suggesting that methods developed for one are also applicable to the other. We thus use the two concepts interchangeably throughout this article.
Regularized regression methods, such as ridge regression (Hoerl and Kennard 1970) or the least absolute shrinkage and selection operator (LASSO) (Tibshirani 1996), are essentially penalized likelihood procedures, where suitable penalty functions are added to the negative log-likelihood to automatically shrink spurious effects (effects of redundant covariates) toward zero, while allowing relevant effects to take values farther from zero.
It has been pointed out that these non-Bayesian shrinkage methods are not suitable for oversaturated models. Zou and Hastie (2005) and Park and Casella (2008) noted that the LASSO cannot select a number of nonzero effects exceeding the sample size. Xu (2003) found that for ridge regression to work, the number of model effects should be in the same order as the number of observations. This is impractical for genomic selection, which capitalizes on the variation due to small-marker effects, the number of which can exceed the sample size, by contrast to QTL mapping where interest lies mostly in a small subset of loci with large effects on the focal phenotype. In connection with the LASSO, the Bayesian LASSO (BL) (Park and Casella 2008; Yi and Xu 2008) has been proposed to overcome this limitation by imposing a selective shrinkage across regression parameters. Xu (2003) also proposed a Bayesian shrinkage method for QTL mapping, which extends ridge regression in a similar fashion.
Although the BL has been successfully applied to QTL mapping (e.g., Yi and Xu 2008) and to GBV estimation (e.g., De los Campos et al. 2009), it relies on a single parameter known as the regularization parameter to simultaneously regulate the overall model sparsity and the extent to which individual regression coefficients are shrunken. However, this is unrealistic when dealing with a large number of predictors whose effect sizes may differ by orders of magnitude. It is therefore natural to ask whether this practice can be relaxed and how such an attempt may impinge on the model performance (e.g., Sun et al. 2010).
Here we propose an extension to the Bayesian LASSO for QTL mapping and unobserved phenotype prediction. Our method, the extended Bayesian LASSO (EBL), introduces locus-specific regularization parameters and utilizes a parameterization that clearly separates the overall model sparsity from the degree of shrinkage of individual regression parameters. We use simulated data to investigate the performance of the EBL relative to the Bayesian LASSO in mapping QTL and in predicting unobserved phenotypes. We also compare the performance of the EBL to the Bayesian adaptive LASSO (BAL) recently proposed by Sun et al. (2010), which also assumes locus-specific regularization parameters.
METHODS
Statistical model:
Let yi (i = 1, … , n) and xij denote, respectively, the value of the phenotypic trait of interest for the ith individual and the genotype code of the ith individual at locus j (j = 1, … , p). The model is developed in the context of experimental crosses derived from two inbred lines such as backcross (BC) or double haploid (DH) progenies with only two possible genotypes at any locus. The dummy variables xij are coded as xij = 0 for one genotype and xij = 1 for the other. We assume an additive model that regresses the phenotypic values of the n individuals on their genotypes at p putative loci, which may be markers or alternatively pseudomarkers (Sen and Churchill 2001; Servin and Stephens 2007). That is,
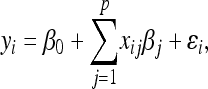 |
(1) |
where β0 is the intercept. Under the genotype coding assumed here, βj represents the genetic effect associated with locus j (j = 1, … , p) or more precisely, the difference (contrast) between the genetic effects associated with the two genotypes for locus j. The residual errors ɛi are assumed to be independent and normally distributed with mean zero and common variance σ2. The model is applicable to other experimental designs, such as F2 intercross progeny under appropriate genotype coding and to population-based samples. For breeding purposes or analysis of family data, model (1) can be extended to include an infinitesimal polygenic term that accounts for relatedness between individuals (see, e.g., De los Campos et al. 2009 or Pikkuhookana and Sillanpää 2009 for more details). In matrix notation, model (1) can be written as
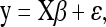 |
(2) |
where y = (y1, … , yn)T, β = (β0, … ,βp)T, ɛ = (ɛ1, … ,ɛn)T, and X is the n × (p + 1) design matrix comprising the genotype profiles of the p loci, with all elements of the first column (which corresponds to the intercept) set to 1. The likelihood for this model is given by
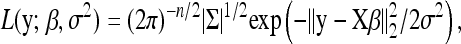 |
(3) |
where Σ = σ−2In is the precision matrix associated with the residual errors, In is the n × n identity matrix, and ‖·‖2 denotes the L2 norm.
Maximum likelihood estimates (MLEs) of β and σ2 are obtained by maximizing the likelihood function L(y; β, σ2) or alternatively, by minimizing the negative likelihood function with respect to β and σ2. However, as already pointed out, when dealing with genome-wide dense sets of markers, the number, p, of predictors can be larger than the sample size n, with most of the markers having weak or no effect on the focal phenotype (e.g., Xu 2003; Zhang and Xu 2005). In such circumstances, regularization methods offer an interesting approach to inducing a sparse model representation by automatically shrinking the effects of redundant covariates (spurious effects) toward zero, while effectively estimating the relevant ones.
Regularization methods and the LASSO:
The rationale of regularization methods is to penalize the log-likelihood function 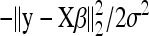 with a suitable nondecreasing function, g(β) > 0, and estimate β by
with a suitable nondecreasing function, g(β) > 0, and estimate β by 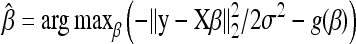 , or equivalently,
, or equivalently,
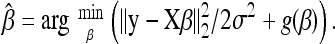 |
(4) |
Several penalty functions have been proposed, including g(β) = λ‖β‖2 (λ > 0), which leads to ridge regression (Hoerl and Kennard 1970; Whittaker et al. 2000; Malo et al. 2008), and g(β) = λ‖β‖1 = λΣj|βj|, which leads to the LASSO estimate
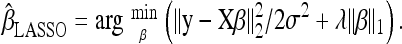 |
(5) |
(Tibshirani 1996; Usai et al. 2009; Wu et al. 2009). The smoothing or regularization parameter, λ, determines the extent of the penalization, with larger values implying a stronger penalization on the Lw norm (w = 1 for LASSO and w = 2 for ridge regression), which results in more regression coefficients being shrunk toward zero. Henceforth, our attention is focused on the LASSO owing to its desirable feature of setting some of the redundant effects to be exactly zero (Tibshirani 1996). Note that if βj is shrunken to zero, then the LASSO objective function, 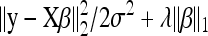 , in Equation 5 becomes independent of βj so that Xj is pruned (discarded) from the model. As λ approaches zero,
, in Equation 5 becomes independent of βj so that Xj is pruned (discarded) from the model. As λ approaches zero, 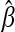 tends to the ordinary least-squares estimate (no shrinkage). The intercept, β0, is not subject to shrinkage since we consider that it is always included into the model. The term
tends to the ordinary least-squares estimate (no shrinkage). The intercept, β0, is not subject to shrinkage since we consider that it is always included into the model. The term 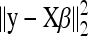 in Equation 5 expresses the least-squares approximation, whereas the penalty function g(β) = λ‖β‖1 is intended to promote sparseness. The LASSO estimate
in Equation 5 expresses the least-squares approximation, whereas the penalty function g(β) = λ‖β‖1 is intended to promote sparseness. The LASSO estimate 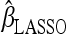 can be interpreted from a Bayesian perspective as the maximum a posteriori (MAP) or posterior mode of the parameter vector β under the prior specification
can be interpreted from a Bayesian perspective as the maximum a posteriori (MAP) or posterior mode of the parameter vector β under the prior specification 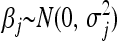 and
and 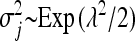 , independently for j = 1, … , p (Tibshirani 1996; Park and Casella 2008; Yi and Xu 2008). Equivalently, this corresponds to assigning to the βj independent Laplacian or double exponential (DE) priors with mean zero and variance 2/λ2. This follows from the fact that, for any x ∈ R, and λ > 0,
, independently for j = 1, … , p (Tibshirani 1996; Park and Casella 2008; Yi and Xu 2008). Equivalently, this corresponds to assigning to the βj independent Laplacian or double exponential (DE) priors with mean zero and variance 2/λ2. This follows from the fact that, for any x ∈ R, and λ > 0,
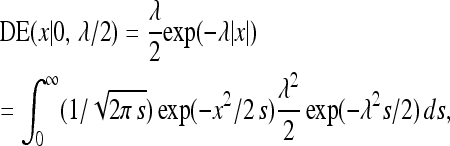 |
(6) |
where DE(x |0, λ/2) denotes the probability density function of the DE distribution with mean 0 and variance 2/λ2, evaluated at x (Park and Casella 2008).
The DE distribution is sharply peaked at the mode and has heavier than Gaussian tails. Setting the prior mode at zero encodes a prior belief of no effect for most of the regression covariates. In addition, the strength of this belief, and hence the degree of model sparsity, is determined by the variance of the DE prior assumed for the regression coefficients, which in turn depends solely on the regularization parameter λ. The LASSO estimate corresponds to the MAP of β when all βj (j = 1, … , p) are independently assigned DE(0, λ/2) priors with λ set to a fixed nonnegative constant. But as pointed out earlier, the number of nonzero effects that the LASSO can select is bounded from above by the sample size. A Bayesian formulation of the LASSO known as the Bayesian LASSO has been proposed to overcome this limitation.
The Bayesian LASSO:
In the BL (Park and Casella 2008; Yi and Xu 2008; De los Campos et al. 2009), each regression parameter, βj (j = 1, … , p), is a priori assumed to be normally distributed around zero with its own variance 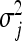 , i.e.,
, i.e., 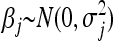 . Consequently, the degree of shrinkage is made locus specific through differences in the variances
. Consequently, the degree of shrinkage is made locus specific through differences in the variances 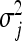 across loci. These variance parameters are independently assigned Exp(λ2/2) priors. With this hierarchical prior specification, the marginal prior of the regression parameters, βj, is DE(0, λ/2), independently for j = 1, … , p, by virtue of Equation 6. Differences in locus-specific variances induce a selective shrinkage with smaller variances implying stronger shrinkage toward zero, as expected for spurious effects. It is clear, however, that the variance 4/λ4 of
across loci. These variance parameters are independently assigned Exp(λ2/2) priors. With this hierarchical prior specification, the marginal prior of the regression parameters, βj, is DE(0, λ/2), independently for j = 1, … , p, by virtue of Equation 6. Differences in locus-specific variances induce a selective shrinkage with smaller variances implying stronger shrinkage toward zero, as expected for spurious effects. It is clear, however, that the variance 4/λ4 of 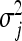 , and hence the amount of shrinkage of all regression parameters is largely influenced by the regularization parameter λ. In the BL, the hyperparameter λ is meant to be estimated alongside the other model parameters and as such, requires a prior distribution e.g., λ ∼ Γ(a, b) for some suitably selected values of a and b. Park and Casella (2008) and Yi and Xu (2008) recognized that the performance of the BL depends critically on the tuning of λ. The hierarchical specification of the priors of the regression coefficients in the BL is graphically depicted in Figure 1a. As can be seen from Figure 1a, all the variances
, and hence the amount of shrinkage of all regression parameters is largely influenced by the regularization parameter λ. In the BL, the hyperparameter λ is meant to be estimated alongside the other model parameters and as such, requires a prior distribution e.g., λ ∼ Γ(a, b) for some suitably selected values of a and b. Park and Casella (2008) and Yi and Xu (2008) recognized that the performance of the BL depends critically on the tuning of λ. The hierarchical specification of the priors of the regression coefficients in the BL is graphically depicted in Figure 1a. As can be seen from Figure 1a, all the variances 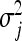 for (j = 1, … , p) are controlled by the hyperparameter λ. This may be unrealistic when dealing with a large number of predictors in which effect sizes may differ greatly, as frequently happens in QTL mapping and in phenotypic value prediction using genome-wide dense sets of markers. Our new method, the extended Bayesian LASSO, relaxes this assumption.
for (j = 1, … , p) are controlled by the hyperparameter λ. This may be unrealistic when dealing with a large number of predictors in which effect sizes may differ greatly, as frequently happens in QTL mapping and in phenotypic value prediction using genome-wide dense sets of markers. Our new method, the extended Bayesian LASSO, relaxes this assumption.
Figure 1.—

Graphical representation of the hierarchical specification of the priors of the locus-specific effects in the BL (a) and the EBL (b), illustrating the roles and scopes of each hyperparameters: λ in the BL and δ and ηj in the EBL.
The extended Bayesian LASSO:
In the vein of the BL, the EBL proceeds by assigning to each regression parameter a Gaussian prior with its own variance, independently. That is, 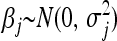 , independently for j = 1, … , p. Each locus-specific variance,
, independently for j = 1, … , p. Each locus-specific variance, 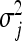 , is further independently assigned an
, is further independently assigned an 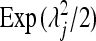 prior. The EBL departs from the BL in that the regularization parameters are locus specific. Crucially for us here, λj is defined in terms of a measure, δ, of model sparsity, which is common to all loci, and a locus-specific deviation, ηj, from δ, which represents the strength of shrinkage specific to locus j. More specifically, we let
prior. The EBL departs from the BL in that the regularization parameters are locus specific. Crucially for us here, λj is defined in terms of a measure, δ, of model sparsity, which is common to all loci, and a locus-specific deviation, ηj, from δ, which represents the strength of shrinkage specific to locus j. More specifically, we let
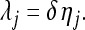 |
(7) |
The common factor δ can, just like λ in the BL, be used to adjust the model to the desired level of sparsity, whereas the locus-specific random deviations, ηj, from δ enforce the difference in the extent of shrinkage across loci. This makes the EBL “doubly adaptive” compared to the adaptiveness of the BL. Figure 1b shows the hierarchical specification for the priors of the regression coefficients in the EBL. The extra layer of the EBL relative to the BL is apparent from Figure 1b. The hyperparameters δ and ηj are also assigned prior distributions and estimated along with the other model parameters. A nice feature of the EBL is that tuning is not critical like in the BL. We discuss this point in more detail further.
APPLICATION
Simulated data analysis:
The performance of the EBL was evaluated on simulated data. The data-generation process used marker data from a well-known data set from the North American Barley Genome Mapping project (Tinker et al. 1996), which has been widely analyzed in mapping studies (e.g., Xu 2003, 2007; Xu and Ja 2007; Yi and Xu 2008). The Barley data, initially intended for analyzing economically important traits in two-row barley (Hordeum vulgare L.), comprise 127 biallelic markers spanning seven chromosomes for 150 DH lines grown in 25 different environments. The markers cover 1270 cM, with an average distance between consecutive markers of 10.5 cM. The original data involved 150 individuals, but five individuals with missing phenotypes (days to heading) were left out. The incentive for using the Barley marker data is to draw close to a real-world situation. The data set also includes some missing genotypes. At the outset, the missing genotypes were filled-in with random draws from Bernoulli (0.5). For a DH population, an individual can take only one of two possible genotypes. The dummy variables xij were coded as xij = 0 for one genotype and xij = 1 for the other. We used the model in (1) to generate phenotypic values assuming a few QTL, namely at loci 4, 25, 50, and 65 with respective effects set to 2.5, −2.5, 4, and −4. We used different values of the residual variance to induce different heritability levels in the data. Heritability, 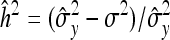 , refers to the proportion of phenotypic variance attributable to genetic factors. Here σ2 is the known residual variance and
, refers to the proportion of phenotypic variance attributable to genetic factors. Here σ2 is the known residual variance and 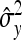 is the empirical phenotypic variance estimated from the data.
is the empirical phenotypic variance estimated from the data.
It is well known that correlations among covariates may greatly affect the performance of variable selection methods. To verify how well our method would perform in the presence of dense marker data, we considered, in addition to the moderately dense Barley marker data, a dense marker data set simulated through the WinQTL Cartographer program (Wang et al. 2006), and used by Sillanpää and Noykova (2008). This data set involves 200 backcross progeny and 102 marker points spanning three chromosomes, with 34 evenly spaced markers on each chromosome. The length of each chromosome was 99 cM, implying a distance of 3 cM between consecutive markers. The methodology described here is straightforwardly applicable to this data set since a backcross can have only two possible genotypes (initially coded as −1 and 1) at each locus. We changed the initial coding to 0 and 1. As before, we assumed only four QTL at loci 25, 60, 70, and 75 with QTL effects of 2.5, −2.5, 4, and −4, respectively. Note that the last two QTL are linked with 15 cM between them and in repulsion (i.e., having effects of opposite signs).
For both the moderately dense Barley marker data and the simulated dense marker data, we generated 50 synthetic data sets of approximately same heritability with the design matrix held fixed and the intercept set to zero without lost of generality. For the reported results, the residual variance was set to 2, yielding an average heritability of 0.8 for the Barley marker data and 0.72 for the simulated dense marker data.
Model fitting and performance evaluation:
We fitted the Bayesian LASSO and extended Bayesian LASSO to the simulated data replicates by Markov chain Monte Carlo (MCMC) (Gilks et al. 1996) simulation through the Bayesian freeware OpenBUGS (Thomas et al. 2006). The BUGS code is available upon request from the authors. The response variable was considered on the original scale to facilitate the comparison between the true effect sizes and their estimates. We used the following, essentially noninformative, priors: β0 ∼ N(0, 100); βj ∼ N(0, 100) for j = 1, …, p; σ2 ∼ Inv − Γ (0.1, 0.1). To mitigate the effect of prior specification on our results, the reported results are based on the noninformative priors λ ∼ Uni (0, 100) in the BL, and δ ∼ Uni (0, 100), ηj ∼ Uni (0, 100) in the EBL. These priors are diffuse (flat). This implies no tuning, which is important if we are to compare the performances of the two models.
Initially, we ran three separate Markov chains for 10,000 iterations each, to assess how fast the chain would converge and how well they would mix (mixing refers to the ease with which the Markov chains explore the full parameter space). For the simulations based on the Barley marker data, 10,000 iterations took 21,483 sec for the BL and 22978 sec for the EBL (i.e., comparable computation time) on an AMD with 1.33 GHz and 768 MB of RAM. For both methods, the chains seemed to reach the stationary distribution after about 1500 iterations. The convergence was assessed through visual inspection of the MCMC traceplots. The sensitivity of the results to the prior specification was assessed by varying the range of the uniform priors for hyperparameters λ in the BL, and δ and ηj in the EBL, but the results were robust to these changes.
After the initial evaluation of the two models, we used simulated data replicates to evaluate their performances in estimating the regression parameters with regard to both the true signals (i.e., QTL) and false signals (i.e., non-QTL), and in predicting unobserved phenotypes. For each replicated data set, we ran 10,000 MCMC iterations and discarded the first 2000 samples as burn-in.
The estimation error on the true and the false signals were respectively evaluated through the statistics
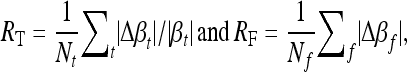 |
where 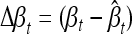 , βt and
, βt and 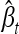 being the true and the estimated effects of locus t, respectively. The summation in the definition of RT is taken over the true signals, and Nt is the number of true signals. RF is defined in a similar manner to RT, but βf = 0, and the formula for RF involves no scaling by the absolute value of the true effect size, which is zero. The number, Nf, of false signals, is given by Nf = (p − Nt).
being the true and the estimated effects of locus t, respectively. The summation in the definition of RT is taken over the true signals, and Nt is the number of true signals. RF is defined in a similar manner to RT, but βf = 0, and the formula for RF involves no scaling by the absolute value of the true effect size, which is zero. The number, Nf, of false signals, is given by Nf = (p − Nt).
The predictive performances of the two models were evaluated through posterior predictive cross-validation. The basic idea of cross-validation (e.g., Picard and Cook 1984) is to fit the model to a subset of the data (the training set) and utilize the remaining data (the test set) to evaluate the model predictive performance. A cross-validation loss function (usually the mean square error or the root mean square error) is required for comparison, and the model associated with the smallest loss is chosen as the best. Here we left out (i.e., we omitted from the training set) two randomly selected phenotypic values in each replicated data set and monitored their posterior predictive means estimated from the MCMC samples. For each replicated data set, we used the posterior predictive means of the omitted data points to compute the root mean square error (RMSE)
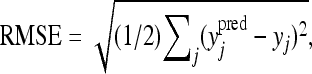 |
where j runs over the indices of the discarded phenotypes, 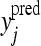 is the posterior predictive mean of the jth discarded phenotype, its actual value being yj.
is the posterior predictive mean of the jth discarded phenotype, its actual value being yj.
To have significance thresholds for distinguishing QTL from non-QTL, we randomly shuffled the phenotypes (permutation without replacement) to artificially destroy the association between the markers and the phenotypes. We fitted the model to each of the ensuing data sets using MCMC simulation. We ran 4000 MCMC iterations, discarding the first 2000 samples as burn-in. For each data set, we monitored the largest absolute posterior mean effect size used as our test statistic. This provided us with an empirical distribution F of the test statistic, and we used the 100 × (1 − α) percentile of as the critical value (Churchill and Doerge 1994). The significance thresholds based on 100 phenotype permutations with α = 0.10 were found to be 0.64 for the BL and 0.67 for the EBL.
RESULTS
The results for the simulations based on the Barley marker data were broadly similar to those based the simulated dense marker data set. In this section, we report only on the performance of the BL and the EBL based on the Barley marker data (an extensive comparison of the EBL to the contending BAL, including for the dense marker data, is provided in the discussion). Figure 2 summarizes the performance of the EBL relative to the BL with regard to the accuracy of effect size estimates on both true and false signals and the accuracy of phenotypic value predictions. Figure 2, bottom right, displays the ratios λ/δ of the locus-independent variance hyperparameters in the BL and the EBL, which is roughly constant over the 50 replicated data sets.
Figure 2.—

Performance evaluation of the EBL and the BL over 50 replicated data sets using the Barley marker data. The results are summarized by the posterior means over 50 replicated data sets. Top left: the estimation errors (differences between the posterior means and known true values) with regard to true signals i.e., QTL. Top right: the estimation errors with regard to false signals i.e., non-QTL. Bottom left: predictive root mean square errors for the BL and the EBL. Bottom right: the ratios of posterior means of λ (in the BL) and δ (in the EBL) over 50 replicated data sets, illustrating the correspondence between the roles of these two locus-independent parameters (λ and δ) in the two methods.
Figure 3 shows the posterior means of QTL effects under the BL and the EBL, along with the true values. The results displayed in Figure 3 suggest that the EBL estimates track the true effects more efficiently than their BL counterparts.
Figure 3.—

Posterior means of estimated QTL effects for the BL (solid squares) and the EBL (solid triangles) at QTL positions, along with their true effects (shaded circles). The plotted values represent the posterior means averaged over 50 replicated data sets based on the Barley marker data.
Figure 4 shows the posterior means of all loci effects averaged over the 50 replicated data sets for the BL and the EBL. For each method, the significance thresholds for declaring QTL are indicated by the horizontal dashed lines.
Figure 4.—

Posterior means of all marker effects averaged over the 50 replicated data sets based on the Barley marker data for the BL and the EBL. The dashed lines indicate the permutation-based QTL significance thresholds.
For a significance level of 10%, the detection probabilities for QTL were roughly 0.87 for the BL and 0.91 for the EBL.
DISCUSSION
Our discussion is structured in subsections relating to different aspects on which the BL and the EBL are being evaluated. We also compare the performance of the EBL to the BAL recently proposed by Sun et al. (2010), which also introduces locus-specific regularization parameters.
Signal detection:
The locus-effect estimates under the EBL were always closer to the true values than their BL counterparts (Figure 3), resulting in lower estimation errors with regard to the true signals (i.e., QTL) and false signals (i.e., non-QTL) (Figure 2). Nevertheless, both the BL and the EBL were effective at identifying the known QTL in our simulation settings, as exemplified by the plots in Figures 3 and 4.
Predictive performance:
The EBL outperformed BL in predictive accuracy, with lower prediction errors (RMSEs displayed in Figure 2). This implies that the EBL is also better suited to breeding value prediction in genomic selection. Our specification of the regularization parameters provides the possibility of incorporating prior information regarding the level of model sparsity through the prior of the overall sparsity parameter, δ, while letting the locus-specific hyperparameters, ηj (j = 1, … , p), take care of keeping the nonzero effects “on” and adjusting their sizes according to their importance. This doubly adaptive feature is appealing in that it allows the model to effectively exert less shrinkage on the effects of important covariates required to be into the model, and more shrinkage on spurious effects. This results in more accurate estimates of marker effects and predictions of phenotypic values as implied by the results of our simulation study. The BL may miss the point as it relies on a single regularization parameter for model sparsity and parameters shrinkage.
Sensitivity to prior specification and separation of QTL from non-QTL loci:
In the BL, the hyperparameters of the prior p(λ) require suitable tuning, with optimal values depending on the data and the number of variables included (Yi and Xu 2008), meaning that tuning is critical to the performance of the BL. The EBL estimates proved to be robust to tuning, owing presumably to the way the priors of the locus-specific hyperparameters λj are specified. In particular, the ratio of the posterior means of the parameters λ and δ in the BL and the EBL, respectively, was found to be roughly constant (Figure 2, bottom right). This indicates that these two parameters equivalently regulate the level of model sparseness in the two models by “borrowing strength” across loci. A consequence of the reliance of the BL on a single smoothing parameter is that placing on this parameter a prior that strongly enforces sparsity will result in all regression parameters being indiscriminately shrunken. This is not a desirable feature for phenotype prediction, as most of the small but relevant parameters will be set to zero.
In the EBL, the locus-specific hyperparameters ηj play a key adaptive role, which makes tuning much less of an issue. It also is obvious that the parameters δ and ηj in the EBL have meaningful interpretations as they operate on different scales: global for the former and local (i.e., locus specific) for the latter. The graphical representation of the hierarchical specifications of the priors for locus-specific effects in the BL and the EBL shown in Figure 1 better illustrates the roles and scopes of the hyperparameters λ in the BL, and (δ and ηj) in the EBL.
It is worth emphasizing that, although the Laplacian distribution is mathematically equivalent to a mixture of an infinite number of Gaussians with exponentially distributed mixing variances as implied in Equation 6, it is computationally more convenient to not integrate out the mixing layer when the required computations can efficiently be carried, for example, through Markov chain Monte Carlo (Gilks et al. 1996). This recommendation seems to oppose the all-too-common practice of analytically integrating out intermediary layers of hierarchical priors in the quest for simplicity. O'Hara and Sillanpää (2009) found that using the Laplace prior directly on the regression parameters in the Bayesian LASSO as obtained by the analytical integration in Equation 6 may result in poor mixing of the MCMC sampler and bad separation of the variables. Like Yi and Xu (2008) and De los Campos et al. (2009), we did not face any mixing problems with the hierarchical specification of the Laplace distribution when implementing the BL and the EBL. The same applies to the Student t-distribution, which is mathematically equivalent to a mixture of an infinite number of Gaussians with inverse gamma-mixing variances.
Statistical identifiability issues:
From a statistical model-fitting perspective, our parameterization of the locus-specific regularization parameters, λj, as a product of a factor, δ, common to all loci and a locus-specific effect, ηj, is crucial for statistical identifiability. If these two components were both indexed by j, they would be confounded and, hence, statistically unidentifiable.
Comparison to the Bayesian adaptive LASSO:
Sun et al. (2010) also introduced locus-specific smoothing parameters in the Bayesian LASSO, but without a separation between model sparsity and parameter shrinkage hyperparameters. They proposed a fully Bayesian method, the BAL, along with an iterative method, the iterative adaptive LASSO, for fitting their model to the data. We compared the performance of the EBL to the BAL on simulated data on the basis of the Barley marker data and the simulated dense marker data set used before. We fitted the BAL to the simulated data replicates using the R library BPrimm (Sun et al. 2010). The BAL has two tuning hyperparameters δ and τ, with a default setting of δ = 1 and τ = 0.01. We used a set of hyperparameter values. With the default setting, the BAL had comparable performance to the EBL on these particular data sets with a good separation between QTL and non-QTL effects as can be seen from Figure 5. The reported results are based on 10,000 MCMC iterations with 4000 burn-in iterations.
Figure 5.—

Performance evaluation of the EBL and the BAL on the Barley marker data (left), and a simulated dense marker data set (right). The results are summarized by boxplots of posterior means over 50 replicated data sets. Top: the estimation errors (differences between the posterior means and known true values) for true signals i.e., QTL. Middle: the estimation errors with regard to false signals i.e., non-QTL. Bottom: predictive root mean square errors.
However, we found that the performance of the BAL may be sensitive to the tuning of the hyperparameters δ and τ, with larger values of τ pointing to bad separation between QTL and non-QTL effects. This is apparent from the results displayed in Figure 6, which depicts typical patterns of posterior means of the marker effects for different hyperparameter values under our simulation setting based on the Barley marker data.
Figure 6.—

Typical patterns of posterior means of marker effects for different hyperparameter values under our simulation setting based on the Barley marker data with QTL at loci 4, 25, 50, and 65 with respective effects 2, −2, 4, and −4. The results are based on 10,000 MCMC iterations, with the first 4000 samples discarded as burn-in.
It is worth giving credit to Sun et al. (2010) for the recommendation of small values of τ. However, too small a value for τ may excessively shrink and eventually set to zero small to moderate effects. As already pointed out above, this might not be desirable for phenotype prediction. The bottom line is that, while Sun et al.'s (2010) approach is an improvement over the Bayesian LASSO, tuning is still an issue therein. The EBL brings more flexibility by separating model sparsity and parameter shrinkage, thereby mitigating the impact of tuning on the model results. In our simulation study, the locus-specific hyperparameter, λj, was efficiently estimated from the data, even when uniform prior with large support such as (0, 100) were placed on δ and ηj, and the results remained robust to changes in the range of these hyperpriors.
Scalability:
It is important to point out that our focus here is on model structure. We are currently engaged in designing practical computational methods that may be needed for large-scale problems. For problems involving a couple of hundreds of predictors (loci), the MCMC-based full Bayesian approach using WinBUGS/OpenBUGS as described here can efficiently be applied. De los Campos et al. (2009) applied a MCMC-based fully Bayesian approach to fit the BL to a data set involving more than 10,000 predictors.
CONCLUSION
In this article we have proposed the extended Bayesian LASSO method for QTL mapping and unobserved phenotype prediction. The EBL introduces locus-specific regularization parameters and is parameterized so as to separate the overall model sparsity to the degree of shrinkage of each marker effect. The explicit separation of these two features allows the EBL to overcome the curse of tuning, which may affect the performance of a similar approach proposed by Sun et al. (2010), which, like the BL, does not include a mechanism for separating model sparsity from parameter shrinkage. We are unaware of any previous attempt to separate these two aspects.
Under our model parameterization, the parameter representing the overall model sparsity is effectively estimated by pooling information (i.e., borrowing strength) across loci, whereas locus-specific effects meant to enforce differential shrinkage across loci are obtained as deviations from the overall parameter. Xu (2003) also proposed the normal Jeffreys' Bayesian shrinkage method, which does not require tuning. However, unlike Xu's approach, the methodology proposed here entertains the possibility of incorporating information relating to the degree of model sparsity through the prior imposed on the corresponding parameter.
Simulations demonstrated the good performance of the EBL with regard to the accuracy of parameter estimates and phenotype predictions, suggesting the potential of this method in QTL/association mapping and phenotype prediction, as well as genomic breeding value estimation and prediction in animal and plant breeding programs.
Acknowledgments
We are grateful for the constructive comments of two anonymous reviewers. This work was supported by a research grant from the Academy of Finland and University of Helsinki's research funds.
References
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De los Campos, G., H. Naya, D. Gianola, J. Crossa, A. Legarra et al., 2009. Predicting quantitative traits with regression models for dense molecular markers and pedigree. Genetics 182 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks, W.R., S. Richardson and D. J. Spiegelhalter, 1996. Markov Chain Monte Carlo in Practice. Chapman & Hall, London.
- Hoerl, A. E., and R. W. Kennard, 1970. Ridge regression: biased estimation for nonorthogonal problems. Technometrics 12 55–67. [Google Scholar]
- Lee, S. H., J. H. J. Van der Werf, B. J. Hayes, M. E. Goddard and P. M. Visscher, 2008. Predicting unobserved phenotypes for complex traits from whole-genome SNP data. PLoS Genet. 4 e1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malo, N., O. Libiger and N. J. Schork, 2008. Accommodating linkage disequilibrium in genetic-association analyses via ridge regression. Am. J. Hum. Genet. 82 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara, R. B., and M. J. Sillanpää, 2009. A review of Bayesian variable selection methods: what, how and which. Bayesian Anal. 4 85–118. [Google Scholar]
- Park, T., and G. Casella, 2008. The Bayesian LASSO. J. Am. Stat. Assoc. 103 681–686. [Google Scholar]
- Picard, R. R., and R. D. Cook, 1984. Cross-validation of regression models. J. Am. Stat. Assoc. 79 575–583. [Google Scholar]
- Pikkuhookana, P., and M. J. Sillanpää, 2009. Correcting for relatedness in Bayesian models for genomic data association analysis. Heredity 103 223–237. [DOI] [PubMed] [Google Scholar]
- Sen, S., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servin, B., and M. Stephens, 2007. Imputation-based analysis of association studies: candidate regions and quantitative traits. PLoS Genet. 3 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpää, M. J., and N. Noykova, 2008. Hierarchical modeling of clinical and expression quantitative trait loci. Heredity 101 271–284. [DOI] [PubMed] [Google Scholar]
- Sun, W., J. G. Ibrahim and F. Zou, 2010. Genome-wide multiple loci mapping in experimental crosses by the iterative penalized regression. Genetics 185 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, A., R.B. O'Hara, U. Ligges and S. Sturtz, 2006. Making BUGS Open. R News 6 12–17. [Google Scholar]
- Tibshirani, R., 1996. Regression shrinkage and selection via LASSO. J. R. Stat. Soc. B 58 267–288. [Google Scholar]
- Tinker, N. A., D. E. Mather, B. G. Rosnagel, K. J. Kasha, and A. Kleinhofs, 1996. Regions of the genome that affect agronomic performance in two-row barley. Crop Sci. 36 1053–1062. [Google Scholar]
- Usai, M. G., D. M. E. Goddard and B. J. Hayes, 2009. LASSO with cross-validation for genomic selection. Genet. Res. 91 427–436. [DOI] [PubMed] [Google Scholar]
- Wang, H., Y-M. Zhang, X. Li, G. L. Masinde, S. Mohan et al., 2005. Bayesian shrinkage estimation of quantitative trait loci parameters. Genetics 170 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S., C. J. Basten, and Z-B. Zeng, 2006. Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC.
- Whittaker, J. C., R. Thompson and M. C. Denham, 2000. Marker-assisted selection using ridge regression. Genet. Res. 75 249–252. [DOI] [PubMed] [Google Scholar]
- Wu, T. T., F. C. Yi, T. Hastie, E. Sobel and K. Lange, 2009. Genome-wide association analysis by LASSO penalized logistic regression. Bioinformatics 25 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C., X. Wang, Z. Li and S. Xu, 2009. Mapping QTL for multiple traits using Bayesian statistics. Genet. Res. 91 23–37. [DOI] [PubMed] [Google Scholar]
- Xu, S., 2003. Estimating polygenic effects using markers of the entire genome. Genetics 163 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S., 2007. An empirical Bayes method for estimating epistatic effects of quantitative trait loci. Biometrics 63 513–521. [DOI] [PubMed] [Google Scholar]
- Xu, S., and Z. Ja, 2007. Genomewide analysis of epistatic effects for quantitative traits in barley. Genetics 175 1955–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., and S. Xu, 2008. Bayesian LASSO for quantitative trait loci mapping. Genetics 179 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y-M., and S. Xu, 2005. A penalized maximum likelihood method for estimating epistatic effects of QTL. Heredity 95 96–104. [DOI] [PubMed] [Google Scholar]
- Zou, H., and T. Hastie, 2005. Regularization and variable selection via elastic net. J. R. Stat. Soc. B 67 301–320. [Google Scholar]