

Abstract
Background
Microarray gene expression data are accumulating in public databases. The expression profiles contain valuable information for understanding human gene expression patterns. However, the effective use of public microarray data requires integrating the expression profiles from heterogeneous sources.
Results
In this study, we have compiled a compendium of microarray expression profiles of various human tissue samples. The microarray raw data generated in different research laboratories have been obtained and combined into a single dataset after data normalization and transformation. To demonstrate the usefulness of the integrated microarray data for studying human gene expression patterns, we have analyzed the dataset to identify potential tissue-selective genes. A new method has been proposed for genome-wide identification of tissue-selective gene targets using both microarray intensity values and detection calls. The candidate genes for brain, liver and testis-selective expression have been examined, and the results suggest that our approach can select some interesting gene targets for further experimental studies.
Conclusion
A computational approach has been developed in this study for combining microarray expression profiles from heterogeneous sources. The integrated microarray data can be used to investigate tissue-selective expression patterns of human genes.
Background
There are many different types of cells in the human body, and similar cells group together to form a tissue with a specialized function. Multiple tissues constitute an organ such as brain, heart or liver. Gene expression variation is the primary determinant of tissue identity and function. Certain genes are expressed specifically or preferentially in a particular tissue. These genes are broadly called tissue-selective genes [1]. Note that tissue specificity is regarded as a special case of tissue selectivity, and tissue-specific genes are expressed only in a particular tissue. It is a fundamental question in biology to understand how selective gene expression underlies tissue development and function. Moreover, tissue-selective genes are implicated in many complex human diseases [2], and identification of these genes may provide valuable information for developing novel biomarkers and drug targets [1].
Tissue-selective expression was traditionally studied at the single-gene level with time-consuming techniques such as Northern blot and in situ hybridization. With the recent development of high-throughput technologies, biologists can perform genome-wide gene expression profiling in various tissues. These high-throughput technologies include Expressed Sequence Tag (EST) sequencing, Serial Analysis of Gene Expression (SAGE), and DNA microarrays. Yu et al. [3] analyzed the NCBI EST database (dbEST) to select a set of human genes that are preferentially expressed in a tissue of interest. The selection was based on the expression enrichment score, which was defined as the ratio between observed and expected number of ESTs for a gene. For the selected tissue-selective genes, regulatory modules were detected by examining the promoter motifs and their relationships with transcription factors. However, EST data are generated mainly for transcript sequence information, and EST counts can only be used as rough estimates of gene expression levels. Siu et al. [4] investigated gene expression patterns in different regions of the human brain by using SAGE, and identified some brain region-selective genes. Kouadjo et al. [5] also used the SAGE strategy to identify housekeeping and tissue-selective genes in fifteen mouse tissues. While SAGE tag counts can provide reliable estimation of gene expression, it is rather inefficient and expensive to use SAGE for profiling a large number of tissue samples with biological replicates.
The DNA microarray technology has been widely used to simultaneously profile the levels of thousands of mRNA transcripts in various tissues, and may hold great promise for elucidating the molecular mechanisms of complex human diseases. Many microarray datasets have been generated for identifying disease-associated biomarkers, classifying disease types, and predicting treatment outcomes. However, only a few microarray studies were designed to investigate human tissue-selective gene expression. Su et al. [6] used custom oligonucleotide arrays to examine the expression patterns of predicted genes across a panel of human and mouse tissues. The NCBI Gene Expression Omnibus (GEO at http://www.ncbi.nlm.nih.gov/geo/) has an Affymetrix microarray dataset for human body index of gene expression (GEO accession: GSE7307). Since each individual dataset does not contain a large number of expression profiles of various tissues, computational methods may be used to integrate the gene expression data from different microarray studies. Greco et al. [7] investigated tissue-selective expression patterns with an integrated dataset of microarray profiles publicly available at the GEO database. The relatively small dataset contained 195 expression profiles from six different microarray studies. The results suggested that gene expression data from Affymetrix GeneChip experiments could be integrated through pre-processing raw data (CEL files) with commonly used methods.
In this study, we have compiled a compendium of 2,968 microarray expression profiles of various human tissues from the NCBI GEO database. These expression profiles have been selected from 131 microarray datasets generated at different laboratories. Our data integration approach includes microarray data normalization, transformation, and quality control. The integrated data have been used to identify brain, liver and testis-selective genes using a new computational method based on both microarray hybridization intensities and detection calls. The results further suggest that the publicly available microarray expression profiles from heterogeneous sources can be integrated into a single dataset for examining gene expression patterns across various tissues.
Methods
Collection and curation of microarray gene expression profiles
Human microarray gene expression data are accumulating in public databases. These expression profiles have been generated for various research objectives, and show significant variations in data quality. To compile a compendium of high-quality microarray profiles for studying gene expression patterns, we manually curated the human microarray data publicly available in the NCBI GEO database (as of November 3, 2009). The following criteria were used to select microarray expression profiles in this study. First, the profiles had to be generated using the Affymetrix HG-U133 Plus 2.0 Array, a platform for complete coverage of the human genome with 54,675 probe sets. This array platform was used by the majority of human gene expression profiles deposited in the GEO database. Second, a detailed description of the microarray profiling study and raw data in CEL file format was available. The description contained important information about a microarray sample (e.g., tissue source, clinical condition, treatment, etc). Third, the expression profiles had to be obtained using normal tissue samples. Microarray profiles of cancer cells or diseased tissues were excluded from selection. Fourth, the tissue sample used for microarray profiling should not be cultured in vitro or treated with any drugs before RNA extraction. No expression profiles of primary or secondary cell cultures were selected for this study.
By following the above criteria, we compiled 3,030 microarray gene expression profiles across a variety of human tissues (Table 1). The number of selected profiles varied among tissues, depending on data availability. An attempt was made to include as many tissues as possible, even though some tissues had only a few expression profiles available in the GEO database. Nevertheless, some tissues had a relatively large number of expression profiles, and were thus particularly suited for identifying tissue-selective genes. For instance, there were 645 brain gene expression profiles (616 profiles after data quality control). These expression profiles were obtained from various regions of postmortem brain such as entorhinal cortex, hippocampus and cerebellum, and could be used to identify genes specifically expressed in neurons.
Table 1.
List of human tissues and microarray expression profiles
| Tissue | # of profiles selected | # of profiles integrated |
|---|---|---|
| Brain (various regions) | 645 | 616 |
| Pituitary gland | 12 | 12 |
| Thyroid gland | 16 | 9 |
| Adrenal gland | 25 | 25 |
| Pancreas | 56 | 55 |
| Skeletal muscle | 122 | 109 |
| Skin | 101 | 101 |
| Adipose tissue | 80 | 80 |
| Retina | 12 | 12 |
| Gingiva | 71 | 71 |
| Salivary gland | 18 | 18 |
| Tongue | 22 | 20 |
| Stomach | 51 | 51 |
| Small intestine | 59 | 59 |
| Colon | 107 | 105 |
| Liver | 117 | 117 |
| Kidney | 73 | 73 |
| Breast | 132 | 132 |
| Ovary | 61 | 59 |
| Uterus | 117 | 117 |
| Placenta | 56 | 56 |
| Umbilical cord | 54 | 54 |
| Testis | 36 | 36 |
| Prostate | 58 | 58 |
| Nasal epithelium | 31 | 31 |
| Airway epithelium | 89 | 89 |
| Lung | 66 | 66 |
| Alveolar macrophage | 88 | 87 |
| Heart | 31 | 31 |
| Tonsil | 13 | 13 |
| Lymph node | 14 | 14 |
| Blood (various cell types) | 413 | 409 |
| Other tissues | 184 | 183 |
Microarray data normalization and integration
Microarray raw data in CEL file format were downloaded from the GEO database, and then normalized by using the dChip software (available at http://www.dchip.org). As a widely used tool for microarray data analysis, dChip can display and normalize CEL files with a model-based approach [8]. For a given group of CEL files, dChip can be used to calculate the model-based expression values and make the qualitative detection calls for each array. The detection call (Present, Marginal or Absent) provides a statistical assessment about whether the perfect matches (PMs) show significantly more hybridization signal than the corresponding mismatches (MMs) in a probe set. Since the detection call and expression level are computed in different ways, a gene transcript with an Absent call may still be given an expression value (although usually low).
One challenging task in this study was to combine the expression profiles of various tissue types and from different microarray studies into a single integrated dataset. As outlined in Figure 1, our approach included the following steps. First, the selected microarray CEL files were organized into different normalization groups, each of which contained expression profiles of the same or similar tissue type. For example, one normalization group was consisted of 117 liver microarray profiles, whereas another group contained 112 expression profiles of six endocrine glands, including pituitary gland (12 profiles), thyroid gland (16 profiles), parathyroid gland (1 profile), thymus gland (2 profiles), adrenal gland (25 profiles) and pancreas (56 profiles). Within a normalization group, the variation of tissue type was thus minimized although the expression profiles were nevertheless obtained from different microarray studies.
Figure 1.

Schematic diagram of microarray data normalization and integration.
Second, each group of microarray profiles was normalized by using the invariant set method [9]. For each normalization group, the expression profile with median overall intensity was chosen as the baseline array, against which the other profiles were normalized at probe intensity level. A subset of PM probes with small rank difference between the profile to be normalized and the baseline array were chosen as the invariant set for fitting a normalization curve. The normalization transformation was then performed for all the probes in the profile based on the curve [9]. While the invariant set normalization method could reduce the variation in microarray profiles from different studies, it might not be applied to an expression dataset with various tissue types. Owing to the biological variation of gene expression across different tissues, a baseline array should be used to normalize the microarray profiles of each tissue type (or similar tissues).
Finally, the normalized microarray profiles were integrated into a single dataset after outlier array exclusion and global median transformation. When fitting the statistical model to a probe set, dChip used an outlier detection algorithm to identify array-outliers whose response pattern for the probe set was significantly different from the consensus probe response pattern in the other arrays [8]. After the model was fitted for all probe sets, the percentage of probe sets detected as array-outliers was calculated for each array. If the percentage exceeded 15%, the array was discarded as an outlier array. In this study, only 62 outlier arrays were detected for all the 3,030 selected expression profiles (Table 1). Global median transformation was then applied to the remaining profiles. Each expression value in a profile was divided by the profile’s median value. The transformation was necessary because the expression profiles from different normalization groups often had different median values. Thus, the integrated dataset had 2,968 expression profiles with the same median value (i.e., 1.00).
Genome-wide identification of tissue-selective genes
In this study, a new computational method has been designed to analyze the integrated microarray data for identifying tissue-selective genes, which refers to the genes specifically or preferentially expressed in a particular tissue. The computational task is not trivial for the following reasons. First, the expression profiles have been compiled from various studies, in which tissues at different ages and in different conditions were used for microarray profiling. Thus, the microarray profiles of the same tissue type should not be considered as biological replicates. Second, some tissue-selective genes can be expressed at certain developmental stages or in specific conditions, and their expression may not be consistently detected in all the microarray profiles of a tissue type. Third, microarray data are inherently noisy. It was thus desired that both the expression values and detection calls of microarray profiles can be utilized for tissue-selective gene identification.
Figure 2 illustrates our approach for genome-wide identification of tissue-selective genes. First, for a given tissue type t, the microarray expression profiles are divided into two sets: experiment set and control set. The experiment set contains the expression profiles of tissue type t, and the control set has the expression profiles of the other tissue types. The experiment set usually has fewer microarray profiles than the control set. For example, to identify brain-selective genes in this study, the experiment set contained 616 expression profiles, whereas the control set had 2,352 expression profiles of the other tissue types such as liver, kidney, muscle, skin, etc.
Figure 2.

Schematic diagram of microarray data analysis for genome-wide identification of tissue-selective genes.
Second, all the human genes (array probe sets) are examined for significant expression in the microarray profiles. The term “significant expression” in this study is used to describe gene expression data that meet the following two criteria: (1) the detection call is Present; and (2) the expression value is no less than a threshold θ (θ ≥ 0). Since there are no negative values in a microarray profile, significant expression would be solely defined by the detection call if θ = 0. For each probe set, the number of significant expression in the experiment set (Se) and that in the control set (Sc) are calculated. Genes that have Se ≥ min and Sc ≤ max are selected for further analyses. The threshold min is used to specify the minimum number of significant expression that should be detected in the experiment set. Considering the noise in microarray data, significant expression may also be detected in the control set, but the number Sc should not exceed max (maximum number of significant expression). The threshold max is set to 0 if no observation of significant expression is allowed in the control set. For a tissue-selective gene, its frequency of significant expression should be higher in the experiment set than in the control set. Score1 is calculated as follows:
(1)
where Ne is the total number of expression profiles in the experiment set, and Nc is the total number of expression profiles in the control set.
Third, for each selected probe set, its expression level in the experiment set is compared with that in the control set. Our assumption is that potential tissue-selective genes should show higher expression in the experiment arrays than in the control arrays. Score2 is calculated as follows:
(2)
where 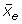 is the mean expression level of the selected probe set in the Se experiment arrays with significant expression, and
is the mean expression level of the selected probe set in the Se experiment arrays with significant expression, and 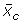 is the mean expression level in control arrays. In this study, the control arrays were sorted according to their expression values for the selected probe set, and the top Se control arrays with the highest expression values were used to compute the mean,
is the mean expression level in control arrays. In this study, the control arrays were sorted according to their expression values for the selected probe set, and the top Se control arrays with the highest expression values were used to compute the mean, 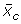 . The probe sets with Score2 ≤ 0 were excluded from consideration for tissue-selective genes.
. The probe sets with Score2 ≤ 0 were excluded from consideration for tissue-selective genes.
Finally, the potential tissue-selective gene targets are prioritized according to the overall score, which is calculated as follows:
(3)
where w1 and w2 are two weights for Score1 and Score2, respectively. In this study, w1 = 1 and w2 = 1 were used to calculate the priority score for each selected probe set. Moreover, the statistical significance of the tissue-selective expression pattern was evaluated by the permutation analysis. The hybridization signals of a probe set, including its expression values and detection calls, were permuted, and then divided into the experiment and control set to calculate the priority score. After one million permutations were performed for each selected probe set, the significance level (p-value) was calculated as the fraction of permutations that gave rise to scores greater than or equal to the actual priority score of the probe set. The p-value thus provided an estimation of the probability for observing the tissue-selective expression pattern by chance.
Results and discussion
A compendium of 2,968 expression profiles of various human tissues have been compiled from 131 microarray studies. These expression profiles have been combined into a single dataset after global normalization, and then used for the genome-wide analysis of tissue-selective gene expression. Although the analysis can be performed for any tissues with available microarray data (Table 1), we present in this paper the results from three case studies on brain, liver and testis-selective gene expression.
Brain-selective gene expression
The human brain is highly complex, and contains 50-100 billion neurons. There are many different brain regions with specific functions. For example, the frontal lobe is involved in higher mental functions and long-term memories, whereas the occipital lobe is the visual processing center. In this study, the microarray expression profiles of different brain regions were combined into the experiment set (616 profiles), and compared with the expression profiles of non-brain tissues in the control set (2,352 profiles). Thus, the brain-selective genes identified in this study might be involved in basic neuron functions such as neural signal processing and transmission via synapses (complex membrane junctions between neurons).
Table 2 shows the top 20 high-scoring genes from one of the analyses with different parameter settings. In this analysis, significant expression was defined by the detection call being Present and the relative expression value no less than 1.00 (array median value). The minimum number of significant expression in the experiment group (min) was set to 62 (~10% of experiment arrays), and the maximum number of significant expression in the control group (max) was set to 24 (~1% of control arrays). With the above parameters, 222 genes have been identified as brain-selective targets with the priority score ranging from 1.18 to 4.69 (see Additional file 1). The permutation analysis suggests that the brain-selective expression patterns of all the selected genes are statistically significant (p < 0.000001). In Figure 3, the gene expression patterns are visualized with the heat map generated by using TM4 MeV [10]. Clearly, the transcripts of the selected genes are predominantly detected in brain samples.
Table 2.
List of high-scoring genes with selective expression in the brain 1
| Probe | Gene | Description | Se | Sc | 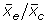 |
Score |
|---|---|---|---|---|---|---|
| 205914_s | GRIN1 | Glutamate receptor, ionotropic, N-methyl D-aspartate 1 | 284 | 0 | 4.54 | 4.69 |
| 236324 | MBP | Myelin basic protein | 211 | 0 | 5.13 | 4.62 |
| 238061 | LGI3 | Leucine-rich repeat LGI family gene 3 | 536 | 1 | 14.77 | 4.48 |
| 205989_s | MOG | Myelin oligodendrocyte glycoprotein | 569 | 2 | 24.03 | 4.42 |
| 206899 | NTSR2 | Neurotensin receptor 2 | 380 | 1 | 16.49 | 4.38 |
| 203540 | GFAP | Glial fibrillary acidic protein | 604 | 14 | 115.10 | 4.28 |
| 244113 | - | cDNA sequence (GB: R44603) | 595 | 3 | 24.55 | 4.27 |
| 231489_x | - | cDNA sequence (GB: H12214) | 551 | 18 | 94.24 | 4.04 |
| 206970 | CNTN2 | Contactin 2 (axonal) | 407 | 1 | 6.67 | 4.02 |
| 224536_s | PCDHGC5 | Protocadherin gamma subfamily C, 5 | 68 | 0 | 3.37 | 3.94 |
| 1556877 | - | cDNA sequence (GB: BC040662) | 364 | 1 | 5.48 | 3.88 |
| 235375_x | TTC9B | Tetratricopeptide repeat domain 9B | 408 | 3 | 13.64 | 3.85 |
| 208320 | CABP1 | Calcium binding protein 1 | 365 | 5 | 18.23 | 3.71 |
| 230255 | GABRD | GABA-A receptor subunit delta | 451 | 8 | 23.42 | 3.70 |
| 235794 | MOBP | Myelin-associated oligodendrocyte basic protein | 596 | 12 | 25.64 | 3.69 |
| 206678 | GABRA1 | GABA-A receptor subunit alpha 1 | 532 | 15 | 35.05 | 3.68 |
| 233471 | PTPN5 | Protein tyrosine phosphatase, non-receptor type 5 | 350 | 2 | 5.52 | 3.57 |
| 1557481_a | C21orf131 | Chromosome 21 open reading frame 131 | 474 | 3 | 5.92 | 3.55 |
| 219642_s | PEX5L | Peroxisomal biogenesis factor 5-like | 415 | 6 | 12.91 | 3.53 |
| 232409_x | FBXL16 | F-box and leucine-rich repeat protein 16 | 227 | 3 | 11.70 | 3.53 |
1 The number of significant expression in the experiment set (Se) and that in the control set (Sc) are shown together with the ratio of the mean expression level in the experiment arrays to the level in the control arrays (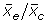 ).
).
Figure 3.

Visualization of tissue-selective gene expression patterns. The normalized expression values were transformed by logarithm (base 10), and each microarray profile had the median value of zero.
Perhaps more importantly, many genes identified in this study have been previously suggested to be expressed specifically or preferentially in the brain. These genes include GRIN1, MBP, LGI3, MOG, NTSR2, GFAP, CNTN2, PCDHGC5, CABP1, GABRD, MOBP and GABRA1 (Table 2). The protein encoded by the GRIN1 gene is a critical subunit of the glutamate receptor channel, and plays a key role in the plasticity of synapses underlying memory and learning [11]. Genetic alterations in GRIN1 have been shown to be associated with Alzheimer's disease [12] and bipolar disorder [13]. In this study, GRIN1 has the highest priority score with significant expression in 284 brain samples but none in the other tissues (Table 2). GABRD and GABRA1 encode two subunits of the GABA-A receptor, which binds the major inhibitory neurotransmitter GABA in the brain [14]. GABA-A receptors are chloride channels that regulate membrane potential, and play structural roles in synapse maturation and stabilization. LGI3 encodes a leucine-rich repeat protein involved in the regulation of neuronal exocytosis [15]. CABP1 is a neuron-specific member of the calmodulin superfamily, and modulates Ca2+-dependent activity of inositol 1, 4, 5-trisphosphate receptors [16]. Both CNTN2 and PCDHGC5 encode immunoglobulin-like proteins important for the establishment and function of neural connections in the brain [17,18]. In addition, MBP, MOG and MOBP encode constituents of the myelin sheath of oligodendrocytes, and GFAP encodes an intermediate filament protein of mature astrocytes in the central nervous system.
However, the expression and function of many other genes selected by the above analysis have not been well documented in the literature. For example, the TTC9B protein contains the tetratricopeptide repeat domain, and is conserved in other mammals, but its function in the brain is still unclear. In this study, the TTC9B gene shows significant expression in 408 out of 616 brain samples (Table 2). By contrast, in only 3 out of 2,352 control samples, significant expression is detected. Moreover, the mean expression level of TTC9B in the brain samples is 13.64-fold higher than that in the other tissues. As shown in Table 2, brain-selective expression patterns have also been demonstrated for four other genes (PTPN5, C21orf131, PEX5L and FBXL16) and three cDNA sequences (R44603, H12214 and BC040662), even though their functions in the brain remain to be characterized. The three sequences were obtained from brain cDNA libraries, but their corresponding genes were not determined. Altogether, the results suggest that the approach developed in this study can be used to not only confirm the brain-selective expression of some known genes, but also identify interesting targets for further experimental studies.
Liver-selective gene expression
The liver plays a key role in metabolism, and its functions include plasma protein synthesis, detoxification, and production of bile necessary for digestion. To identify liver-selective genes, the microarray data were grouped into the experiment set consisting of 117 liver expression profiles and the control set containing 2,851 profiles of non-liver tissues. The parameters for the analysis are as follows: θ = 1.00 (array median value), min = 23 (~20% of liver arrays), and max = 29 (~1% of control arrays), where θ is the relative intensity threshold for significant expression, min is the minimum number of significant expression in the experiment set, and max is the maximum number of significant expression in the control set. There are 69 gene targets identified for potential liver-selective expression, and the priority score ranges from 1.64 to 5.88 (see Additional file 2). Based on the permutation analysis, the liver-selective expression patterns of all the selected genes are statistically significant (p < 0.000001). The expression patterns of these genes are shown in Figure 3.
Interestingly, 17 of the top 20 high-scoring genes listed in Table 3 are previously known to be expressed predominantly in the liver. In particular, nine genes (MASP2, CFHR5, CFHR3, CRP, SERPINC1, F2, CFHR4, APOA5 and MBL2) are highly expressed in the liver, and their protein products are secreted to blood plasma. MASP2, CFHR5, CFHR3, CRP, CFHR4 and MBL2 play important roles in the innate immune defense against pathogens [19]. SERPINC1 and F2 are involved in regulating the blood coagulation cascade [20]. APOA5 encodes an apolipoprotein important for the regulation of plasma triglyceride level, a major risk factor for coronary artery disease [21]. Six of the known liver-selective genes encode metabolic enzymes involved in cholesterol catabolism and bile acid biosynthesis (CYP7A1), the urea cycle (ARG1), glyoxylate detoxification (AGXT), and the oxidation of alcohols (ADH4) and other compounds (CYP2C8 and HAO1). In addition, HGFAC encodes a peptidase involved in hepatocyte growth factor activation, and C14orf68 encodes a liver-specific mitochondrial carrier protein. The other three high-scoring genes (SLC17A2, ASPG and TDO2) have not been previously shown to be expressed preferentially in the liver.
Table 3.
List of high-scoring genes with selective expression in the liver 1
| Probe | Gene | Description | Se | Sc | 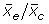 |
Score |
|---|---|---|---|---|---|---|
| 210798_x | MASP2 | Mannan-binding lectin serine peptidase 2 | 116 | 0 | 26.61 | 5.88 |
| 208088_s | CFHR5 | Complement factor H-related 5 | 110 | 1 | 95.56 | 5.41 |
| 1554459_s | CFHR3 | Complement factor H-related 3 | 116 | 5 | 103.31 | 4.77 |
| 37020 | CRP | C-reactive protein, pentraxin-related | 110 | 18 | 228.26 | 4.53 |
| 210049 | SERPINC1 | Serpin peptidase inhibitor, clade C, 1 | 117 | 18 | 213.02 | 4.53 |
| 205754 | F2 | Coagulation factor II (thrombin) | 117 | 13 | 135.57 | 4.47 |
| 207406 | CYP7A1 | Cytochrome P450, 7A1 | 56 | 1 | 19.52 | 4.43 |
| 207097_s | SLC17A2 | Solute carrier family 17, member 2 | 117 | 6 | 45.67 | 4.34 |
| 207874_s | CFHR4 | Complement factor H-related 4 | 115 | 18 | 138.54 | 4.33 |
| 207027 | HGFAC | HGF activator | 112 | 4 | 22.37 | 4.18 |
| 220224 | HAO1 | Hydroxyacid oxidase 1 | 117 | 18 | 86.38 | 4.14 |
| 231703_s | ADH4 | Alcohol dehydrogenase 4 (class II), pi | 89 | 3 | 18.26 | 4.12 |
| 224243 | APOA5 | Apolipoprotein A-V | 109 | 3 | 14.26 | 4.10 |
| 207256 | MBL2 | Mannose-binding lectin 2, soluble | 117 | 18 | 74.97 | 4.07 |
| 219903_s | CYP2C8 | Cytochrome P450, 2C8 | 61 | 1 | 7.03 | 4.02 |
| 1557226_a | ASPG | Similar to asparaginase | 95 | 2 | 8.27 | 3.98 |
| 231702 | TDO2 | Tryptophan 2,3-dioxygenase | 84 | 3 | 13.10 | 3.95 |
| 231662 | ARG1 | Arginase, liver | 102 | 14 | 40.21 | 3.85 |
| 210326 | AGXT | Alanine-glyoxylate aminotransferase | 117 | 8 | 14.95 | 3.73 |
| 237765 | C14orf68 | Chromosome 14 open reading frame 68 | 117 | 13 | 23.52 | 3.71 |
1 The number of significant expression in the experiment set (Se) and that in the control set (Sc) are shown together with the ratio of the mean expression level in the experiment arrays to the level in the control arrays (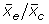 ).
).
Testis-selective gene expression
When compared with brain and liver tissues, many other tissues have fewer number of microarray expression profiles available (Table 1). The microarray dataset has only 36 expression profiles of the testis, which produces sperm and male sex hormones. To identify testis-selective genes, these 36 expression profiles (experiment set) were compared with 2,932 microarray profiles of non-testis tissues (control set) by using the following parameters: θ = 1.00 (array median value), min = 7 (~20% of testis arrays), and max = 29 (~1% of control arrays). The analysis resulted in 581 gene targets with the priority score ranging from 1.35 to 6.05 (see Additional file 3). The testis-selective expression patterns of these targets were found to be statistically significant by permutation testing (p < 0.000001). Figure 3 shows the expression patterns of the testis-selective gene targets.
As listed in Table 4, the top 20 high-scoring targets include five known testis-selective genes (C9orf11, TNP2, TSSK3, ACTL7B and NT5C1B). The C9orf11 gene encodes a vesicle membrane protein involved in the biogenesis of acrosome, a cap-like structure that covers the anterior half of the head in the spermatozoa [22]. TNP2 encodes a chromosomal transition protein for the conversion of nucleosomal chromatin to the compact form found in the sperm nucleus [23]. TSSK3 encodes a protein kinase expressed exclusively in the testis, and may be involved in signal transduction during male germ cell development or mature sperm function [24]. ACTL7B and NT5C1B are expressed preferentially in the testis, but their exact functions are still unknown.
Table 4.
List of high-scoring genes with selective expression in the testis 1
| Probe | Gene | Description | Se | Sc | 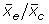 |
Score |
|---|---|---|---|---|---|---|
| 1554981 | C9orf11 | Chromosome 9 open reading frame 11 | 19 | 0 | 71.87 | 6.05 |
| 207736_s | TNP2 | Transition protein 2 (histone to protamine) | 18 | 0 | 52.45 | 5.89 |
| 231563 | - | cDNA sequence (GB: BF508261) | 19 | 0 | 49.40 | 5.88 |
| 237319 | C2orf53 | Chromosome 2 open reading frame 53 | 14 | 0 | 36.33 | 5.62 |
| 1552395 | TSSK3 | Testis-specific serine kinase 3 | 17 | 0 | 22.10 | 5.49 |
| 1562864 | - | cDNA sequence (GB: BC033504) | 15 | 0 | 22.90 | 5.45 |
| 243494 | - | cDNA sequence (GB: AI204633) | 19 | 0 | 15.59 | 5.38 |
| 243143 | FAM24A | Family with sequence similarity 24, A | 18 | 0 | 15.27 | 5.35 |
| 231362 | - | cDNA sequence (GB: AI423933) | 19 | 0 | 14.14 | 5.34 |
| 1560494_a | CPXCR1 | CPX chromosome region, candidate 1 | 17 | 0 | 14.38 | 5.30 |
| 236661 | IQCF6 | IQ motif containing F6 | 18 | 0 | 12.53 | 5.26 |
| 220498 | ACTL7B | Actin-like 7B | 18 | 0 | 11.49 | 5.23 |
| 1556740 | - | cDNA sequence (GB: AA398245) | 12 | 0 | 16.70 | 5.21 |
| 1554368 | NT5C1B | 5'-nucleotidase, cytosolic IB | 19 | 1 | 99.85 | 5.19 |
| 241527 | - | cDNA sequence (GB: AI799028) | 16 | 0 | 11.57 | 5.18 |
| 1561704 | - | cDNA sequence (GB: BC041892) | 11 | 0 | 16.40 | 5.17 |
| 241518 | - | cDNA sequence (GB: AA428659) | 11 | 0 | 16.11 | 5.16 |
| 242925 | RNF148 | Ring finger protein 148 | 19 | 1 | 91.88 | 5.15 |
| 1554855 | PARK2 | E3 ubiquitin-protein ligase parkin | 10 | 0 | 16.74 | 5.13 |
| 1556207_a | - | cDNA sequence (GB: BC035261) | 17 | 0 | 8.65 | 5.08 |
1 The number of significant expression in the experiment set (Se) and that in the control set (Sc) are shown together with the ratio of the mean expression level in the experiment arrays to the level in the control arrays (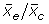 ).
).
The other high-scoring targets have not been previously shown to be testis-selective genes. PARK2 is known to be expressed in the brain, and mutations in this gene cause Parkinson disease [25]. The results from this study suggest that the highest expression of PARK2 appears to occur in the testis (Table 4). There are five other genes (C2orf53, FAM24A, CPXCR1, IQCF6 and RNF148) whose expression and function in the testis have not been well documented in the literature. In addition, the high-scoring targets include nine cDNA sequences. Interestingly, all the sequences except BC033504 and AI423933 were obtained from testis cDNA libraries (BC033504 from a brain library and AI423933 from a glioblastoma library). Considering the relative small sample size of testis expression profiles, it is uncertain whether all the selected probe sets represent true testis-selective genes. However, the targets with high priority scores should provide a good starting point for experimental studies on testis-selective gene expression and function.
Conclusion
A comprehensive microarray dataset has been compiled in this study for genome-wide analysis of human tissue-selective gene expression. The dataset contains 2,968 expression profiles of various normal tissues from 131 microarray studies. A new computational method has been designed to identify tissue-selective genes using both microarray intensity values and detection calls. To demonstrate that the integrated microarray data can be used to investigate human gene expression patterns, we have examined the lists of potential brain, liver and testis-selective genes. Notably, many of the high-scoring targets are actually known tissue-selective genes, suggesting that the approach developed in this study works effectively. Furthermore, the approach can be used to identify some interesting targets with tissue-selective expression patterns. These targets may be used for further experimental studies on human gene expression and function.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LW conceived and designed the study, conducted the data analysis, and drafted the manuscript. AKS and CES contributed to result interpretation and manuscript preparation.
Supplementary Material
List of brain-selective gene targets
List of brain-selective gene targets. The full list of potential brain-selective genes identified in this study.
List of liver-selective gene targets. The full list of potential liver-selective genes identified in this study.
List of testis-selective gene targets. The full list of potential testis-selective genes identified in this study.
Contributor Information
Liangjiang Wang, Email: liangjw@clemson.edu.
Anand K Srivastava, Email: anand@ggc.org.
Charles E Schwartz, Email: ceschwartz@ggc.org.
Acknowledgment
This work is supported by the CSREES/USDA, under project number SC-1700355. Publication of this supplement was made possible with support from the International Society of Intelligent Biological Medicine (ISIBM).
This article has been published as part of BMC Genomics Volume 11 Supplement 2, 2010: Proceedings of the 2009 International Conference on Bioinformatics & Computational Biology (BioComp 2009). The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2164/11?issue=S2.
References
- Klee EW. Data mining for biomarker development: a review of tissue specificity analysis. Clin Lab Med. 2008;28(1):127–143. doi: 10.1016/j.cll.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Lage K, Hansen NT, Karlberg EO, Eklund AC, Roque FS, Donahoe PK, Szallasi Z, Jensen TS, Brunak S. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci USA. 2008;105(52):20870–20875. doi: 10.1073/pnas.0810772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Lin J, Zack DJ, Qian J. Computational analysis of tissue-specific combinatorial gene regulation: predicting interaction between transcription factors in human tissues. Nucleic Acids Res. 2006;34(17):4925–4936. doi: 10.1093/nar/gkl595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu IM, Lal A, Riggins GJ. A database for regional gene expression in the human brain. Gene Expr Patterns. 2001;1(1):33–38. doi: 10.1016/S1567-133X(01)00006-0. [DOI] [PubMed] [Google Scholar]
- Kouadjo KE, Nishida Y, Cadrin-Girard JF, Yoshioka M, St-Amand J. Housekeeping and tissue-specific genes in mouse tissues. BMC Genomics. 2007;8:127. doi: 10.1186/1471-2164-8-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101(16):6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco D, Somervuo P, Di Lieto A, Raitila T, Nitsch L, Castrén E, Auvinen P. Physiology, pathology and relatedness of human tissues from gene expression meta-analysis. PLoS One. 2008;3(4):e1880. doi: 10.1371/journal.pone.0001880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98(1):31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2(8):research0032.1–0032.11. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- Henson MA, Roberts AC, Salimi K, Vadlamudi S, Hamer RM, Gilmore JH, Jarskog LF, Philpot BD. Developmental regulation of the NMDA receptor subunits, NR3A and NR1, in human prefrontal cortex. Cereb Cortex. 2008;18(11):2560–2573. doi: 10.1093/cercor/bhn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Lippa CF, Zhu J, Lin Q, Rosso AL. Disruption of glutamate receptors at Shank-postsynaptic platform in Alzheimer's disease. Brain Res. 2009;1292:191–198. doi: 10.1016/j.brainres.2009.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundo E, Tharmalingham S, Neves-Pereira M, Dalton EJ, Macciardi F, Parikh SV, Bolonna A, Kerwin RW, Arranz MJ, Makoff AJ, Kennedy JL. Evidence that the N-methyl-D-aspartate subunit 1 receptor gene (GRIN1) confers susceptibility to bipolar disorder. Mol Psychiatry. 2003;8(2):241–245. doi: 10.1038/sj.mp.4001218. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Scheiffele P. GABA and neuroligin signaling: linking synaptic activity and adhesion in inhibitory synapse development. Curr Opin Neurobiol. 2008;18(1):77–83. doi: 10.1016/j.conb.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Wevers A, Schröder H, Grzeschik KH, Derst C, Brodtkorb E, de Vos R, Steinlein OK. The LGI1 gene involved in lateral temporal lobe epilepsy belongs to a new subfamily of leucine-rich repeat proteins. FEBS Lett. 2002;519(1-3):71–76. doi: 10.1016/S0014-5793(02)02713-8. [DOI] [PubMed] [Google Scholar]
- Li C, Chan J, Haeseleer F, Mikoshiba K, Palczewski K, Ikura M, Ames JB. Structural insights into Ca2+-dependent regulation of inositol 1,4,5-trisphosphate receptors by CaBP1. J Biol Chem. 2009;284(4):2472–2481. doi: 10.1074/jbc.M806513200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlou O, Theodorakis K, Falk J, Kutsche M, Schachner M, Faivre-Sarrailh C, Karagogeos D. Analysis of interactions of the adhesion molecule TAG-1 and its domains with other immunoglobulin superfamily members. Mol Cell Neurosci. 2002;20(3):367–381. doi: 10.1006/mcne.2002.1105. [DOI] [PubMed] [Google Scholar]
- Wu Q, Maniatis T. A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell. 1999;97(6):779–790. doi: 10.1016/S0092-8674(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Zipfel PF. Complement and immune defense: from innate immunity to human diseases. Immunol Lett. 2009;126(1-2):1–7. doi: 10.1016/j.imlet.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Kalafatis M, Egan JO, van't Veer C, Cawthern KM, Mann KG. The regulation of clotting factors. Crit Rev Eukaryot Gene Expr. 1997;7(3):241–280. doi: 10.1615/critreveukargeneexpr.v7.i3.40. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, Krauss RM, Rubin EM. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294(5540):169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- Li YC, Hu XQ, Zhang KY, Guo J, Hu ZY, Tao SX, Xiao LJ, Wang QZ, Han CS, Liu YX. Afaf, a novel vesicle membrane protein, is related to acrosome formation in murine testis. FEBS Lett. 2006;580(17):4266–4273. doi: 10.1016/j.febslet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Wykes SM, Krawetz SA. The structural organization of sperm chromatin. J Biol Chem. 2003;278(32):29471–29477. doi: 10.1074/jbc.M304545200. [DOI] [PubMed] [Google Scholar]
- Bucko-Justyna M, Lipinski L, Burgering BM, Trzeciak L. Characterization of testis-specific serine-threonine kinase 3 and its activation by phosphoinositide-dependent kinase-1-dependent signalling. FEBS J. 2005;272(24):6310–6323. doi: 10.1111/j.1742-4658.2005.05018.x. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Haass C. How does parkin ligate ubiquitin to Parkinson's disease? EMBO Rep. 2004;5(7):681–685. doi: 10.1038/sj.embor.7400188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of brain-selective gene targets
List of brain-selective gene targets. The full list of potential brain-selective genes identified in this study.
List of liver-selective gene targets. The full list of potential liver-selective genes identified in this study.
List of testis-selective gene targets. The full list of potential testis-selective genes identified in this study.