

Abstract
MEKK1 is a ubiquitously expressed mitogen activated protein kinase that is involved in tissue remodeling in a variety of settings including carotid artery blood flow cessation, wound healing and breast adenocarcinoma intravasation. Here, we have tested the function of MEKK1 in genetic hypertrophic cardiomyopathy (HCM). MEKK1 was genetically deleted in C57Bl6/J mice expressing a mutant β-myosin heavy chain (HCM-MEKK1-/-). The absence of MEKK1 in HCM resulted in a more pronounced hypertrophy when compared to HCM mice with the MEKK1 gene intact without further increases in atrial natriuretic factor and β-myosin heavy chain (MyHC) expression and fibrosis. Since MEKK1 is required for the induction of several tissue proteases, we tested the hypothesis that cardiac enlargement of HCM- MEKK1-/- mice was due to altered expression of urokinase-type plasminogen activator (uPA), JunB, matrix-metalloproteinase (MMP), and tissue inhibitors of MMPs (TIMPs). Because of its role in preventing apoptosis, we also tested the loss of MEKK1 on apoptotic mediators Bcl-2, cytochrome C, caspase-9 and caspase-3. uPA expression was decreased while Jun B, MMP-9, caspase-9 and caspase-3 activities were elevated in HCM- MEKK1-/- hearts when compared to MEKK1-/-, wild-type (WT) and HCM mice. Bcl-2 and Cyt C expression was elevated only in HCM mice. We conclude that the absence of MEKK1 induces a more pronounced cardiac hypertrophy to HCM through altered expression of proteases implicated in cardiac remodeling and increased apoptosis.
Keywords: Hypertrophic cardiomyopathy, ventricular remodeling, apoptosis, urokinase-type plasminogen activator, matrix-metalloproteinase, caspase-3
INTRODUCTION
Mitogen-activated protein kinases (MAPKs) are stimulated in the heart by biomechanical, chemical and cellular stress, and by dysregulated activation of G protein coupled receptors (for a review see ref. 1). The MAPKs consist of three distinct signaling cascades and the serine/threonine kinases that make up each arm are extracellular signal-regulated protein kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK. Stimulation of the α1-adrenergic receptor (α1-AR), which is coupled to Gαq, activates MAPK/ERK kinase (MEK) kinase-1 (MEKK1) 2. MEKK1 functions as a MAPKKK in the JNK and ERK1/2 pathways 3. MEKK1 can modulate the transcriptional regulation of cardiac hypertrophy, including induction of atrial natriuretic factor (ANF), a well-known indicator of pathological cardiac hypertrophy. 4, 5, 6 This can occur through JNK-dependent phosphorylation of c-Jun to enhance the transcription of activator protein-1 (AP-1) responsive genes including ANF. 2
As a key regulator of AP-1 dependent transcription, MEKK1 plays a central role in tissue remodeling in response to stress. Both JNK and ERK1/2 activity are required for formation of an AP-1 complex for the regulation of urokinase-type plasminogen activator (uPA) expression, a serine protease required for tissue remodeling. 7, 8 Inducible uPA expression is controlled by MEKK1 and is dependent on JNK and ERK1/2 activities. 9 Upon proteolytic cleavage, the active form of uPA, plasmin, targets matrix metalloproteinases (MMPs) both of which have been implicated in cardiac repair and remodeling after cardiac injury. 10
Cardiac remodeling, defined as changes in cardiac size, cellular ultrastructure, and genetic expression similarly involves the integration of MAPK signaling pathways. A direct role for MEKK1 in cardiac disease was examined using transgenic mice that overexpressed Gαq and exhibit significant pathological cardiac remodeling and dysfunction. Deletion of MEKK1 in these mice prevented the hypertrophy and dysfunction normally seen. 5, 11 Thus, the loss of MEKK1 in this setting was preventative. However, in a model of pressure overload, hearts lacking MEKK1 still undergo cardiac hypertrophy but also display enhanced apoptosis. 12 This latter observation suggests that MEKK1 is not required for hypertrophy but is critical for the protection against cell death due to this particular pathological cardiac stimulus.
From these two observations, the protective role of MEKK1 in cardiac pathological stimuli is unclear. We hypothesized that MEKK1 is not required for cardiac hypertrophy but is required for protection against pathological remodeling due to HCM. We previously reported that male HCM mice expressing a mutant α-myosin heavy chain (MyHC) transgene exhibit pathologic cardiac remodeling including cardiac hypertrophy, interstitial fibrosis, and induction of ANF. 13, 14, 15 These mice also display changes in Bcl-2 (an inhibitor of apoptosis) and procaspase-9 (an effector of apoptosis) expression, and caspase-3 activity that indicate an increase in apoptosis. To address this hypothesis, we generated mice that harbor the HCM transgene and have global disruption of the MEKK1 gene (HCM-MEKK1-/-). In HCM-MEKK1-/- mice, cardiac mass as an indicator of cardiac hypertrophy was measured, and the induction of ANF as a general indicator of cardiac pathology was determined. We demonstrate that the loss of MEKK1 in mice with HCM leads to increased cardiac hypertrophy through differential regulation of uPA, matrix-metalloproteinase (MMP), tissue inhibitors of MMPs, and mediators of apoptosis, Bcl-2, caspase 9 and caspase-3 in HCM-MEKK1-/- mice. The findings support an important role for MEKK1 by controlling the expression of proteases and apoptotic mediators critical in the protection against hypertrophy-induced remodeling due to HCM.
METHODS
Experimental animals
MEKK1-/- animals 16 were crossed with HCM mice13 resulting in HCM mice that were MEKK1+/-. HCM and WT controls used for this study were generated from this cross. MEKK1+/- animals that were positive for the HCM transgene were backcrossed with MEKK1-/- animals to generate the experimental group (HCM-MEKK1-/-) and MEKK1-/- without the HCM transgene as littermate controls. Only male mice were used for this study as the HCM animals demonstrate significant sex dimorphisms. 13, 15 Animals were euthanized following each protocol and hearts were excised and equally sectioned for the assays described below. All of the animals were handled according to approved protocols of the University of Colorado and conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE)
Heart extracts from above were analyzed by SDS-PAGE for cardiac MyHC content as previously described.17
Gelatin Zymography of MMP activity
Cardiac extracts were mechanically disrupted in ice-cold lysis buffer (0.02 ml/g tissue): (in mmol/L) Tris(hydroxymethyl)-aminomethane (20); NaCl (137); EDTA (0.2); EGTA (0.5); Triton X-100 (1%); Glycerol (10%) (pH 7.4). Samples were then prepared and subjected to gelatin zymography (30-50 µg total protein) to measure MMP activity according to previous methods. 18 Each sample was normalized to the total quantifiable amount of protein loaded in each respective lane.
Analysis of fibrosis
Histological analysis and quantitation were performed as previously described with slight modification.14 Samples were processed, embedded in paraffin, sectioned, and stained Masson's trichrome stain by Premier Histology (Boulder, CO). At least 8-10 random fields per heart were digitized and then analyzed by blinded observers using ImageJ 1.34s (NIH;http://rsb.info.nih.gov/ij/). Fibrosis was expressed as a percent of the total image area. For each sample, the images were averaged and used as the representative percent fibrosis of the entire heart.
Western blot analysis
Preparation of heart samples for Western immunoblotting were performed as previously described.19 All antibodies were obtained commercially from Cell Signaling Technology (Caspase 9, JunB, uPA), Santa Cruz Biotechnology (TIMP-1 and TIMP-3), or BD Transduction Laboratories (Bcl-2). Membranes were stripped and stained with Coomassie blue for calculation of total loaded protein (30-50 μg total protein loaded onto each gel depending on antibody). The stained blots were then scanned and quantified using Image J image processing program. This value was used to normalize each sample.
Caspase-3 activity assay
Frozen hearts extracts were prepared as described above caspase-3 activity analysis. Caspase-3 activity was determined by monitoring the rate of cleavage of a fluorogenic caspase-3 specific substrate (Acetyl-AspGluValAsp-AMC; Calbiochem) as described previously. 14, 15 Rates were then normalized to total protein.
Data and statistical analysis
Results are presented as mean ± SEM. The percent change in cardiac mass was determined by comparing the mass of each experimental animal to the mean cardiac mass of the control group. The difference in cardiac mass was then expressed as a percent change from control animals for each respective animal. The differences between groups were analyzed with a one-way ANOVA followed by a Student's t test using a Tukey's HSD post hoc test to assess differences among mean values.
RESULTS
Cardiac Adaptation to Pathologic Stimuli
MEKK1-/- mice were bred to HCM mice expressing the mutant MyHC transgene that results in cardiac hypertrophy and significant histopathology. 13 The animal morphometrics are summarized in Table 1. All animals were males at 4 months of age. MEKK1-/- mice were indistinguishable from WT mice as reported previously. 16 This includes comparisons of cardiac mass and cardiac mass adjusted for body weight (HW/BW). Both absolute and normalized (HW/BW) cardiac mass are significantly greater in HCM-MEKK1-/- and HCM mice compared to MEKK1-/- and WT mice. Similarly, absolute and normalized cardiac mass are significantly greater in HCM-MEKK1-/- mice compared to HCM mice. HCM mice developed cardiac hypertrophy relative to WT mice that was similar in magnitude to that previously determined in age-matched HCM male mice. 17
Table 1. Animal Morphometries.
The impact of pathologic hypertrophic stimuli on absolute and normalized cardiac mass. All animals were sacrificed at 4 months of age. Heart weight (HW) was normalized to body weight (HW/BW) by dividing cardiac mass (in mg) by body weight (in g). (Significance determined as p<0.05 from indicated group as follows: a, MEKK1-/- and WT animals; b, HCM animals.)
| Cardiac mass (mg) | Body mass (g) | HW/BW (mg/g) | |
|---|---|---|---|
| MEKK1-/- (n=20) | 131.0 ± 2.4 | 28.1 ± 0.4 | 4.65 ± 0.05 |
| HCM-MEKKT-/- (n=8) | 172.7 ± 10.3a,b | 30.3 ± 0.9 | 5.71 ± 0.32a,b |
| WT (n=15) | 141.7 ± 3.3 | 29.9 ± 0.8 | 4.75 ± 0.07 |
| HCM (n=13) | 152.2 ± 3.9a | 29.5 ± 0.8 | 5.17 ± 0.08a |
To better illustrate the relative change in cardiac mass due to each stimulus, we calculated the percent change in normalized mass from each respective control group (Table 2). HCM-MEKK1-/- mice were compared to MEKK1-/- mice, and HCM mice were compared to WT mice. The magnitude of hypertrophy seen in HCM-MEKK1-/- mice relative to MEKK1-/- controls is significantly greater (22.6±6.4%) than that determined in HCM compared to WT mice with an intact MEKK1 pathway (8.8±1.7%). Again, the percent increase in cardiac mass observed in HCM hearts is similar to that from previous studies. 14, 15, 17
Table 2. Relative Cardiac Hypertophy.
Relative cardiac hypertrophy due the HCM transgene. The percent change in cardiac mass due to the HCM transgene was determined in mice expressing the HCM transgene with and without MEKK1. The change in cardiac mass was compared to each respective control (*p<0.05 from HCM group).
| HCM-MEKK1-/- | HCM | |
|---|---|---|
| % Change in HW | 22.6 ± 6.4* | 8.84 ± 1.7 |
Expression of Pathological Markers
Pathologic cardiac hypertrophy is accompanied by the induction of genes normally expressed during fetal development. 4, 20 The HCM mouse represents a chronic pathological model, and, typically, the hearts of HCM male mice express more β-MyHC protein, the myosin motor normally expressed during development, relative to WT mice by 4 months of age 17 and that amount doubles by 8-10 months of age. 14, 15, 17 HCM-MEKK1-/- hearts exhibit the same 5-fold elevation (5.5±1.4%) of β-MyHC protein relative to α-MyHC when compared to MEKK1-/- control hearts (1.1±1.3%; Figure 1). HCM mice with an intact MEKK1 pathway express a similar increase in β-MyHC protein (5.8±0.3%) compared to WT (1.2±0.1%; Figure 1), paralleling the pathological increase observed previously. 17 Interestingly, HCM-MEKK1-/- hearts are much larger than those of HCM animals (Table 2), but β-MyHC is similarly increased in the presence or absence of MEKK1 expression. The findings suggest that increased expression of β-MyHC can be dissociated from the increased cardiac hypertrophy induced by HCM in a null MEKK1 background.
Figure 1.

Impact of HCM on the expression of pathological markers in MEKK1-/- hearts. A: (top panel) Representative silver-stained gels illustrating 2 dilutions of an individual sample in each of the experimental groups studied. Bottom panel (left): Bar graph representation of the relative α-MyHC expression; (right) Bar graph representation of the relative β-MyHC expression. (MEKK1-/-: n=4; HCM-MEKK1-/-: n=5; WT: n=4 and HCM: n=3; *p<0.05 from measured values in MEKK1-/- and WT controls.)
Since we have previously shown that ANF expression is significantly increased by about 2-3 fold in HCM male mice at 4 and 8 months of age 13, 21, we examined whether the absence MEKK1 impacted the expression of ANF in the context of HCM. There is a similar, significant, 2 to 2.5-fold induction of ANF in HCM-MEKK1-/- compared to MEKK1-/- mice (data not shown). The quantitative elevation of ANF expression in HCM-MEKK1-/- hearts was similar to that of HCM hearts compared to WT hearts. 13, 21 These findings demonstrate that HCM mice lacking MEKK1 still display elevated ANF expression.
Urokinase-type Plasminogen Activator Expression
Remodeling of the extracellular matrix (ECM) and regulation of reparative processes such as wound healing are dependent on uPA. 9, 22 uPA expression requires JNK and ERK1/2 activation, both of which are regulated by MEKK1 in response to specific stimuli. 3, 9 Considering that cardiac hypertrophy due to HCM, which is enhanced in HCM-MEKK1-/- hearts, involves significant ventricular remodeling. 14, 15 we measured uPA expression in HCM-MEKK1-/- hearts compared to MEKK1-/- mice. As shown in Figure 2, uPA expression is significantly lower in HCM-MEKK1-/- hearts when compared to MEKK1-/-, age-matched WT and HCM hearts. Interestingly, uPA expression in HCM and control WT hearts is similar, indicating that uPA expression is not significantly altered in HCM at 4 months of age. uPA expression in the MEKK1-/- hearts is intermediate between WT or HCM hearts and the HCM-MEKK1-/- hearts, consistent with MEKK1 being required for regulating uPA expression in pathological settings. The induction of uPA mRNA parallels the changes in protein expression (data not shown).
Figure 2.
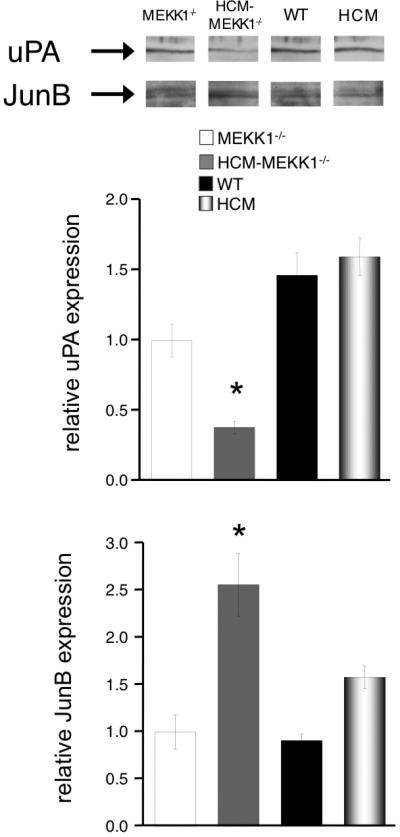
Regulation of urokinase-type plasminogen activator (uPA) expression. uPA and Jun B protein expression was measured by Western Blotting using specific antibodies. Bar graph representation summarizes the expression of uPA (top panel) and the expression of Jun B (bottom panel). Expression levels of each group studied are normalized to total protein loaded on the gel. (MEKK1-/-: n=4; HCM-MEKK1-/-: n=5; WT: n=5; HCM: n=5; *p<0.05 from measured values in MEKK1-/- controls.)
It has previously been shown that upregulation of JunB expression occurs in MEKK1-/- cells forming an inhibitory AP-1 complex that negatively regulates uPA expression. 23 Accordingly, we illustrate an upregulation of JunB expression concomitant with the downregulation of uPA in HCM-MEKK1-/- hearts (Figure 2; bottom panel), Interestingly, there are no significant differences in JunB expression between MEKK1-/-, WT or HCM hearts.
Matrix-Metalloproteinase and Tissue Inhibitors of Metalloproteinase Expression
ECM remodeling during cardiac disease states largely depends on matrix metalloproteinases (MMPs). 18, 24 Therefore, we measured MMP activity by gelatin zymography. The 92- and 72-kDa digested bands correspond to MMP-9 and MMP-2, respectively. MMP-9 activity is 1.5-fold elevated in HCM-MEKK1-/- hearts compared to HCM, or MEKK1-/- or control hearts (Figure 3). In contrast, MMP-2 but not MMP-9 is elevated in HCM with an intact MEKK1 pathway. These findings indicate that MEKK1 regulates the expression profiles of MMPs in a pathophysiological setting such as HCM. I think we should address the concern about the opposite behavior of MMP2 and 9 as requested by one of the reviewers. I have now read the discussion, and maybe what's there is enough.
Figure 3.

MMP-2 and MMP-9 activity. MMP-2 and MMP-9 activity was determined by gelatin zymography from cardiac extracts. The 92kDa band represents MMP-9 and the 72 kDa band represents MMP-2. Quantitation of MMP activity was achieved by in gel gelatinase activity. (MEKK1-/-: n=4; HCM-MEKK1-/-: n=5; WT: n=5; HCM: n=5; *p<0.05 from measured values in all groups studied.)
To explore the determinants of this differential MMP activity, we measured the expression levels of critical regulators for MMPs called tissue inhibitors of metalloproteinases (TIMPs), specifically TIMP-1 and TIMP-3. The hearts from HCM-MEKK1-/- mice have significantly lower TIMP-1 and TIMP-3 expression when compared to MEKK1-/- controls (Figure 4). TIMP-1 expression in HCM-MEKK1-/- hearts is significantly lower than NTG (WT) but not HCM hearts. However, TIMP-3 expression is significantly lower in the hearts of HCM-MEKK1-/- mice compared to either WT or HCM hearts.
Figure 4.

Expression levels of TIMP-1 and TIMP-3. A: (top panel) Representative immunoblot of TIMP-1 expression from each group studied; (bottom panel) bar graph representation of summarized expression levels relative to MEKK1-/- hearts. B: (top panel) Representative immunoblot of TIMP-3 expression from each group studied; (bottom panel) bar graph representation of summarized expression levels relative to MEKK1-/- hearts. (MEKK1-/-: n=4; HCM-MEKK1-/-: n=6; WT: n=5; HCM: n=5; †p<0.05 from MEKK1-/- and WT, only; *p<0.05 from measured values in all groups studied,)
Fibrosis
Considering that MMP-2, MMP-9, TIMP-1 and TIMP-3 are differentially regulated in HCM-MEKK1-/- compared to HCM hearts, we hypothesized that the loss of MEKK1 in HCM would increase the amount of fibrosis compared to HCM with an intact MEKK1 pathway. Hearts of HCM male mice had been previously shown to exhibit significant fibrosis and myocellular disarray. 21, 25 As illustrated in Figure 5, hearts from HCM-MEKK1-/- mice show significant fibrosis (4.13 ±0.47%) as measured by Masson's trichrome staining compared to MEKK1-/- mice (0.36 ±0.17%); myocyte disarray, although not quantified, is clearly greater in HCM-MEKK1-/- hearts compared to MEKK1-/- hearts. The amount of fibrosis in HCM-MEKK1-/- hearts is not different from HCM males, which also exhibit pronounced (3.01±0.51%) fibrosis (and myocyte disarray) compared to NTG hearts (0.35 ±0.16%; Figure 5).
Figure 5.
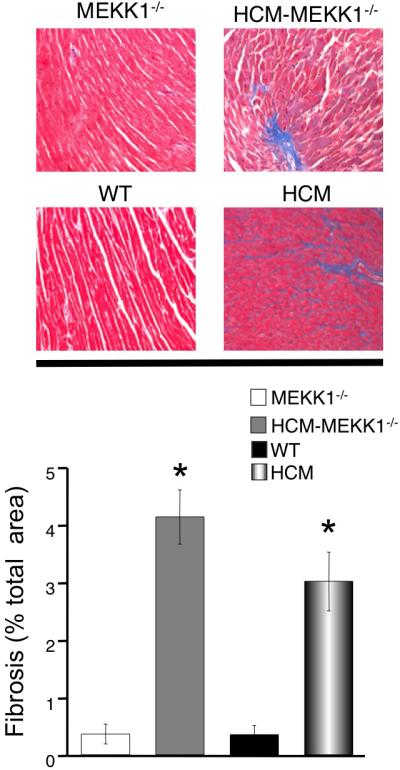
Cardiac fibrosis in each experimental group studied. Fibrosis was quantified in each of the experimental groups (top panel) and summarized in the bar graph (bottom panel). Fibrosis was visualized using Masson's trichrome and quantified as described. (n=3 for each experimental group; *p<0.05 from measured values in WT and MEKK1-/-controls.)
Apoptosis
A critical point in cardiac disease progression is the transition from a compensated hypertrophic state to a decompensated dilated heart. The remodeling of the heart associated with this decompensation and wall thinning/dilation can be hastened and/or caused by an increase in the rate of myocardial cell loss, or apoptosis. We have previously documented a propensity towards increased apoptosis in HCM male hearts compared to WT controls at 8 months of age. 14, 15 In addition, MEKK1-/- mice that underwent aortic banding displayed elevated levels of apoptosis and inflammatory lesions suggesting a protective role for MEKK1 signaling during a pathologic hypertrophic stimulus. 12 Therefore, we measured mediators of apoptosis including, cytochrome C (Cyt C; an apoptotic signal), Bcl-2 (an anti-apoptotic mediator), caspase 9 and caspase 3 (end-effectors of apoptosis).
Activation of caspase 9 occurs by proteolytic cleavage of the larger (47kDa), inactive pro- form to one of two smaller (39 kDa and 37 kDa), active forms. 26 Expression of procaspase 9 (inactive) was significantly reduced in the hearts of HCM-MEKK1-/- mice from all other groups. More importantly, the ratio of cleaved caspase 9 to procaspase 9 was 2-fold elevated in the hearts of HCM-MEKK1-/- mice from all other groups studied (Figure 6a). Taken together, these data suggest an increased rate of procaspase 9 cleavage and enhanced apoptotic activity due to caspase 9 in the hearts of HCM-MEKK1-/- mice compared to all other groups.
Figure 6.

Expression and activity of apoptotic mediators. A: (top panel) Representative immunoblot of Caspase 9 expression from each group studied; (bottom panel) bar graph representation of summarized expression levels of cleaved (39 kDa and 37 kDa) caspase 9 (active)/pro- (47 kDa) caspase 9 (inactive) relative to MEKK1-/- hearts; B: Caspase-3 activity in cardiac extracts from each group. Caspase-3 activity as an indicator of apoptosis was determined by monitoring the rate of cleavage of a fluorogenic caspase-3 specific substrate in each experimental group studied. Values were normalized to MEKK1-/- controls. C: Representative immunoblot of Blc-2 expression from each group studied; (bottom panel) bar graph representation of summarized expression levels relative to MEKK1-/- hearts. D: Representative immunoblot of Cyt C expression from each group studied; (bottom panel) bar graph representation of summarized expression levels relative to MEKK1-/- hearts. Expression levels of each group studied are normalized to total protein loaded on the gel. (MEKK1-/-: n=4; HCM-MEKK1-/-: n=6; WT: n=5; HCM: n=5.; *p<0.05 from measured values in all groups studied.)
We also measured apoptotic activity by monitoring the rate of cleavage of a fluorogenic caspase-3 specific substrate in whole heart homogenates from MEKK1-/-, HCM-MEKK1-/-, WT and HCM hearts. 14, 15 In this study, caspase-3 activity was normalized to that in MEKK1-/- animals and was used as a measure of relative apoptosis. As illustrated in Figure 6c, relative caspase-3 activity in HCM-MEKK1-/-hearts was approximately 4–fold higher when compared to MEKK1-/- and WT controls. Interestingly, caspase-3 activity in HCM hearts was not different from WT levels at 4 months of age despite significantly elevated caspase-3 activity in HCM mice at 8 months of age. 14, 15
Interestingly, there are no differences in Cyt C or Bcl-2 expression between WT, MEKK1-/- and HCM-MEKK1-/- hearts. Moreover, opposite to what we found in the hearts of 8-month HCM mice, Bcl-2 and Cyt C expression is significantly elevated in the hearts of HCM mice when compared to the hearts of WT, MEKK1-/- and HCM-MEKK1-/- mice (Figure 6b).
DISCUSSION
Hypertrophic signaling cascades include a diverse collection of protein kinases including the MAPKs, which can elicit an increase in heart size. 27 Global deletion of MEKK1 eliminates cardiac hypertrophy and hemodynamic dysfunction in transgenic mice that overexpress Gαq in the heart 5, 11 but not in mice that undergo pressure overload. 12 Therefore, it was difficult to predict a priori the impact of a genetic mutation that leads to HCM in MEKK1-/- mice.
The absence of MEKK1 in HCM mice (HCM-MEKK1-/-) resulted in significantly enlarged hearts over MEKK1-/- or HCM controls whether compared by absolute, adjusted, or percent change in cardiac mass. 13, 17 We hypothesized that the enlarged hearts of HCM-MEKK1-/- mice were indicative of a more hastened, pathological ventricular remodeling. Contrary to this hypothesis, the hearts of HCM male mice lacking MEKK1 demonstrated an amount of ANF and β-MyHC expression and fibrosis that was equivalent to that of HCM mice despite this greater relative heart size. 14, 15
When alternative signaling pathways associated with ventricular remodeling were examined, interesting differences were apparent. A reduction in uPA expression was observed in the hearts of HCM-MEKK1-/- mice when compared to all other groups. The reduced uPA expression in HCM mice lacking MEKK1 seems likely to be related to changes in the composition of the AP-1 transcription complex. Upregulation of JunB expression occurs in MEKK1-/- cells forming an inhibitory AP-1 complex that negatively regulates uPA expression. 23 Furthermore, recent findings indicate that JunD is able to negatively regulate AP-1 activity in response to PE induced cardiac hypertrophy. 28 Thus, a pro-hypertrophic model like HCM in the absence of MEKK1 would be hypothesized to stimulate JunD expression and to enhance JunB expression, both of which promote inhibitory AP-1 complexes and thus a reduction in uPA expression. 23, 29 Our data indicating enhanced expression of Jun B concomitant with decreased uPA expression support this contention.
Although consistent MEKK1-dependent regulation of uPA, the reduced uPA expression in HCM-MEKK1-/- mice does not correlate with the increase in MMP-9 activity when compared to MEKK1-/- controls. Interestingly, we show that HCM hearts with an intact MEKK1 pathway have elevated MMP-2, but not MMP-9, activity when compared to HCM-MEKK1-/- mice. The data in HCM mice retaining the MEKK1 pathway is consistent with previous studies in which MMP-2 activity was associated with non-ischemic hypertrophic cardiomyopathy in humans. 18, 30 Yet, it is unclear why HCM-MEKK1-/- mice display elevated MMP-9 activity only.
We reasoned that the differential MMP activation described above was a result of selective regulation by TIMPs. Although TIMP-1 expression was significantly reduced in HCM animals lacking MEKK1 when compared to MEKK1-/- and WT mice, it was not different from HCM animals. However, TIMP-3 expression was significantly reduced when compared to all groups including HCM mice. These data directly relate to the finding that selective disruption of TIMP-3 in mice results in dilated cardiomyopathy. 31, 32 In mice lacking TIMP-3, MMP-9 was concomitantly elevated paralleling the results found in HCM-MEKK1-/- mice. Moreover, genetic deletion of MMP-9 with a concomitant increase in TIMP-1, opposite to what we observed in HCM-MEKK1-/- mice, is associated with an improved phenotype. 33 The cause of this reduction in TIMP expression in HCM-MEKK1-/- mice is not known but may be explained by the finding that transcriptional regulation of TIMPs can be mediated by AP-1 sites, which are known to be MEKK1-dependent. 34 Therefore, it is likely that MMP-9 activity is associated with maladaptive cardiac hypertrophy related to the pathological transition that occurs with HCM whereas MMP-2 activation is associated with adaptive hypertrophy. 35, 36
In addition to aberrant matrix remodeling, myocellular apoptosis can exacerbate cardiac pathology and may contribute to the enlarged cardiac phenotype seen in HCM-MEKK1-/- mice. Bcl-2 acts as an anti-apoptotic intermediate by preventing the release of mitochondrial membrane proteins, such as cytochrome c, that perpetuates the death signal. 26 Cytochrome c forms an activation complex with apoptotic protein-activating factor-1 and caspase-9 that activates downstream effector caspases including caspase-3. We previously demonstrated elevated caspase-3 activity levels in HCM mice at 8 months of age, corresponding to a decompensated, pathological hypertrophic state 14, 15, a similar finding in HCM-MEKK1-/- hearts. In addition, the hearts of HCM-MEKK1-/-mice display a reduction in pro-capsase 9 relative the all other groups suggesting increased conversion of the pro-, inactive, to cleaved, active form of caspase 9. This is further supported by the finding that HCM-MEKK1-/- hearts demonstrate a significantly increased ratio of cleaved to pro- form of caspase 9.
Interestingly, we found an elevated amount of Bcl-2, an anti-apoptotic mediator, and Cyt C in the hearts of HCM with the MEKK1 pathway intact. This is different from what was previously reported in the same model at 8 months of age 14, 15, 37 suggesting a compensatory response to prevent apoptosis in HCM mice at 4 months of age. The lack of elevated caspase 9 or caspase 3 activity in HCM mice is consistent with the enhanced expression of the anti-apoptotic mediator, Bcl-2 and, concurrently, Cyt C. Bcl-2 and Cyt C levels in the hearts of HCM-MEKK1-/- mice were not different from MEKK1-/-or WT controls perhaps indicative of a transition to increased apoptosis. More conclusive studies regarding apoptosis and the development of the HCM-MEKK1-/- phenotype will be the subject of future investigations.
Based on the differential activation of matrix metalloproteinases consistent with a pathological phenotype and upregulation of pro-apoptotic mediators, we propose that the more pronounced hypertrophic response in HCM mice lacking MEKK1 is indicative of an exacerbation of the HCM phenotype. It has been shown that MEKK1 is involved in mediating the antiapoptotic effect of Bcr-Abl. 38 Moreover, MEKK1-/- mice that underwent aortic banding displayed elevated levels of apoptosis and inflammatory lesions indicating a protective role for MEKK1 signaling during this pathologic hypertrophic stimulus. 12 Thus, it is suggested that the presence of MEKK1 in HCM hearts presumably opposes the progression of the HCM pathology in younger mice.
In conclusion, these studies shed light on the complexity of the MAPK signaling controlled by the different MKKKs, of which there are more than 20 in mouse and humans. The ability of MEKK1-/- mice to undergo more extensive hypertrophy in the context of and HCM mutation is supported by previous studies. 12, 39 A noted limitation of these studies is a complete characterization of the hypertrophy, i.e., whether the increase in cardiac mass is due to an enlargement of cardiac myocytes or some additional components not identified in the histological examinations. Another limitation is the lack of functional significance associated with the pronounced cardiac hypertrophy. This will have to be addressed in future studies. Nevertheless, we suggest that the loss of MEKK1 expression results in a more severe pathologic state and increased expression of ventricular remodeling factors and apoptosis. Future investigations need to quantify the contribution of different MKKKs and downstream MAPK networks controlled by the repertoire of MKKKs expressed in cardiac myocytes and other cell types in the heart that control resultant cardiac hypertrophy in response to different types of pathologic hypertrophic stimuli.
Footnotes
FUNDING
This work was supported by NIH grant to L.A. Leinwand (HL 56510) and G.L. Johnson (GM30324 and DK37871) and by a National Research Service Award and a Mentored Research Scientist Development Award from the NIH awarded to J.P. Konhilas (F32 HL 70509 and K01 AR052840).
CONFLICTS OF INTEREST
There are no conflicts of interest to report.
REFERENCES
- 1.Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11(2):211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 2.Lange-Carter CA, Pleiman CM, Gardner AM, Blumer KJ, Johnson GL. A divergence in the map kinase regulatory network defined by mek kinase and raf. Science. 1993;260(5106):315–319. doi: 10.1126/science.8385802. [DOI] [PubMed] [Google Scholar]
- 3.Yujiri T, Sather S, Fanger GR, Johnson GL. Role of mekk1 in cell survival and activation of jnk and erk pathways defined by targeted gene disruption. Science. 1998;282(5395):1911–1914. doi: 10.1126/science.282.5395.1911. [DOI] [PubMed] [Google Scholar]
- 4.Nadal-Ginard B, Mahdavi V. Molecular basis of cardiac performance. Plasticity of the myocardium generated through protein isoform switches. J Clin Invest. 1989;84(6):1693–1700. doi: 10.1172/JCI114351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minamino T, Yujiri T, Terada N, et al. Mekk1 is essential for cardiac hypertrophy and dysfunction induced by gq. Proc Natl Acad Sci U S A. 2002;99(6):3866–3871. doi: 10.1073/pnas.062453699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorn GW, 2nd, Brown JH. Gq signaling in cardiac adaptation and maladaptation. Trends Cardiovasc Med. 1999;9(1-2):26–34. doi: 10.1016/s1050-1738(99)00004-3. [DOI] [PubMed] [Google Scholar]
- 7.Karin M. The regulation of ap-1 activity by mitogen-activated protein kinases. Philos Trans R Soc Lond B Biol Sci. 1996;351(1336):127–134. doi: 10.1098/rstb.1996.0008. [DOI] [PubMed] [Google Scholar]
- 8.De Cesare D, Vallone D, Caracciolo A, et al. Heterodimerization of c-jun with atf-2 and c-fos is required for positive and negative regulation of the human urokinase enhancer. Oncogene. 1995;11(2):365–376. [PubMed] [Google Scholar]
- 9.Witowsky J, Abell A, Johnson NL, Johnson GL, Cuevas BD. Mekk1 is required for inducible urokinase-type plasminogen activator expression. J Biol Chem. 2003;278(8):5941–5946. doi: 10.1074/jbc.M212363200. [DOI] [PubMed] [Google Scholar]
- 10.Heymans S, Luttun A, Nuyens D, et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5(10):1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 11.D'Angelo DD, Sakata Y, Lorenz JN, et al. Transgenic galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A. 1997;94(15):8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sadoshima J, Montagne O, Wang Q, et al. The mekk1-jnk pathway plays a protective role in pressure overload but does not mediate cardiac hypertrophy. J Clin Invest. 2002;110(2):271–279. doi: 10.1172/JCI14938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vikstrom KL, Factor SM, Leinwand LA. Mice expressing mutant myosin heavy chains are a model for familial hypertrophic cardiomyopathy. Mol Med. 1996;2(5):556–67. [PMC free article] [PubMed] [Google Scholar]
- 14.Konhilas JP, Watson PA, Maass A, et al. Exercise can prevent and reverse the severity of hypertrophic cardiomyopathy. Circ Res. 2006;98(4):540–548. doi: 10.1161/01.RES.0000205766.97556.00. [DOI] [PubMed] [Google Scholar]
- 15.Stauffer BL, Konhilas JP, Luczak ED, Leinwand LA. Soy diet worsens heart disease in mice. J Clin Invest. 2006;116(1):209–216. doi: 10.1172/JCI24676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yujiri T, Ware M, Widmann C, et al. Mek kinase 1 gene disruption alters cell migration and c-jun nh2- terminal kinase regulation but does not cause a measurable defect in nf- kappa b activation. Proc Natl Acad Sci U S A. 2000;97(13):7272–7277. doi: 10.1073/pnas.130176697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olsson MC, Palmer BM, Stauffer BL, Leinwand LA, Moore RL. Morphological and functional alterations in ventricular myocytes from male transgenic mice with hypertrophic cardiomyopathy. Circ Res. 2004;94(2):201–207. doi: 10.1161/01.RES.0000111521.40760.18. [DOI] [PubMed] [Google Scholar]
- 18.Spinale FG, Coker ML, Heung LJ, et al. A matrix metalloproteinase induction/activation system exists in the human left ventricular myocardium and is upregulated in heart failure. Circulation. 2000;102(16):1944–1949. doi: 10.1161/01.cir.102.16.1944. [DOI] [PubMed] [Google Scholar]
- 19.Konhilas JP, Maass AH, Luckey SW, et al. Sex modifies exercise and cardiac adaptation in the mouse. Am J Physiol Heart Circ Physiol. 2004 doi: 10.1152/ajpheart.00292.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mercadier JJ, Lompre AM, Wisnewsky C, et al. Myosin isoenzyme changes in several models of rat cardiac hypertrophy. Circ Res. 1981;49(2):525–532. doi: 10.1161/01.res.49.2.525. [DOI] [PubMed] [Google Scholar]
- 21.Luckey SW, Mansoori J, Fair K, et al. Blocking cardiac growth in hypertrophic cardiomyopathy induces cardiac dysfunction and decreased survival only in males. Am J Physiol Heart Circ Physiol. 2007;292(2):H838–845. doi: 10.1152/ajpheart.00615.2006. [DOI] [PubMed] [Google Scholar]
- 22.Cuevas BD, Winter-Vann AM, Johnson NL, Johnson GL. Mekk1 controls matrix degradation and tumor cell dissemination during metastasis of polyoma middle-t driven mammary cancer. Oncogene. 2006;25(36):4998–5010. doi: 10.1038/sj.onc.1209507. [DOI] [PubMed] [Google Scholar]
- 23.Cuevas BD, Uhlik MT, Garrington TP, Johnson GL. Mekk1 regulates the ap-1 dimer repertoire via control of junb transcription and fra-2 protein stability. Oncogene. 2005;24(5):801–809. doi: 10.1038/sj.onc.1208239. [DOI] [PubMed] [Google Scholar]
- 24.Heymans S, Schroen B, Vermeersch P, et al. Increased cardiac expression of tissue inhibitor of metalloproteinase-1 and tissue inhibitor of metalloproteinase-2 is related to cardiac fibrosis and dysfunction in the chronic pressure-overloaded human heart. Circulation. 2005;112(8):1136–1144. doi: 10.1161/CIRCULATIONAHA.104.516963. [DOI] [PubMed] [Google Scholar]
- 25.Olsson MC, Palmer BM, Leinwand LA, Moore RL. Gender and aging in a transgenic mouse model of hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2001;280(3):H1136–1144. doi: 10.1152/ajpheart.2001.280.3.H1136. [DOI] [PubMed] [Google Scholar]
- 26.Thornberry NA, Lazebnik Y. Caspases: Enemies within. Science. 1998;281(5381):1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 27.Molkentin JD, Dorn GW., 2nd Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- 28.Hilfiker-Kleiner D, Hilfiker A, Castellazzi M, et al. Jund attenuates phenylephrine-mediated cardiomyocyte hypertrophy by negatively regulating ap-1 transcriptional activity. Cardiovasc Res. 2006;71(1):108–117. doi: 10.1016/j.cardiores.2006.02.032. [DOI] [PubMed] [Google Scholar]
- 29.Pollack PS, Pasquarello LM, Budjak R, et al. Differential expression of c-jun and jund in end-stage human cardiomyopathy. J Cell Biochem. 1997;65(2):245–253. doi: 10.1002/(sici)1097-4644(199705)65:2<245::aid-jcb9>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 30.Noji Y, Shimizu M, Ino H, et al. Increased circulating matrix metalloproteinase-2 in patients with hypertrophic cardiomyopathy with systolic dysfunction. Circ J. 2004;68(4):355–360. doi: 10.1253/circj.68.355. [DOI] [PubMed] [Google Scholar]
- 31.Fedak PW, Altamentova SM, Weisel RD, et al. Matrix remodeling in experimental and human heart failure: A possible regulatory role for timp-3. Am J Physiol Heart Circ Physiol. 2003;284(2):H626–634. doi: 10.1152/ajpheart.00684.2002. [DOI] [PubMed] [Google Scholar]
- 32.Fedak PW, Smookler DS, Kassiri Z, et al. Timp-3 deficiency leads to dilated cardiomyopathy. Circulation. 2004;110(16):2401–2409. doi: 10.1161/01.CIR.0000134959.83967.2D. [DOI] [PubMed] [Google Scholar]
- 33.Ducharme A, Frantz S, Aikawa M, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106(1):55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: Influence on cardiac form and function. Physiol Rev. 2007;87(4):1285–1342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 35.Torre-Amione G. Immune activation in chronic heart failure. Am J Cardiol. 2005;95(11A):3C–8C. doi: 10.1016/j.amjcard.2005.03.006. discussion 38C-40C. [DOI] [PubMed] [Google Scholar]
- 36.Di Girolamo N, Indoh I, Jackson N, et al. Human mast cell-derived gelatinase b (matrix metalloproteinase-9) is regulated by inflammatory cytokines: Role in cell migration. J Immunol. 2006;177(4):2638–2650. doi: 10.4049/jimmunol.177.4.2638. [DOI] [PubMed] [Google Scholar]
- 37.Watson PA, Reusch JE, McCune SA, et al. Restoration of creb function is linked to completion and stabilization of adaptive cardiac hypertrophy in response to exercise. Am J Physiol Heart Circ Physiol. 2007;293(1):H246–259. doi: 10.1152/ajpheart.00734.2006. [DOI] [PubMed] [Google Scholar]
- 38.Nawata R, Yujiri T, Nakamura Y, et al. Mek kinase 1 mediates the antiapoptotic effect of the bcr-abl oncogene through nf-kappab activation. Oncogene. 2003;22(49):7774–7780. doi: 10.1038/sj.onc.1206901. [DOI] [PubMed] [Google Scholar]
- 39.Braz JC, Bueno OF, Liang Q, et al. Targeted inhibition of p38 mapk promotes hypertrophic cardiomyopathy through upregulation of calcineurin-nfat signaling. J Clin Invest. 2003;111(10):1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]