

Abstract
Endothelial production of nitric oxide is critical to the regulation of vascular responses, including vascular tone and regional blood flow, leukocyte–endothelial interactions, platelet adhesion and aggregation, and vascular smooth muscle cell proliferation. A relative deficiency in the amount of bioavailable vascular NO results in endothelial dysfunction, with conditions that are conducive to the development of atherosclerosis: thrombosis, inflammation, neointimal proliferation, and vasoconstriction. This review focuses on mouse models of endothelial dysfunction caused by direct genetic modification of the endothelial nitric oxide synthase (eNOS) gene. We first describe the cardiovascular phenotypes of eNOS knockout mice, which are a model of total eNOS gene deficiency and thus the ultimate model of endothelial dysfunction. We then describe S1177A and S1177D eNOS mutant mice as mouse models with altered eNOS phosphorylation and therefore varying degrees of endothelial dysfunction. These include transgenic mice that carry the eNOS S1177A and S1177D transgenes, as well as knockin mice in which the endogenous eNOS gene has been mutated to carry the S1177A and S1177D mutations. Together, eNOS knockout mice and eNOS S1177 mutant mice are useful tools to study the effects of total genetic deficiency of eNOS as well as varying degrees of endothelial dysfunction caused by eNOS S1177 phosphorylation.
Keywords: Nitric oxide, Endothelium, Vascular dysfunction, Transgenic mouse, Cerebral circulation
Introduction
In this article, we describe mutant mouse models of endothelial dysfunction specifically caused by total or partial deficiency in activity of endothelial nitric oxide synthase (eNOS). As the primary interface between the blood and body tissues, the endothelium regulates several important aspects of vascular responses [1–4]. First, it regulates vascular tone and regional blood flow, as it produces factors involved in vasodilation, including nitric oxide (NO) and prostacyclin, and vasoconstriction, including endothelin. Second, it modulates adhesion and aggregation of platelets, thus affecting propensity or resistance to thrombosis. Third, the endothelium expresses surface adhesion molecules that influence interactions with leukocytes and monocytes, affecting recruitment of inflammatory cells and atherosclerotic plaque formation. Finally, it modulates vascular smooth muscle cell proliferation in response to vascular injury. A relative deficiency in the amount of bioavailable vascular NO results in endothelial dysfunction, with conditions that are conducive to the development of atherosclerosis: thrombosis, inflammation, neointimal proliferation, and vasoconstriction.
Regulation of NO production by eNOS
Physiologic endothelial function requires both basal and stimulated production of NO by eNOS. The eNOS enzyme utilizes L-arginine as substrate for generation of NO from the terminal guanidine nitrogen, producing L-citrulline from the rest of the molecule. eNOS activity is regulated at multiple steps, including transcription; substrate availability; interactions with calcium, calmodulin, and enzymatic cofactors such as FAD, FMN, NADPH, and BH4; protein–protein interactions with hsp90 and caveolins; and post-translational modifications. eNOS is also regulated by dimerization and precise subcellular localization to caveolae [5, 6]. In many processes, the downstream target of NO is soluble guanylate cyclase (sGC). NO binds to the heme moiety of sGC, activating it and resulting in increased cyclic guanosine monophosphate (cGMP) production. cGMP mediates vascular smooth muscle relaxation [7, 8], as well as effects on platelet aggregation [9]. Once generated, NO may also interact with superoxide to form peroxynitrite anion, effectively removing it from its physiologic functions (Fig. 1).
Fig. 1.

Biological roles of endothelial NO: inhibition of platelets aggregation, modulation of leukocyte–endothelial interactions, modulation of smooth muscle cell proliferation, and maintenance of vascular tone. eNOS is activated by physiological and metabolic stimuli, shear stress, and receptor-dependent agonists. S1177 phosphorylation site is an important regulatory mechanism for eNOS activity. eNOS catalytic activity depends on the intracellular localization to caveolae and the protein–protein interactions with caveolins and hsp-90. NO activates cGMP, inducing PKG-dependent relaxation of smooth muscle cells and increasing luminal diameter and blood flow. P phosphorylation; S1177 serine 1177, Hsp-90 heat shock protein-90, PKG serine–threonine-specific protein kinase G
eNOS is activated by various physiological mediators, including mechanical shear stress [3, 10–12], estrogens [13], insulin [14], acetylcholine [15], and other receptor-dependent agonists [10]. eNOS is post-translationally phosphorylated at several serine (S) and threonine (T) residues, including T497, S617, S635, and S1177, using the human amino acid sequence numbering [16]. Phosphorylation of eNOS at the human S1177 residue is an important mechanism for physiologic regulation of NO production. eNOS phosphorylation at this site is increased by estrogens and statins. It is decreased in diabetes, hypertension, metabolic syndrome, and obesity, as well as by interleukin-6 (IL-6), tumor necrosis factor α (TNFα), and resistin [17, 18].
The corresponding positions for the human eNOS S1177 phosphorylation site differ in other species. The possibility for confusion is compounded by the fact that there are other serine residues in close proximity. The sequence for the 1203 amino acid human eNOS gene, starting at position 1171, reads SRIRTQSFSL. Within this sequence, S1177, indicated in boldface and underlined, is the serine residue that is phosphorylated. Comparison of the sequences of the eNOS gene in other species shows that the corresponding residue is S1176 in the murine sequence and S1179 in the bovine sequence. This is relevant because when the bovine sequence is used in transgenic constructs, it is the bovine S1179 site that is modified, corresponding to the serine at the 1177 position in the human sequence. For the remainder of this paper, we will always refer to this site as S1177, using the human sequence for numbering.
Endothelial dysfunction
Endothelial dysfunction, broadly defined, occurs when the endothelium fails to serve its normal physiologic and protective functions [3, 19–22]. This may occur because the endothelium is damaged or missing, as in the case of denuded endothelium in coronary arteries subjected to angioplasty or percutaneous intervention. It may also occur when the normal responses of the endothelium are altered, for example, by oxidative stress, hyperglycemia, advanced glycation products, free fatty acids, or inflammatory cytokines. Endothelial dysfunction is an important final common pathway between many cardiovascular risk factors—such as hypertension, obesity, diabetes, and dyslipidemia—and the development of atherosclerosis. Metabolic diseases such as atherosclerosis, hypertension, diabetes, obesity, hyperlipidemia, hypercholesterolemia, and metabolic syndrome impair endothelial NO availability, resulting in endothelial dysfunction.
Endothelial dysfunction can be measured by assessing physiologic vasodilator responses [20]. Vascular dilation to mediators such as acetylcholine or increased blood flow depends on NO produced by the endothelium. In animal models, vascular reactivity can be measured in isolated vessel segments using myography of vessel rings or vessel segments. In man, endothelial function can be measured in the cardiac catheterization laboratory by monitoring the effect of intracoronary acetylcholine infusion. In the clinical setting, flow-mediated dilation (FMD) can be measured by using ultrasound to quantitate the response of brachial artery diameter to reflow after a period of vascular occlusion by a blood pressure cuff. Although they measure responses in different vascular beds, FMD correlates with the catheterization laboratory measurements of coronary circulation responses. Furthermore, FMD predicts future cardiovascular events. Thus, FMD is a commonly used method to assess endothelial function, and impaired FMD is taken to reflect endothelial dysfunction.
The mechanism of endothelial dysfunction is insufficient endothelial NO availability [19]. This can be caused by reduced eNOS gene expression, L-arginine deficiency [23], inhibition of NOS by asymmetric dimethylarginine [24], uncoupling of eNOS and tetrahydrobiopterin deficiency [25], mechanical damage of endothelium, metabolic intoxication by free fatty acids and inflammatory cytokines IL-6 and TNFα [17], alteration of endothelial intracellular PI3-Akt signaling by hyperglycemia and advanced glycation products [20], and scavenging vascular NO by superoxide anion [26]. Two important causes of endothelial dysfunction include a reduction in eNOS phosphorylation at S1177 [27, 28] and rapid reaction of NO with superoxide to form peroxynitrite anion [29].
Many mouse models of human disease manifest endothelial dysfunction, including leptin receptor-deficient (db/db) mice [30], leptin-deficient (ob/ob) mice [31], and genetic knockouts of apoE [32], Akt1 [33, 34], superoxide dismutase [35], sGC [36], and glutathione peroxidase [37]. Endothelium-specific overexpressions of GTP cyclohydrolase I [38] and SIRT1 [39] have been shown to ameliorate endothelial dysfunction in mouse models of atherosclerosis and diabetes. This review focuses on mouse models of endothelial dysfunction caused by direct genetic modification of the eNOS gene. We first describe the cardiovascular phenotypes of eNOS knockout mice, which are a model of total eNOS gene deficiency and thus the ultimate model of endothelial dysfunction. We then describe S1177A and S1177D mutant mice as models that demonstrate altered eNOS phosphorylation, and therefore varying degrees of endothelial dysfunction.
eNOS knockout mice
eNOS knockout mice were generated as the first model of direct endothelial dysfunction by disrupting the region that encodes for the NADPH ribose and adenine binding sites, which are essential for eNOS catalytic activity [15]. These mice demonstrate the absence of eNOS mRNA and enzymatic activity, but are fertile and have normal anatomy. Other lines of eNOS knockout mice have also been generated [40–42], and all of them show similar phenotypes.
eNOS knockout mice show the phenotypes caused by total genetic absence of eNOS. This includes lack of endothelium-derived relaxing factor (EDRF) activity and hypertension [15], increased platelet aggregation [9], leukocyte–endothelial adhesion [43], increased vascular smooth muscle cell proliferation [44], and increased propensity to thrombosis [43], stroke [45, 46], and atherosclerosis [47]. eNOS knockout mice do not demonstrate ischemic preconditioning [48], and show abnormalities in mitochondrial function and biogenesis [49, 50], and insulin resistance [51, 52].
It is possible that other NOS isoforms may compensate for lack of eNOS in the eNOS knockout mice. To address this question, double and triple NOS knockout mice have been generated. Mice that lack both neuronal nitric oxide synthase (nNOS) and eNOS display abnormalities in hippocampal long-term potentiation, a model for learning and memory [53]. Mice that lack all three NOS isoforms show nephrogenic diabetes insipidus [54] as well as spontaneous myocardial infarction [55].
Vascular reactivity and hypertension
eNOS knockout mice lack EDRF activity as measured by vascular relaxation in response to acetylcholine and other endothelium-dependent vasodilators, both in isolated arteries [15] and in vivo [8]. This is important because in the development of atherosclerotic vascular disease, functional impairment of endothelium-dependent relaxation occurs before morphological changes of vessels and is the first sign of endothelial dysfunction. The fact that eNOS knockout mice are hypertensive proves the essential role of endothelial NO in the physiologic regulation of blood pressure. Absence of eNOS cannot be compensated by cardiac, neuronal, or hormonal homeostatic mechanisms, and hypertension occurs despite these counterregulatory mechanisms [56, 57]. eNOS knockout mice paradoxically manifest decreased blood pressure after nonspecific inhibition of NO by L-NA, indicating an important role for neuronal NOS (likely in perivascular nerves, see Fig. 2)in the maintenance of blood pressure [58].
Fig. 2.

Effect of eNOS mutations on CBF deficit. a Laser speckle contrast images in mice subjected to middle cerebral artery occlusion. Blue areas indicate regions with ≤30% residual blood flow after 60 min of ischemia. b The area of cortex with≤20% (black) or 21–30% (blue) residual blood flow as compared with preischemic baseline. *P<0.05, comparing the area with ≤20% of residual blood flow of eNOS knockout and S1177A transgenic mice with WT and S1177D transgenic mice
Interactions with leukocytes, platelets, and vascular smooth muscle
eNOS knockout mice demonstrate the essential roles of endothelial NO in leukocyte–endothelial interactions, which are important for both inflammatory responses and for development of atherosclerotic plaques. Absence of eNOS increases expression of endothelial P-selectin and thereby increases leukocyte rolling and adhesion to the endothelial surface [43]. Similarly, platelet aggregation, hemostasis [9], coagulability [43], and thrombosis [59, 60] are increased in the absence of eNOS. NO-induced inhibition of platelet aggregation is mediated by sGC in platelets [7, 36].
The roles of eNOS in regulating vascular smooth muscle proliferation are demonstrated by the phenotype of eNOS knockout mice after vascular injury [61]. Cuff injury of the femoral artery results in neointima formation, and this response is greatly exaggerated in eNOS knockout mice as compared with wild-type mice [60].
Together, neutrophil infiltration, platelet aggregation, and smooth muscle proliferation accelerate vascular complications and reduce tissue perfusion and endothelial dysfunction. Thus, it is not surprising that apoE/eNOS double knockout mice demonstrate accelerated atherosclerosis compared to apoE knockout mice. In addition, apoE/eNOS double knockout mice show evidence of ischemic heart disease, left ventricular dysfunction, and aortic aneurysm, making them a useful model that mimics many aspects of human atherosclerotic cardiovascular disease [62]. Thus, eNOS deficiency causes endothelial dysfunction, characterized by impaired vascular reactivity, followed by abnormal vascular tone, impaired tissue perfusion, and hypertension. Effects on platelet aggregation and adhesion, leukocyte–endothelial interactions, and vascular smooth muscle proliferation likely underlie the increased propensity to atherosclerosis seen in man when there is endothelial dysfunction, particularly in the setting of diabetes and metabolic syndrome.
Effects on cardiac function, pulmonary vasculature, and mitochondria
eNOS maintains basal vasorelaxation in the lungs and protects the lung by attenuating pulmonary hypertension. Endothelial dysfunction in eNOS knockout mice increases hypoxic pulmonary hypertension [63].
NO is produced in the heart by eNOS present in the cardiac endothelium and cardiac myocytes. eNOS knockout mice show enhanced inotropic and lusitropic responses under basal conditions [64] and in response to isoproterenol [65]. These results indicate that eNOS normally blunts the inotropic response to β-adrenergic stimulation [58]. Furthermore, eNOS deficient mice demonstrate enhanced left ventricular dysfunction and post-myocardial infarction remodeling by diminished myocyte hypertrophy [66].
eNOS is essential to mitochondrial biogenesis, as eNOS knockout mice show fewer mitochondria, decreased expression of mitochondrial genes, and less mitochondrial respiration both under basal conditions and after cold exposure and exercise [49]. Furthermore, NO produced by eNOS plays important roles in the differentiation and proliferation of brown adipocytes, which are important to thermogenesis and energy metabolism [50, 67–70]. These results provide evidence that abnormalities in eNOS function may underlie the metabolic changes that occur in obesity and diabetes.
Cerebral blood flow regulation and stroke
Hypertension and impaired vasorelaxation are the primary underlying mechanisms of increased vulnerability of eNOS knockout mice to stroke injury [45, 46, 71]. Using the middle cerebral artery occlusion model of stroke, eNOS knockout mice show larger areas of ischemia and correspondingly smaller penumbra zone (Figs. 2, 3) [46, 71]. This results in worse post-stroke neurological deficits and larger cerebral infarct volumes as compared with wild-type mice (Fig. 5) [46].
Fig. 3.
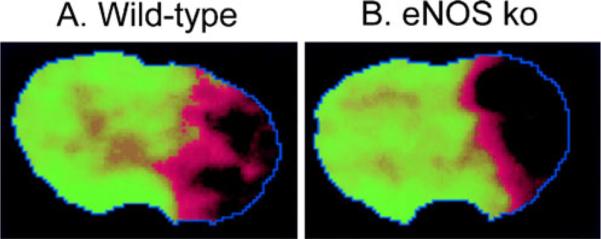
a Temporal correlation mapping image shows blood flow after focal cerebral ischemia in wild-type mouse. Normal hemodynamic status is shown as green. Regions with abnormal CBF are represented with a red-to-black sliding color scale. The core appears black, and the penumbra is the reddish rim surrounding the core. An extensive hemodynamic penumbra exist in wild-type mice 30 min after focal ischemia. b The hemodynamic deficit in eNOS knockout mice after focal ischemia is more severe. The core is larger, and the penumbra is correspondingly smaller compared with wild-type mice
Fig. 5.

Effect of S1177 mutations on cerebral infarct volume. Mice were subjected to 1 h of focal brain ischemia followed by reperfusion for 23 h. The brains were cut into coronal sections and stained using 2,3,5-triphenyltetrazolium chloride. a Abscissa shows the distance (millimeter) from the rostral surface of the brain. For each group, n=7 mice. *P<0.05 versus S1177A transgenic mice, †P<0.05 versus eNOS KO mice. b Infarct volumes were determined by the indirect method, which corrects for edema. *P<0.05 versus infarct volume from S1177A mutant and from eNOS knockout mice
eNOS plays a major role in the regulation of cerebral blood flow (CBF). Cerebral vasorelaxation to acetylcholine is impaired in eNOS knockout mice [8]. Interestingly, despite absence of eNOS and resulting hypertension, eNOS knockout mice have the same level of resting CBF as WT mice have. This is likely due to autoregulation and the compensatory action of perivascular neuronal NOS.
eNOS knockout mice have been instrumental in defining the roles of eNOS in CBF regulation during hyperbaric oxygenation [8]. The physiologic response is to protect the brain by vasoconstriction against dangerously increased reactive oxygen species and hyperbaric oxygen. Quantitative CBF measurements demonstrate that the first phase cerebral vasoconstriction response to hyperbaric oxygen is mediated by endothelial NO inactivation by superoxide anion, causing decreased blood flow. Later, the second phase vasodilator response is due to NO-dependent hyperbaric oxygen vasodilation, resulting in seizures. The second phase depends on NO produced by both eNOS and nNOS. The likely mechanism triggering NO production in this phase is increased concentration of nNOS [72] and eNOS [73] mediated by hsp-90 binding. During prolonged HBO exposure, reactive oxygen species initiate NOS stimulation, increased protein–protein interactions between hsp90 and NOS isoforms and intracellular calcium entry [74]. These data show the important role of endothelial NO production in cerebral vasoconstriction and in hyperoxic hyperemia preceding oxygen-induced seizures [75].
Ischemic preconditioning is a phenomenon in which brief ischemic events protect the brain from subsequent severe ischemic insult [76, 77]. eNOS knockout mice do not show ischemic preconditioning [48], demonstrating an essential role for eNOS in the protection. Resting CBF and CBF changes during preconditioning do not mediate the difference between preconditioned WT and eNOS knockout mice. The exact mechanisms underlying the protective role of eNOS in ischemic preconditioning are still not known.
eNOS S1177 mutant mice
Transgenic S1177A and S1177D mice
Although eNOS knockout mice are an extreme example of endothelial dysfunction in that they totally lack eNOS, there are no known human examples of eNOS gene deficiency. Rather, patients who manifest endothelial dysfunction show a relative deficiency in bioavailable endothelial NO. One of the mechanisms for this is abnormalities in phosphorylation of the eNOS enzyme at the S1177 residue. Indeed, S1177 phosphorylation of eNOS is one of most important and intensively investigated regulatory mechanisms for eNOS enzymatic activity. Estrogens, statins, insulin, leptin, adiponectin, VEGF, and shear stress promote S1177 eNOS phosphorylation and activation. In contrast, eNOS phosphorylation is decreased in patients with hypertension, hyperlipidemia, diabetes, and metabolic syndrome.
eNOS S1177 is a target substrate for Akt kinase [27, 28, 78, 79]. In addition, S1177 is known to be phosphorylated by several other kinases, including AMP kinase [80], protein kinase A (PKA) and protein kinase G (PKG) [81], and calmodulin-dependent kinase II (CaMKII) [82]. The phosphatases that regulate dephosphorylation of S1177 have not been identified. Mechanistically, increased NOS activity appears to be due to enhanced electron flux through the reductase domain and reduced calmodulin dissociation upon S1177 phosphorylation [83]. By crystallography, the carboxyl tail of eNOS normally blocks electron transfer from the reductase domain [84]. Phosphorylation of S1177 results in a negative charge, swinging the entire FMN domain to allow enhanced electron transfer through the reductase domain, activating the enzyme. Physiologically, shear stress, VEGF, and insulin activate eNOS by S1177 phosphorylation, permitting greater eNOS activation to take place at any level of calcium without substantially changing the affinity of calcium-activated calmodulin for eNOS.
Mutant forms of eNOS have been used to study the role of eNOS S1177 phosphorylation. Substitution of alanine in place of S1177 (S1177A) produces a form of eNOS that cannot be phosphorylated, since it lacks the hydroxyl group on the side chain of serine. Substitution of aspartate in place of S1177 (S1177D) mimics the increased catalytic activity caused by phosphorylation, since the aspartate side chain contains a negatively charged carboxyl group that mimics the negatively charged phosphate group of eNOS phosphorylated at S1177 [83, 85]. S1177A and S1177D mutations both are active in endothelial cells [85], but S1177A eNOS does not demonstrate Akt-dependent NO release, while S1177D shows enhanced NO production even in the absence of Akt [27, 28]. S1177D eNOS shows reduced calmodulin dissociation and enhanced electron flux via the reductase domain [83].
We generated transgenic mice carrying the S1177A and S1177D mutations in an eNOS cDNA transgene [46]. We bred these mice with eNOS knockout mice to obtain mice that only express S1177A or S1177D mutant eNOS forms. The expression and subcellular localization of the S1177A and S1177D mutant forms of eNOS were documented by Western blotting and en face immunostaining of arteries. There was no difference in protein level expression and subcellular localization of eNOS in either mutant strains (Fig. 4). Unphosphorylatable S1177A transgenic mice showed attenuated vascular relaxation to acetylcholine. Using the middle cerebral artery occlusion model of stroke, S1177A mice showed increased infarct volume, as compared with S1177D and WT mice [46] (Fig. 5).
Fig. 4.

Subcellular localization of eNOS S1177 mutation in transgenic mice. a En face immunostaining of carotid arteries shows localization of eNOS in perinuclear Golgi. VE-cadherin (VE-cad) staining shows the outlines of the endothelial membranes. b En face immunostaining of carotid arteries shows perinuclear location of the transgene hemagglutinin tag in the perinuclear Golgi in a pattern similar to eNOS immunostaining in wild-type mice. Original magnification ×600
Laser speckle flowmetry assesses CBF by measuring blood flow with high spatial and temporal resolution. This provides a two dimensional determination of real-time flow deficit [86]. eNOS knockout mice developed larger cortical ischemic areas compared with wild-type and S1177D mice. The S1177A mutation did not improve cortical blood flow whereas the S1177D eNOS transgene reduces the ischemic area to the size seen in wild-type mice (Fig. 2).
S1177 transgenic mice have been useful to study the involvement of eNOS S1177 phosphorylation in maintaining pulmonary relaxation and protecting against hyperoxic pulmonary injury. Comparison of three strains of mice having different level of eNOS activity (eNOS knockout, S1177A transgenic, and S1177 D transgenic mice) shows that pulmonary vascular protection correlates directly with eNOS activity [87]. S1177A transgenic mice demonstrated profound oxygen-induced pulmonary injury, while S1177D transgenic mice were protected.
Knockin S1177A and S1177D mice
After generating S1177 transgenic mice on eNOS knockout background, we made knockin mice that carry the same mutations. We replaced the endogenous eNOS gene with a mutant gene carrying the S1177D and S1177A mutations [28]. These mice are currently being characterized. As an initial step, we bred the knockin mice with Akt1 knockout mice to test whether the mutations could overcome the effects of Akt1 deficiency [88].
Acetylcholine-induced production of cGMP was diminished in aortas of S1177A/Akt1 mutant mice and increased in S1177D/Akt1 mutant mice as compared with WT mice. Measurements of NO by nitrite production showed that the S1177D mutation maintains NO production despite deficiency of Akt1.
Wound healing experiments demonstrated a critical role of eNOS as Akt substrate for postnatal pathological angiogenesis [88]. S1177A/Akt1 mutant mice show attenuated wound healing and increased ischemic limb injury after ligation of femoral artery, while S1177D/Akt1 mutant mice demonstrated faster wound healing and resistance to ischemic injury. Recovery of blood flow, revascularization, clinical score, and recruitment of pericytes was better in S1177/Akt1 mutant mice (comparable with WT) as compared with S1177A/Akt1 mutants (comparable with Akt1 knockout mice). Taken together, these results prove that wound healing and post-ischemic angiogenesis mainly depends on S1177 eNOS phosphorylation.
The mRNA expressions of HIF1α-responsive genes, including VEGF-A, VEGF receptor (Flk-1), and angiopoietin-like 4 protein were increased in S1177D/Akt1 mutants and wild-type mice as compared with Akt1 deficient and S1177A/Akt1 mutant mice. HIF1α abundance in ischemic muscles of S1177D/Akt1 deficient mice was higher as compared with S1177A/Akt1 deficient mice. These results indicate that the phosphomimetic S1177D mutation was able to rescue defective postnatal angiogenesis of Akt1 knockout mice by stabilization of HIF1α and increased production of responsive genes.
Conclusion
eNOS knockout and apoE/eNOS double knockout mice are models of endothelial dysfunction due to total absence of endothelial NO production. S1177A and S1177A/Akt1 mutant mice are models with partial endothelial dysfunction, which are more clinically relevant in that they mimic the deficiency in eNOS S1177 phosphorylation seen in man. Modulation of eNOS phosphorylation on serine 1177 is a promising approach to define the roles of its phosphorylation on endothelial dysfunction. As such, S1177 mutant mice may lead to proof of concept for eNOS phosphorylation as a target for the treatment and prevention of cardiovascular disease associated with conditions that show endothelial dysfunction, including diabetes, metabolic syndrome, and obesity.
References
- 1.Gimbrone MA., Jr Endothelial dysfunction and atherosclerosis. J Card Surg. 1989;4:180–183. doi: 10.1111/j.1540-8191.1989.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 2.Gimbrone MA., Jr Vascular endothelium: an integrator of pathophysiologic stimuli in atherosclerosis. Am J Cardiol. 1995;75:67B–70B. doi: 10.1016/0002-9149(95)80016-l. [DOI] [PubMed] [Google Scholar]
- 3.Gimbrone MA, Jr, Topper JN, Nagel T, Anderson KR, Garcia-Cardena G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann NYAcad Sci. 2000;902:230–239. doi: 10.1111/j.1749-6632.2000.tb06318.x. discussion 239–40. [DOI] [PubMed] [Google Scholar]
- 4.Ross R. Atherosclerosis is an inflammatory disease. Am Heart J. 1999;138:S419–S420. doi: 10.1016/s0002-8703(99)70266-8. [DOI] [PubMed] [Google Scholar]
- 5.Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem. 1996;271:22810–22814. doi: 10.1074/jbc.271.37.22810. [DOI] [PubMed] [Google Scholar]
- 6.Shaul PW. Regulation of endothelial nitric oxide synthase: location, location, location. Annu Rev Physiol. 2002;64:749–774. doi: 10.1146/annurev.physiol.64.081501.155952. [DOI] [PubMed] [Google Scholar]
- 7.Mergia E, Friebe A, Dangel O, Russwurm M, Koesling D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J Clin Invest. 2006;116:1731–1737. doi: 10.1172/JCI27657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atochin DN, Demchenko IT, Astern J, Boso AE, Piantadosi CA, Huang PL. Contributions of endothelial and neuronal nitric oxide synthases to cerebrovascular responses to hyperoxia. J Cereb Blood Flow Metab. 2003;23:1219–1226. doi: 10.1097/01.WCB.0000089601.87125.E4. [DOI] [PubMed] [Google Scholar]
- 9.Freedman JE, Sauter R, Battinelli EM, Ault K, Knowles C, Huang PL, Loscalzo J. Deficient platelet-derived nitric oxide and enhanced hemostasis in mice lacking the NOSIII gene. Circ Res. 1999;84:1416–1421. doi: 10.1161/01.res.84.12.1416. [DOI] [PubMed] [Google Scholar]
- 10.Barton M. Endothelial dysfunction and atherosclerosis: endothelin receptor antagonists as novel therapeutics. Curr Hyper-tens Rep. 2000;2:84–91. doi: 10.1007/s11906-000-0064-5. [DOI] [PubMed] [Google Scholar]
- 11.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 12.Moens AL, Goovaerts I, Claeys MJ, Vrints CJ. Flow-mediated vasodilation: a diagnostic instrument, or an experimental tool? Chest. 2005;127:2254–2263. doi: 10.1378/chest.127.6.2254. [DOI] [PubMed] [Google Scholar]
- 13.Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, Sessa WC, Bender JR. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res. 2000;87:677–682. doi: 10.1161/01.res.87.8.677. [DOI] [PubMed] [Google Scholar]
- 14.Rask-Madsen C, Ihlemann N, Krarup T, Christiansen E, Kober L, Nervil Kistorp C, Torp-Pedersen C. Insulin therapy improves insulin-stimulated endothelial function in patients with type 2 diabetes and ischemic heart disease. Diabetes. 2001;50:2611–2618. doi: 10.2337/diabetes.50.11.2611. [DOI] [PubMed] [Google Scholar]
- 15.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 16.Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- 17.Kim F, Gallis B, Corson MA. TNF-alpha inhibits flow and insulin signaling leading to NO production in aortic endothelial cells. Am J Physiol Cell Physiol. 2001;280:C1057–C1065. doi: 10.1152/ajpcell.2001.280.5.C1057. [DOI] [PubMed] [Google Scholar]
- 18.Shen YH, Zhang L, Gan Y, Wang X, Wang J, LeMaire SA, Coselli JS, Wang XL. Up-regulation of PTEN (phosphatase and tensin homolog deleted on chromosome ten) mediates p38 MAPK stress signal-induced inhibition of insulin signaling. A cross-talk between stress signaling and insulin signaling in resistin-treated human endothelial cells. J Biol Chem. 2006;281:7727–7736. doi: 10.1074/jbc.M511105200. [DOI] [PubMed] [Google Scholar]
- 19.Huang PL. Unraveling the links between diabetes, obesity, and cardiovascular disease. Circ Res. 2005;96:1129–1131. doi: 10.1161/01.RES.0000170705.56583.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang PL. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol Metab. 2009;20:295–302. doi: 10.1016/j.tem.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech. 2009;2:231–237. doi: 10.1242/dmm.001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 23.Bode-Boger SM, Boger RH, Kienke S, Junker W, Frolich JC. Elevated L-arginine/dimethylarginine ratio contributes to enhanced systemic NO production by dietary L-arginine in hypercholesterolemic rabbits. Biochem Biophys Res Commun. 1996;219:598–603. doi: 10.1006/bbrc.1996.0279. [DOI] [PubMed] [Google Scholar]
- 24.Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol. 2000;20:2032–2037. doi: 10.1161/01.atv.20.9.2032. [DOI] [PubMed] [Google Scholar]
- 25.Cosentino F, Luscher TF. Tetrahydrobiopterin and endothelial function. Eur Heart J. 1998;19(Suppl G):G3–G8. [PubMed] [Google Scholar]
- 26.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 28.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 30.Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153:1127–1128. doi: 10.1126/science.153.3740.1127. [DOI] [PubMed] [Google Scholar]
- 31.Coleman DL, Hummel KP. The influence of genetic background on the expression of the obese (Ob) gene in the mouse. Diabetologia. 1973;9:287–293. doi: 10.1007/BF01221856. [DOI] [PubMed] [Google Scholar]
- 32.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 34.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 35.Matzuk MM, Dionne L, Guo Q, Kumar TR, Lebovitz RM. Ovarian function in superoxide dismutase 1 and 2 knockout mice. Endocrinology. 1998;139:4008–4011. doi: 10.1210/endo.139.9.6289. [DOI] [PubMed] [Google Scholar]
- 36.Buys ES, Cauwels A, Raher MJ, Passeri JJ, Hobai I, Cawley SM, Rauwerdink KM, Thibault H, Sips PY, Thoonen R, Scherrer-Crosbie M, Ichinose F, Brouckaert P, Bloch KD. sGC (alpha)1(beta)1 attenuates cardiac dysfunction and mortality in murine inflammatory shock models. Am J Physiol Heart Circ Physiol. 2009;297:H654–H663. doi: 10.1152/ajpheart.00367.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torzewski M, Ochsenhirt V, Kleschyov AL, Oelze M, Daiber A, Li H, Rossmann H, Tsimikas S, Reifenberg K, Cheng F, Lehr HA, Blankenberg S, Forstermann U, Munzel T, Lackner KJ. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:850–857. doi: 10.1161/01.ATV.0000258809.47285.07. [DOI] [PubMed] [Google Scholar]
- 38.Alp NJ, Mussa S, Khoo J, Cai S, Guzik T, Jefferson A, Goh N, Rockett KA, Channon KM. Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression. J Clin Invest. 2003;112:725–735. doi: 10.1172/JCI17786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng W, Liu G, Wei YS, Cai H, Liu DP, Liang CC. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res. 2008;80:191–199. doi: 10.1093/cvr/cvn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B, Scherrer U. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104:342–345. doi: 10.1161/01.cir.104.3.342. [DOI] [PubMed] [Google Scholar]
- 42.Cook S, Hugli O, Egli M, Vollenweider P, Burcelin R, Nicod P, Thorens B, Scherrer U. Clustering of cardiovascular risk factors mimicking the human metabolic syndrome X in eNOS null mice. Swiss Med Wkly. 2003;133:360–363. doi: 10.4414/smw.2003.10239. [DOI] [PubMed] [Google Scholar]
- 43.Lefer DJ, Jones SP, Girod WG, Baines A, Grisham MB, Cockrell AS, Huang PL, Scalia R. Leukocyte-endothelial cell interactions in nitric oxide synthase-deficient mice. Am J Physiol. 1999;276:H1943–H1950. doi: 10.1152/ajpheart.1999.276.6.H1943. [DOI] [PubMed] [Google Scholar]
- 44.Huang PL. Lessons learned from nitric oxide synthase knockout animals. Semin Perinatol. 2000;24:87–90. doi: 10.1016/s0146-0005(00)80064-6. [DOI] [PubMed] [Google Scholar]
- 45.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 46.Atochin DN, Wang A, Liu VW, Critchlow JD, Dantas AP, Looft-Wilson R, Murata T, Salomone S, Shin HK, Ayata C, Moskowitz MA, Michel T, Sessa WC, Huang PL. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. J Clin Invest. 2007;117:1961–1967. doi: 10.1172/JCI29877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuhlencordt PJ, Rosel E, Gerszten RE, Morales-Ruiz M, Dombkowski D, Atkinson WJ, Han F, Preffer F, Rosenzweig A, Sessa WC, Gimbrone MA, Jr, Ertl G, Huang PL. Role of endothelial nitric oxide synthase in endothelial activation: insights from eNOS knockout endothelial cells. Am J Physiol Cell Physiol. 2004;286:C1195–C1202. doi: 10.1152/ajpcell.00546.2002. [DOI] [PubMed] [Google Scholar]
- 48.Atochin DN, Clark J, Demchenko IT, Moskowitz MA, Huang PL. Rapid cerebral ischemic preconditioning in mice deficient in endothelial and neuronal nitric oxide synthases. Stroke. 2003;34:1299–1303. doi: 10.1161/01.STR.0000066870.70976.57. [DOI] [PubMed] [Google Scholar]
- 49.Nisoli E, Clementi E, Carruba MO, Moncada S. Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circ Res. 2007;100:795–806. doi: 10.1161/01.RES.0000259591.97107.6c. [DOI] [PubMed] [Google Scholar]
- 50.Nisoli E, Clementi E, Tonello C, Sciorati C, Briscini L, Carruba MO. Effects of nitric oxide on proliferation and differentiation of rat brown adipocytes in primary cultures. Br J Pharmacol. 1998;125:888–894. doi: 10.1038/sj.bjp.0702131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jonk AM, Houben AJ, de Jongh RT, Serne EH, Schaper NC, Stehouwer CD. Microvascular dysfunction in obesity: a potential mechanism in the pathogenesis of obesity-associated insulin resistance and hypertension. Physiol (Bethesda) 2007;22:252–260. doi: 10.1152/physiol.00012.2007. [DOI] [PubMed] [Google Scholar]
- 52.Serne EH, de Jongh RT, Eringa EC, IJ RG, Stehouwer CD. Microvascular dysfunction: a potential pathophysiological role in the metabolic syndrome. Hypertension. 2007;50:204–211. doi: 10.1161/HYPERTENSIONAHA.107.089680. [DOI] [PubMed] [Google Scholar]
- 53.Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- 54.Morishita T, Tsutsui M, Shimokawa H, Sabanai K, Tasaki H, Suda O, Nakata S, Tanimoto A, Wang KY, Ueta Y, Sasaguri Y, Nakashima Y, Yanagihara N. Nephrogenic diabetes insipidus in mice lacking all nitric oxide synthase isoforms. Proc Natl Acad Sci USA. 2005;102:10616–10621. doi: 10.1073/pnas.0502236102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakata S, Tsutsui M, Shimokawa H, Suda O, Morishita T, Shibata K, Yatera Y, Sabanai K, Tanimoto A, Nagasaki M, Tasaki H, Sasaguri Y, Nakashima Y, Otsuji Y, Yanagihara N. Spontaneous myocardial infarction in mice lacking all nitric oxide synthase isoforms. Circulation. 2008;117:2211–2223. doi: 10.1161/CIRCULATIONAHA.107.742692. [DOI] [PubMed] [Google Scholar]
- 56.Matsuda T, Bates JN, Lewis SJ, Abboud FM, Chapleau MW. Modulation of baroreceptor activity by nitric oxide and S-nitrosocysteine. Circ Res. 1995;76:426–433. doi: 10.1161/01.res.76.3.426. [DOI] [PubMed] [Google Scholar]
- 57.Scrogin KE, Veelken R, Luft FC. Sympathetic baroreceptor responses after chronic NG-nitro-L-arginine methyl ester treatment in conscious rats. Hypertension. 1994;23:982–986. doi: 10.1161/01.hyp.23.6.982. [DOI] [PubMed] [Google Scholar]
- 58.Liu VW, Huang PL. Cardiovascular roles of nitric oxide: a review of insights from nitric oxide synthase gene disrupted mice. Cardiovasc Res. 2008;77:19–29. doi: 10.1016/j.cardiores.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Radomski MW, Palmer RM, Moncada S. Modulation of platelet aggregation by an L-arginine-nitric oxide pathway. Trends Pharmacol Sci. 1991;12:87–88. doi: 10.1016/0165-6147(91)90510-y. [DOI] [PubMed] [Google Scholar]
- 60.Moroi M, Zhang L, Yasuda T, Virmani R, Gold HK, Fishman MC, Huang PL. Interaction of genetic deficiency of endothelial nitric oxide, gender, and pregnancy in vascular response to injury in mice. J Clin Invest. 1998;101:1225–1232. doi: 10.1172/JCI1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mooradian DL, Hutsell TC, Keefer LK. Nitric oxide (NO) donor molecules: effect of NO release rate on vascular smooth muscle cell proliferation in vitro. J Cardiovasc Pharmacol. 1995;25:674–678. [PubMed] [Google Scholar]
- 62.Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 63.Steudel W, Scherrer-Crosbie M, Bloch KD, Weimann J, Huang PL, Jones RC, Picard MH, Zapol WM. Sustained pulmonary hypertension and right ventricular hypertrophy after chronic hypoxia in mice with congenital deficiency of nitric oxide synthase 3. J Clin Invest. 1998;101:2468–2477. doi: 10.1172/JCI2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O'Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 65.Gyurko R, Kuhlencordt P, Fishman MC, Huang PL. Modulation of mouse cardiac function in vivo by eNOS and ANP. Am J Physiol Heart Circ Physiol. 2000;278:H971–H981. doi: 10.1152/ajpheart.2000.278.3.H971. [DOI] [PubMed] [Google Scholar]
- 66.Scherrer-Crosbie M, Ullrich R, Bloch KD, Nakajima H, Nasseri B, Aretz HT, Lindsey ML, Vancon AC, Huang PL, Lee RT, Zapol WM, Picard MH. Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation. 2001;104:1286–1291. doi: 10.1161/hc3601.094298. [DOI] [PubMed] [Google Scholar]
- 67.Giordano A, Tonello C, Bulbarelli A, Cozzi V, Cinti S, Carruba MO, Nisoli E. Evidence for a functional nitric oxide synthase system in brown adipocyte nucleus. FEBS Lett. 2002;514:135–140. doi: 10.1016/s0014-5793(02)02245-7. [DOI] [PubMed] [Google Scholar]
- 68.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 69.Saha SK, Kuroshima A. Nitric oxide and thermogenic function of brown adipose tissue in rats. Jpn J Physiol. 2000;50:337–342. doi: 10.2170/jjphysiol.50.337. [DOI] [PubMed] [Google Scholar]
- 70.Kikuchi-Utsumi K, Gao B, Ohinata H, Hashimoto M, Yamamoto N, Kuroshima A. Enhanced gene expression of endothelial nitric oxide synthase in brown adipose tissue during cold exposure. Am J Physiol Regul Integr Comp Physiol. 2002;282:R623–R626. doi: 10.1152/ajpregu.00310.2001. [DOI] [PubMed] [Google Scholar]
- 71.Lo EH, Hara H, Rogowska J, Trocha M, Pierce AR, Huang PL, Fishman MC, Wolf GL, Moskowitz MA. Temporal correlation mapping analysis of the hemodynamic penumbra in mutant mice deficient in endothelial nitric oxide synthase gene expression. Stroke. 1996;27:1381–1385. doi: 10.1161/01.str.27.8.1381. [DOI] [PubMed] [Google Scholar]
- 72.Thom SR, Fisher D, Zhang J, Bhopale VM, Ohnishi ST, Kotake Y, Ohnishi T, Buerk DG. Stimulation of perivascular nitric oxide synthesis by oxygen. Am J Physiol Heart Circ Physiol. 2003;284:H1230–H1239. doi: 10.1152/ajpheart.01043.2002. [DOI] [PubMed] [Google Scholar]
- 73.Cabigas BP, Su J, Hutchins W, Shi Y, Schaefer RB, Recinos RF, Nilakantan V, Kindwall E, Niezgoda JA, Baker JE. Hyperoxic and hyperbaric-induced cardioprotection: role of nitric oxide synthase 3. Cardiovasc Res. 2006;72:143–151. doi: 10.1016/j.cardiores.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 74.Thom SR, Bhopale V, Fisher D, Manevich Y, Huang PL, Buerk DG. Stimulation of nitric oxide synthase in cerebral cortex due to elevated partial pressures of oxygen: an oxidative stress response. J Neurobiol. 2002;51:85–100. doi: 10.1002/neu.10044. [DOI] [PubMed] [Google Scholar]
- 75.Demchenko IT, Atochin DN, Boso AE, Astern J, Huang PL, Piantadosi CA. Oxygen seizure latency and peroxynitrite formation in mice lacking neuronal or endothelial nitric oxide synthases. Neurosci Lett. 2003;344:53–56. doi: 10.1016/s0304-3940(03)00432-4. [DOI] [PubMed] [Google Scholar]
- 76.Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, et al. `Ischemic tolerance' phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- 77.Perez-Pinzon MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J Cereb Blood Flow Metab. 1997;17:175–182. doi: 10.1097/00004647-199702000-00007. [DOI] [PubMed] [Google Scholar]
- 78.Gallis B, Corthals GL, Goodlett DR, Ueba H, Kim F, Presnell SR, Figeys D, Harrison DG, Berk BC, Aebersold R, Corson MA. Identification of flow-dependent endothelial nitric-oxide synthase phosphorylation sites by mass spectrometry and regulation of phosphorylation and nitric oxide production by the phosphatidylinositol 3-kinase inhibitor LY294002. J Biol Chem. 1999;274:30101–30108. doi: 10.1074/jbc.274.42.30101. [DOI] [PubMed] [Google Scholar]
- 79.Michell BJ, Griffiths JE, Mitchelhill KI, Rodriguez-Crespo I, Tiganis T, Bozinovski S, de Montellano PR, Kemp BE, Pearson RB. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr Biol. 1999;9:845–848. doi: 10.1016/s0960-9822(99)80371-6. [DOI] [PubMed] [Google Scholar]
- 80.Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- 81.Butt E, Bernhardt M, Smolenski A, Kotsonis P, Frohlich LG, Sickmann A, Meyer HE, Lohmann SM, Schmidt HH. Endothelial nitric-oxide synthase (type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J Biol Chem. 2000;275:5179–5187. doi: 10.1074/jbc.275.7.5179. [DOI] [PubMed] [Google Scholar]
- 82.Cai H, Liu D, Garcia JG. CaM Kinase II-dependent pathophysiological signalling in endothelial cells. Cardiovasc Res. 2008;77:30–34. doi: 10.1093/cvr/cvm010. [DOI] [PubMed] [Google Scholar]
- 83.McCabe TJ, Fulton D, Roman LJ, Sessa WC. Enhanced electron flux and reduced calmodulin dissociation may explain “calcium-independent” eNOS activation by phosphorylation. J Biol Chem. 2000;275:6123–6128. doi: 10.1074/jbc.275.9.6123. [DOI] [PubMed] [Google Scholar]
- 84.Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- 85.Scotland RS, Morales-Ruiz M, Chen Y, Yu J, Rudic RD, Fulton D, Gratton JP, Sessa WC. Functional reconstitution of endothelial nitric oxide synthase reveals the importance of serine 1179 in endothelium-dependent vasomotion. Circ Res. 2002;90:904–910. doi: 10.1161/01.res.0000016506.04193.96. [DOI] [PubMed] [Google Scholar]
- 86.Ayata C, Dunn AK, Gursoy OY, Huang Z, Boas DA, Moskowitz MA. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab. 2004;24:744–755. doi: 10.1097/01.WCB.0000122745.72175.D5. [DOI] [PubMed] [Google Scholar]
- 87.Demchenko IT, Atochin DN, Gutsaeva DR, Godfrey RR, Huang PL, Piantadosi CA, Allen BW. Contributions of nitric oxide synthase isoforms to pulmonary oxygen toxicity, local vs. mediated effects. Am J Physiol Lung Cell Mol Physiol. 2008;294:L984–L990. doi: 10.1152/ajplung.00420.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schleicher M, Yu J, Murata T, Derakhshan B, Atochin D, Qian L, Kashiwagi S, Di Lorenzo A, Harrison KD, Huang PL, Sessa WC. The Akt1-eNOS axis illustrates the specificity of kinase-substrate relationships in vivo. Sci Signal. 2009;2:ra41. doi: 10.1126/scisignal.2000343. [DOI] [PMC free article] [PubMed] [Google Scholar]