

Abstract
Lipid droplets are complex and dynamic intracellular organelles that play an essential role in cholesterol and lipid homeostasis and profoundly affect cellular structure and function. Variations in lipid droplet composition exist between different cell types, but whether there are differences in the mechanisms of lipid droplet accumulation remains to be elucidated. Here we report that the gene P311, previously identified to have a function in neuronal regeneration and a potential role in distal lung generation, regulates lipid droplet accumulation. P311 up-regulates several classes of genes associated with lipid synthesis, significantly increases intracellular cholesterol and triglyceride levels, and increases intracellular lipid droplets. Interestingly, P311 expression is not necessary for lipogenesis in the well-established NIH3T3-L1 model of adipogenic differentiation. Instead, we demonstrate a novel role for P311 in an alternative pathway of lipid droplet accumulation induced by the regeneration-inducing molecule retinoic acid.
Supplementary key words: P311, lipid droplet accumulation, retinoic acid
INTRODUCTION
Lipid droplets, also known as lipid bodies, are intracellular inclusions that generally consist of a core of neutral lipids, predominantly triglycerides and steryl esters, surrounded by a monolayer of phospholipids and proteins (Martin and Parton, 2005). Although typically associated with the phenotype and functions of adipocytes and steroidogenic cells, lipid droplets are also found in many other cell types and typically form in response to elevated fatty-acid levels (Pol et al., 2004). The size, composition, and function of these lipid droplets may vary, however, according to cell type or metabolic state (Martin and Parton, 2006). Initially thought of as inert intracellular lipid stores, these cytosolic lipid droplets have recently been shown to be dynamic, motile organelles that participate in a variety of cellular functions. Lipid droplets regulate the storage and turnover of neutral lipids and cholesterol esters that can be used for energy metabolism, membrane synthesis, steroid synthesis, and synthesis of lipid mediators. Defects in intracellular lipid homeostasis are found in many diseases such as obesity, insulin resistance, diabetes, atherosclerosis, and lipid-storage diseases. Although functionally-linked to vital cellular processes, basic information regarding the biology of the lipid droplet is still poorly understood, including potential differences in the factors which stimulate their formation in alternative cellular contexts.
P311 is a gene originally discovered as an abundant transcript at sites of active embryonic and postnatal neurogenesis (Studler et al., 1993) and has been subsequently shown to induce neurite extension in cultured neurons and nerve regeneration in vivo (Fujitani et al., 2004). It encodes an 8-Kda, 68-aa protein with no structural motifs suggestive of its molecular functions. It has also been identified in other cell types, such as glioblastoma cells (Mariani et al., 2001), chondrocytes (Sironen et al., 2000), and has been shown to induce myofibroblast differentiation in vitro (Pan et al., 2002). We identified P311 as a gene substantially up-regulated during lung alveolarization, a stage of distal lung development that generates the respiratory gas exchange surface (Zhao et al., 2006). This stage of lung development is associated with an abundance of lipid-containing fibroblasts (McGowan and Torday, 1997). Interestingly, the known expression pattern and functions of P311 in the neuronal system and the distal lung parallel that of retinoic acid – a powerful hormone well known for its ability to stimulate regeneration in the nervous system (Maden and Hind, 2003) as well as of the distal lung structure (Maden and Hind, 2003; Maden and Hind, 2004). In this study we sought to further investigate the cellular and molecular functions of P311. We report here that P311 up-regulates classes of genes associated with lipid-uptake and synthesis, increases cellular levels of cholesterol and triglycerides, and increases intracellular lipid droplets. Interestingly, we find that P311 specifically mediates lipid droplet accumulation induced by retinoic acid in lung fibroblasts. Our data thus demonstrate a novel function for P311 in lipid accumulation and implicate its functions in retinoic acid regulation of lipid droplet biogenesis.
RESULTS
Genes related to lipid synthesis and metabolism are up-regulated by P311
To explore the potential cellular functions of P311, we stably expressed P311 in C3H10T1/2 mouse embryonic fibroblasts under the tetracycline-regulated conditional expression system. Semi-quantitative RT-PCR confirmed induction of exogenous P311 transcript levels in P311 stable cells upon doxycycline administration (Fig. 1A). In addition, a low basal level of P311 transcripts without exposure to doxycycline was observed (Fig. 1A). These basal levels were sufficient to cause a morphological change in P311 stable cells seeded at a higher cell density (Fig. 1B–E). To determine gene expression changes that might be responsible for this alteration in cell morphology induced by P311, we examined the global transcript expression profile of C3H10 P311 stable cells seeded at the higher cell density compared to C3H10 control stable cells. Microarray analysis revealed 579 genes statistically and significantly up-regulated at least two-fold in the P311 stable cells. To determine the functional significance of these regulated genes, we used Ensemble attribute profile clustering data (Semeiks et al., 2006) to functionally classify these genes. The analysis revealed that genes associated with the synthesis and metabolism of lipid and cholesterol are the primary classes of genes significantly up-regulated by P311. Specifically, these classes of genes encode proteins that function in multiple aspects of lipid physiology, including the transport of fatty acids, the synthesis and metabolism of sterols and steroid molecules, and the accumulation of cholesterol esters (Table 1).
Figure 1. P311 expression induces morphological changes in cells seeded at high cell density.

A: C3H10 cells containing the Tet-On transactivator and a tet-responsive empty vector (control) or myc-tagged P311 (mP311-myc) demonstrate low levels of background P311 transcripts in P311 stable cells without doxycycline and induction of transcripts with doxycycline by semiquantitative RT-PCR. Beta-actin transcripts were used as a loading standard. B&C: Control cells seeded at high cell density (106 cells/100mm) have the typical spindle shape of C3H10T1/2 cells in culture. D&E: P311 stable cells seeded at the same high cell density show a different morphology. Bars in B, D = 40µm. Bars in C, E = 10µm.
Table I.
Lipid metabolism genes upregulated (at least two-fold) in P311-stable C3H10 cells
| Function | Symbol | Gene name | Fold increase |
|---|---|---|---|
|
Sterol /Steroid Biosynthesis |
CYP51A1 | cytochrome P450, family 51, subfamily A, polypeptide 1 |
4.20 |
| Dhcr24 | 24-dehydrocholesterol reductase | 2.31 | |
| EBP | emopamil binding protein | 2.43 | |
| FDPS | farnesyl diphosphate synthase | 3.43 | |
| Hmgcs1 | 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 1 | 3.53 | |
| HSD17B7 | hydroxysteroid (17-beta) dehydrogenase 7 | 2.75 | |
| IDI1 | isopentenyl-diphosphate delta isomerase | 11.71 | |
| LSS | lanosterol synthase | 2.77 | |
| NSDHL | NAD(P) dependent steroid dehydrogenase-like | 3.10 | |
| Sc4mol | sterol-C4-methyl oxidase-like | 4.32 | |
| Sc5d | sterol-C5-desaturase | 2.95 | |
| Stard4 | StAR-related lipid transfer (START) domain contain. 4 | 4.72 | |
|
Synthesis of triglycerols |
DGAT2 | Diacylglycerol acyltransferase 2 | 2.77 |
|
Transport and Internalization of lipid |
ABCD3 | ATP-binding cassette sub-family D (ALD) member 3 | 2.20 |
| CROT | carnitine O-octanoyltransferase | 2.00 | |
| FAAH | fatty acid amide hydrolase | 3.07 | |
|
Accumulation of cholesterol/ cholesterol ester |
ACAT2 | acetyl-Coenzyme A acetyltransferase 2 | 2.53 |
| LDLR | low density lipoprotein receptor | 2.57 | |
|
Prevention of oxidation |
CAT | catalase | 3.97 |
| GSTA2 | glutathione S-transferase A2 | 7.41 | |
| MT1A | metallothionein 1A | 2.93 | |
| MT2A | metallothionein 2A | 2.07 |
P311 increases lipid accumulation
Since the microarray data suggest a role for P311 in lipid metabolism, we determined whether lipid synthesis and accumulation were altered in P311 stable cells. We added radio-labeled fatty acids to control and P311 stable cells and assayed for the amount incorporated into cellular triglycerides. We found that there was an approximately three-fold higher level of labeled triglycerides in P311 stable cells relative to control cells (Fig. 2A). Total cellular cholesterol was also elevated to approximately three-fold in the P311 stable cells following fatty acid administration (Fig. 2B). In the process of performing these biochemical assays we observed the visible accumulation of lipid droplets following fatty acid supplementation in P311 stable cells. To better visualize these lipid droplets we stained C3H10 P311 stable and control cells following addition of fatty acids with Oil-Red-O. In contrast to control cells, P311 stable cells exhibit strong Oil-Red-O staining (Fig. 2C&D), indicating a significantly increased ability to synthesize lipid droplets. These lipid droplets exhibited some perinuclear clustering but also localized throughout the cytoplasm. Interestingly, these droplets did not associate with adipophilin and TIP47, markers typically associated with lipid droplets in adipocytes (Figure 2E–L) and we did not observe changes in the transcript expression of these markers (data not shown). These data indicate that the cellular expression of P311 leads to an increased ability to synthesize lipids and accumulate lipid droplets.
Figure 2. P311 increases lipid accumulation.

A&B: C3H10 control and P311 stable cells were pulsed with fatty acid complexes, lipids extracted and measured for relative levels of triglycerides (A) and cholesterol (B). There is a substantial (approximately three-fold) increase in cholesterol and triglyceride levels in P311 stable cells. Mean and standard deviation from triplicate samples are shown. The differences between P311 and control samples are statistically significant (P<0.05). C&D: Staining with the lipophilic dye Oil-Red-O of C3H10 control (C) and P311 stable (D) cells pulsed with fatty acid complexes for 24 hours demonstrates the presence of lipid droplets in P311 stable cells (D). E–L: Phase contrast (E, I, G, K) and epifluorescence (F, J, H, L) images of C3H10 P311 stable cells (E–H) and NIH3T3-L1 adipocytes (I–L) immunostained with either adipophilin (E, F, I, J) or TIP47 (G, K, H, L) antibody demonstrating staining of lipid droplets in NIH3T3-L1 adipocytes with both adipophilin and TIP47 antibody, but no staining of lipid droplets in C3H10 P311 with either antibody. Bar in C&D = 5µm.
P311 gene silencing does not affect lipid accumulation in NIH3T3-L1 cells
Since cellular expression of P311 induces lipid synthesis and accumulation we asked whether P311 might have a function in adipogenesis. We assayed for changes in P311 gene expression during the adipogenic induction of NIH3T3-L1 cells, an in vitro model of adipocyte differentiation. We observed a substantial initial decrease followed by a gradual increase in P311 transcript expression (Fig. 3A). To determine whether P311 has a role in this process, lentiviruses expressing short hairpin RNAs (shRNAs) targeting the P311 transcript were used to silence P311 transcript expression (Ventura et al., 2004). Quantification of P311 expression in NIH3T3-L1 cells infected with control and P311 shRNA-expressing lentiviruses by real-time RT-PCR demonstrated a substantial decrease in P311 transcript expression by the P311 shRNA (Fig. 3B). The infected cells were subsequently induced toward adipogenic differentiation. Accumulation of large lipid droplets in nearly all control and P311 shRNA lentiviral-infected cells was observed (Fig. 3C–F), and quantification by Nile Red staining followed by flow cytometry demonstrated no significant decrease in intracellular lipids in P311 shRNA lentivirus-infected cells relative to control lentivirus-infected cells (Fig. 3G). These results indicate that P311 gene silencing has no significant effects on adipogenic differentiation and/or lipogenesis in this system. Thus typical levels of P311 expression are not required for lipid accumulation in this in vitro model of adipogenesis.
Figure 3. P311 gene silencing does not affect lipid accumulation in NIH3T3-L1 cells.

A: P311 RNA expression levels during adipogenic differentiation of NIH3T3-L1 cells as determined by quantitative real-time RT-PCR. There is nearly a complete decrease in P311 transcripts at the beginning of induction (Day 2) and a gradual increase to base-line levels at the completion of differentiation (Day 8). Mean and standard deviation from triplicate samples are shown. Asterisks indicate statistically significant (P < 0.01) pair wise comparison between Day 0 and Day 2 samples (*) and between Day 0 and Day 4 samples (**). There is no significant difference between Day 0 and Day 8 samples. B: NIH3T3-L1 preadipocytes infected with control lentivirus or lentivirus expressing a short hairpin RNA (shRNA) targeting P311 were examined for relative P311 RNA expression levels by quantitative real-time RT-PCR. There is an approximately 90% decrease in P311 transcripts in P311 shRNA lentivirus-infected cells relative to control lentivirus-infected cells. Mean and standard deviation from triplicate samples are shown. The differences between P311 shRNA and control samples are statistically significant (P<0.05). C–F: Phase contrast (C&E) and epifluorescence (D&F) images of control (C&D) and P311 shRNA lentivirus-infected NIH3T3-L1 (E&F) cells following induction of adipogenic differentiation demonstrate that both control and P311 shRNA lentivirus-infected cells (marked by GFP) accumulate lipid droplets (arrows) to similar extent. Bar = 10µm. G: The relative amount of lipids in control versus P311 shRNA lentivirus-infected NIH3T3-L1 cells following induction of adipogenic differentiation, assessed by Nile Red staining followed by flow cytometry, indicates no significant change in lipid accumulation in the P311 shRNA lentivirus-infected population. Control and P311 shRNA lentivirus-infected NIH3T3-L1 cells were sorted for GFP and Nile Red fluorescence. The mean fluorescence intensities of the Nile Red channel in the GFP+ populations were determined. Values are normalized to the average of the mean fluorescence intensities of the control GFP+ samples. Mean and standard deviation from triplicate samples are shown.
P311 is a retinoic acid-responsive gene in lung fibroblasts
Since P311 did not have a significant role in a conventional model of adipogenesis, yet causes increased cellular lipid uptake and accumulation when expressed in C3H10 cells, we sought to determine the identity and significance of signaling pathways that regulate P311 and its functions. We examined whether fatty acids (FA) regulate P311 transcript expression, as FA is known to stimulate lipogenesis through the peroxisome proliferators activated receptor (PPAR) pathway (Green, 1995). In addition, we speculated that retinoic acid (RA) might also modulate P311 transcript expression, as there is evidence suggesting P311 might be associated with the RA signaling pathway: 1) P311 expression is high in brain regions with high RA signaling (Studler et al., 1993); 2) P311 promotes neuronal regeneration - an effect also attributed to RA (Maden and Hind, 2003); and 3) RA signaling is highly active during lung alveolarization (Chytil, 1996), a period when P311 expression is highly increased (Zhao et al., 2006). We therefore isolated lung fibroblasts from one-week-old mice, as both lipid accumulation and retinoid signaling are very active in this cell type during the early postnatal period (Chen et al., 1998; Kaplan et al., 1985; Liu et al., 1993; Vaccaro and Brody, 1978) and examined the influence of FA and RA treatment on P311 expression in these cells. Treatment of lung fibroblasts from one week-old mice with FA alone did not induce P311 transcript expression, as assayed by real-time quantitative RT-PCR (Fig. 4A). RA, however, induced P311 transcript expression approximately 2.7 fold after one week and the addition of fatty acids did not further affect the degree of P311 induction by RA (Fig. 4A). The induction of P311 expression by RA occurs by 48 hours and is dose-dependent (Fig. 4B). These data demonstrate that P311 expression is regulated by retinoic acid.
Figure 4. P311 is a retinoic acid-responsive gene in lung fibroblasts.

A: Quantitative RT-PCR showing induction of P311 mRNA in lung fibroblasts isolated from early postnatal mice following treatment with retinoic acid (RA, 1µM), but not with fatty acids (FA, 0.1µM) for one week. Mean and standard deviation from triplicate samples are shown. Asterisks indicate statistically significant (P<0.01) pair wise comparison between 1uM RA-treated and untreated samples (*) and between 1uM RA+FA-treated and untreated samples (**). B: Quantitative RT-PCR showing that treatment with a higher concentration of FA (FA, 1µM) for 48 hours also did not induce P311 mRNA in lung fibroblasts isolated from early postnatal mice, and that the induction of P311 mRNA by RA is dose-dependent. Note that treatment with 1µM RA at 48 hours induced P311 mRNA to a similar extent as treatment for 1 week as shown in panel A. Mean and standard deviation from triplicate samples are shown. Asterisks indicate statistically significant (P<0.01) pair wise comparison between 200nM RA+FA-treated and 12.5nM RA+FA-treated samples (*) and between 1uM RA+FA-treated and 12.5nM RA+FA-treated samples (**).
Retinoic acid increases lipid accumulation in lung fibroblasts
The induction of P311 expression by RA in lung fibroblasts and the ability of P311 to increase lipid droplet accumulation in C3H10 cells led us to hypothesize that RA might stimulate lipid droplet accumulation in lung fibroblasts. This would be consistent with the presence of active retinoid signaling and intracellular lipid accumulation in the lung mesenchyme during alveolarization. We therefore determined whether RA induces intracellular lipid accumulation in lung fibroblasts isolated from one week-old mice. We found that a large portion of freshly isolated lung fibroblasts at this stage contain lipid droplets (data not shown), consistent with previous observations that there is a high abundance of lipid containing fibroblasts in the lung during alveolarization. However, maintenance of these lung fibroblasts for at least one week in culture without fatty acid supplementation depleted most cells of their lipids (data not shown). Re-supplementation of fatty acids to these cells resulted in accumulation of intracellular lipids as assayed by Nile Red staining followed by flow cytometry (Fig. 5), suggesting increased levels of fatty acid metabolism in lung fibroblasts at the alveolarization stage of lung development (Abumrad et al., 1981; Abumrad et al., 1984; Dutta-Roy, 2000). Treatment of the cells with RA caused a further accumulation of intracellular lipids (Fig. 5), demonstrating that retinoic acid stimulates lipid accumulation in these lung fibroblasts.
Figure 5. Retinoic acid increases lipid accumulation in lung fibroblasts.

A: Lung fibroblasts cultured for one week to deplete endogenous lipid droplets were exposed to fatty acid complexes (0.1µM) and retinoic acid (1µM) for one week, stained with Nile Red and analyzed by flow cytometry. The shift in mean fluorescence intensity with FA and RA treatment indicates increased Nile Red staining and hence increased cellular lipids. B: Graph of mean fluorescence intensities normalized to the average mean fluorescence intensity of untreated samples. The mean and standard deviation from triplicate samples are shown. Asterisks indicate statistically significant (P < 0.01) pair wise comparison between FA treated and untreated samples (*) and between RA treated and untreated samples (**).
P311 gene silencing affects retinoic acid-mediated lipid accumulation
We next determined whether P311 expression was required for RA-mediated lipid accumulation in lung fibroblasts. We silenced P311 expression by the lentiviral-mediated shRNA approach. Quantification of P311 expression by real-time RT-PCR in lung fibroblasts infected with either control or P311 shRNA-expressing lentiviruses demonstrated a significant knockdown of P311 transcripts in the P311 shRNA lentivirus-infected cells (Fig. 6A). Control or P311 shRNA lentivirus-infected cells were then treated with RA in the presence of fatty acids, and intracellular lipid accumulation assayed by Nile Red staining. A higher fatty acid concentration was used in these experiments as it decreases the time to observe lipid droplet accumulation induced by RA. P311 shRNA-lentivirus-infected lung fibroblasts exposed to RA and fatty acids accumulated substantially less lipid droplets compared to control lentivirus-infected cells (Fig. 6B). Quantification by flow cytometry indicated an approximately 85% decrease in intracellular lipids in P311 shRNA lentivirus-infected cells relative to control lentivirus-infected cells (Fig. 6C). The incomplete inhibition of lipid accumulation in cells infected with P311 shRNA lentivirus might be due to the incomplete knockdown of P311 expression. Overall, these data demonstrate that the knockdown of P311 expression has a significant effect on the induction of lipid accumulation by RA in lung fibroblasts in vitro and suggest that P311 mediates the effects of RA in this process.
Figure 6. P311 gene silencing affects retinoic acid-mediated lipid accumulation in lung fibroblasts.
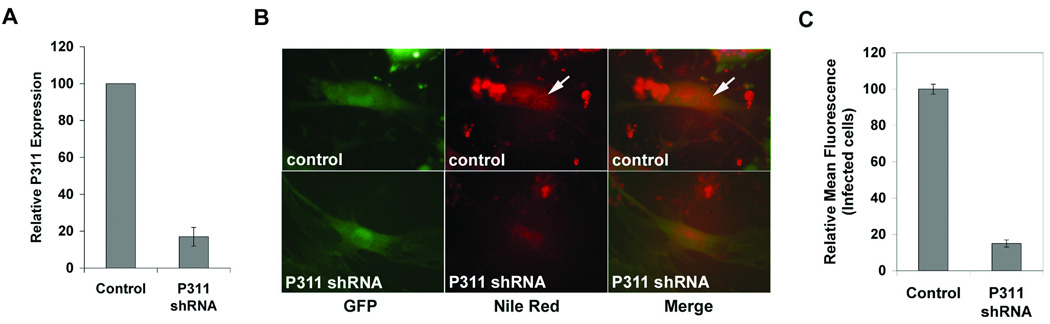
A: Lung fibroblasts cultured for one week and infected with control lentivirus or P311 shRNA lentivirus were examined for relative P311 RNA expression levels by quantitative real-time RT-PCR. There is an approximately 80% decrease in P311 transcripts in P311 shRNA lentivirus-infected cells relative to control lentivirus-infected cells. Mean and standard deviation from triplicate samples are shown. The differences between P311 shRNA and control samples are statistically significant (P<0.01). B: Epifluorescence images of control and P311 shRNA lentivirus-infected cells exposed to fatty acid complexes (1µM) and RA (1µM) for 48 hours, showing that P311 shRNA lentivirus-infected cells (marked by co-expression of GFP) accumulated less lipid droplets (stained with Nile Red) relative to control lentivirus-infected cells (arrows). C: The relative amount of lipids in control versus P311 shRNA lentivirus-infected cells, assessed by Nile Red staining followed by flow cytometry, indicates an approximately 85% decrease in lipid accumulation in the P311 shRNA lentivirus-infected population. FA (1µM) and RA (1µM) treated control and P311 shRNA lentivirus-infected cells were sorted for GFP and Nile Red fluorescence after 48 hours. The mean fluorescence intensities of the Nile Red channel in the GFP+ populations were determined. Values are normalized to the average of the mean fluorescence intensities of the control GFP+ samples. Mean and standard deviation from triplicate samples are shown. The differences between P311 shRNA and control samples are statistically significant (P<0.01).
DISCUSSION
P311 has been previously identified to be differentially regulated in many different cellular contexts and implicated in diverse cellular functions, including cell proliferation, migration, and differentiation. However, the molecular function of P311 is still unknown. In the present study we identify a novel function for P311, demonstrating its ability to up-regulate genes associated with lipid synthesis and metabolism, increase cellular levels of triglycerides and cholesterol, and induce lipid droplet accumulation. Since lipid droplets regulate many cellular processes, this function of P311 may contribute to the pleiotropic effects of P311 expression. Interestingly, P311 is not required for lipid accumulation during adipogenic differentiation, but instead has an important role in the induction of lipid droplet accumulation mediated by retinoic acid. Our data thus identify the presence of distinct pathways in the accumulation of lipid droplets and places P311 within the context of cellular functions mediated by retinoic acid.
Our laboratory previously identified P311 as a gene that potentially regulates alveolarization, as the expression of P311 is markedly increased in the lung during alveolar development and is decreased in mice impaired in distal lung development (Zhao et al., 2006). In this context, it is significant that P311 induces cellular lipid droplet accumulation and that it mediates this effect of retinoic acid in lung fibroblasts, since during alveolarization there is both a high abundance of lipid containing interstitial cells (termed lipofibroblasts) (McGowan, 2002; McGowan and Torday, 1997) as well as high retinoic acid signaling in the lungs (Chen et al., 1998; Kaplan et al., 1985; Liu et al., 1993; Vaccaro and Brody, 1978). The function of P311 in alveolar development may thus be to induce or mediate retinoic acid-induction of lipofibroblast formation. On the other hand, P311 has also been reported to direct the transition of fibroblasts to myofibroblasts (Pan et al., 2002), which are smooth-muscle alpha actin-containing cells located at sites requiring an active contractile function including the tip of the alveolar septa (Vaccaro and Brody, 1978). Our cellular expression of P311 under the tetracycline-conditional system does not seem to induce a myofibroblast phenotype, as we found no change in smooth-muscle alpha-actin expression in our P311 stable cells (Leung et al, unpublished observations). This difference in cellular response is possibly due to differences in P311 expression levels, as tetracycline-induced conditional expression is relatively weaker compared to the cytomegalovirus (CMV) promoter used in the previous study (Pan et al., 2002). In addition, our observation of lipid accumulation in P311-stable cells is under conditions of higher cell confluency, where there might be differences in cell state and signaling.
It is also intriguing to consider the potential relationship between the lipid-containing lipofibroblasts and the smooth-muscle alpha actin-containing myofibroblasts. These two cell types are located at close proximity during the development of lung alveoli; myofibroblasts at the tips of newly formed septa and lipofibroblasts situated in close proximity at the base of the septa (Vaccaro and Brody, 1978). Lipofibroblasts also share characteristics with the myofibroblasts, including the synthesis of extracellular matrix structural components such as collagen and elastin (McGowan and Torday, 1997). In the liver, hepatic stellate cells are known to exist in a quiescent lipid-containing state until their activation to myofibroblasts (Sato et al., 2003). Thus lipid-containing P311 stable cells might represent an intermediate cell-type in the pathway to myofibroblast development.
Retinoic acid is a potent hormone that is essential for the morphogenesis and function of many tissues. It has pleiotropic cellular effects, including the regulation of cell proliferation, differentiation, and survival. Previous data indicate that retinoic acid can also significantly affect lipid accumulation, depending on tissue and cell-type. Retinoic acid increases lipid accumulation in macrophages (Inazawa et al., 2003), Ob1771 preadipocytes (Safonova et al., 1994), and murine embryonic stem cells (Dani et al., 1997). On the other hand, retinoic acid inhibits lipid accumulation in NIH3T3-L1 preadipocytes (Schwarz et al., 1997). We found that retinoic acid significantly increases lipid accumulation in lung fibroblasts. In the lungs, retinoic acid is important for alveolar development, as defects in retinoic acid signaling are associated with defects in distal lung formation (McGowan et al., 2000; Snyder et al., 2005; Yang et al., 2003). Retinoic acid can also stimulate alveolar regeneration. Treatment of adult rodents with emphysema induced by dexamethasone or elastase with retinoic acid causes the formation of new alveoli (Hind and Maden, 2004; Massaro and Massaro, 1997), (McGowan, 2002). The cellular and molecular bases of retinoic acid functions during alveolarization are unknown. Our data implicate the regulation of intracellular lipid droplet accumulation as a potential function of retinoic acid signaling during lung development and regeneration, and demonstrate that P311 is an important component of this process. Both retinoic acid and P311 have also been shown to induce neurite outgrowths and nerve regeneration (Fujitani et al., 2004; Maden and Hind, 2003). It will be of significant interest for future studies to determine whether the regulation of cellular lipid stores is an aspect of retinoic acid and P311 functions in the neuronal system as well.
Lipid droplets are increasingly recognized as important intracellular organelles that regulate diverse cellular functions (Martin and Parton, 2005). Studies of lipid droplet biogenesis and function have traditionally focused on adipocytes and hepatocytes, which regulate lipid storage in response to whole body lipid homeostasis (Badman and Flier, 2007; Yan et al., 2007). However, almost all cell types can form lipid droplets, which are essential for the regulation of cellular lipid homeostasis and function. Lipid droplets are the major storage sites of retinyl esters, the precursors of vitamin A, in liver hepatic stellate cells (Wake and Sato, 1993). They are sites of arachidonic acid metabolism, the pathway of eicosanoids and prostanoids synthesis, in leucocytes (Wan et al., 2007) and macrophages (Dvorak et al., 1983), and sources of cholesterol used in steroid hormone synthesis in steroidogenic cells in the adrenal cortex (Toth et al., 1997), testes, and ovaries (Murphy, 2001). In Drosophila embryos lipid droplets are depots for the storage of proteins, including histones (Cermelli et al., 2006). Although the regulation of lipid droplet formation in adipocytes and hepatocytes has been extensively studied, its regulation in other cell types has not been well characterized. Our data identify differences between the regulation of lipid accumulation in adipocytes and that in fibroblasts. Whereas P311 promotes lipid droplet accumulation in fibroblasts, the silencing of P311 gene expression did not significantly delay or alter the formation of lipid bodies in the accumulation of lipids in NIH3T3-L1 preadipocytes, a well-established model for adipogenesis (Green and Meuth, 1974), suggesting that P311 is not required for this process. This importantly implicates mechanistic differences in the accumulation of lipid droplets that depend on the stimulus and cell type. Our study identifies a novel gene, P311, as part of this mechanistic difference, showing that it has a distinct role in lipid droplet accumulation mediated by retinoic acid.
MATERIALS AND METHODS
Cells and construction of P311 expression vector
The C3H10T1/2 fibroblast (American Type Culture Collection, Manassas, Virginia, USA) and PT67 retroviral packaging cell lines (BD Clontech, Mountain View, CA) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 4.5g/L glucose with 10% fetal bovine serum, 100U/ml penicillin G and 100µg/ml streptomycin. For routine subculture, control and stable cells were seeded at 2.5 × 105 cells/100mm. The pREV-TRE:mP311-myc plasmid was constructed as follows. The cDNA of the coding region of mouse P311 was conjugated with a myc tag through PCR amplification with modified primers (sense: 5’-TTT GGA TCC GCC GCC ACC ATG GTT TAC TAC CCA GAA-3’; antisense 5-’AAA ATC GAT TTA CAG ATC CTC TTC TGA GAT GAG TTT TTG TTC AAA AGG GTG GAG GTA-3’), digested with BamHI and ClaI, and cloned into the pREV-TRE retroviral tetracycline-responsive element vector (BD Clontech, Mountain View, CA). The recombinant protein produced by this construct is the full-length murine P311 with a myc tag at the C terminus.
Generation of stable cell lines with tetracycline regulatable P311 expression
The stable C3H10T1/2 cell lines containing the Tet-On transactivator and TRE:mP311-myc transgenes were generated by either sequential transfection or double infection. To obtain stable cell pools generated by transfection, the cells were transfected first with the pREV-Tet-On plasmid (BD Clontech, Mountain View, CA). Following selection, single stable Tet-On clones were transiently transfected with the TRE-Luciferase plasmid (BD Clontech) and induced with 1µg/mL doxycycline (Sigma, St. Louis, MO) to identify those clones with the highest induced:uninduced ratio of luciferase activity. The clone with highest ratio was subsequently transfected with either pREV-TRE or pREV-TRE:mP311-myc and selection performed under 200µg/mL G418 and 200µg/mL hygromycin (Invitrogen, Carlsbad, California, USA) for two weeks to select for C3H10T1/2 stable cells containing both pREV-Tet-On and either pREV-TRE (control cells) or pREV-TRE:mP311-myc (P311 stable cells). To obtain stable cell pools generated by double infection, Tet-On and TRE-mP311-myc plasmids were each transfected into the PT67 retroviral packaging cell line. Retroviral-containing media was harvested two days later. Polybrene (Sigma, St. Louis, MO) was added to the medium at a final concentration of 4µg/mL and filtered through a 0.45 µm cellulose acetate syringe filter (Fisher, Hampton, NH). Retroviral supernatants containing Tet-On and TRE-mP311-myc were used to co-infect C3H10T1/2 cells at 70% confluency. After 2 days cells were passaged at 1:10 dilution and treated with 200µg/mL G418 and 200µg/mL hygromycin for one month to select for C3H10T1/2 stable cells containing both pREV-Tet-On and pREV-TRE:mP311-myc.
Microarray analysis
Total RNA was extracted from four biological samples of C3H10 Tet-On/TRE-REV stable cells and C3H10 Tet-On/TRE mP311-myc cells using the RNeasy mini kit (Qiagen, Valencia, CA). After verifying the quality of the RNA samples on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA), the samples were amplified for one round using T7 RNA polymerase from the MessageAmpII aRNA Kit (Ambion, Austin, TX). Cy3 or Cy5 was incorporated into the amplified RNA products using amino allyl-modified dUTPs. Fluorescently-labeled amplified RNAs were hybridized to DNA microarrays using SlideHyb Glass Array Hybridization Buffer (Ambion, Austin, TX). Arrays were produced by the UCSF Sandler Microarray facility using the Mouse Exonic Evidence Based Oligonucleotide (MEEBO) set, a 70-mer oligonucleotide gene set that contains over 25,000 genes, including alternatively spliced exons. After hybridization, arrays were washed and scanned using an Axon GenePix 4000B scanner (Molecular Devices Corporation, Sunnyvale, CA) and images were processed using GenePix 5.0 software. The “print-tip loess” normalization and single channel quantile normalization was used to correct for within-array dye and spatial effects in order to facilitate comparison between arrays. The functions in the library marrayNorm of the R/Bioconductor package were used to perform these normalizations. After normalization, a log ratio (C3H10 Tet-On mP311-myc/C3H10 Tet-On TRE-REV) was determined for each probe on the array. Statistical analyses were performed using the functions in the library limma of the R/Bioconductor package to obtain a B (log posterior odds ratio) value and those genes with a B value greater than one and with a two-fold or greater change in gene expression in P311 stable cells were further assessed by Ensemble attribute profile clustering (Semeiks et al., 2006).
Triglycerides and Cholesterol Assays
To measure total cellular cholesterol, cells were treated with fatty acid complexes consisting of 1mM of Na oleate and 1% BSA. The next day, cellular lipids were extracted with 2mL of hexane:isopropanol (3:2 vol:vol) per well of a 6-well plate. The lipid extract was collected into 12×75mm glass tubes, the wells rinsed with an additional 1mL of hexane:isopropanol (3:2), and the combined extracts dried under nitrogen and dissolved in 200 µL of isopropanol containing 10% Triton X-100. The levels of free cholesterol were quantified with the Cholesterol Quantitation Kit (BioVision, Mountain View, CA) according to the manufacturer’s instructions. To measure triglyceride synthesis and accumulation, cells were cultured in 6-well plates until confluency and supplemented for 24 h with medium containing 1 mM of Na oleate, 1% BSA, and 2µCi of [14C] oleic acid as a tracer. The next day cells were placed on ice and washed three times with ice cold PBS 0.2% BSA, two times for 10 minutes with ice cold PBS 0.2% BSA, and two times with ice cold PBS. A recovery standard of 0.02 µCi of [3H] triolein was added in each well and lipids were extracted with 2mL of hexane:isopropanol (3:2 vol:vol) per well of a 6-well plate as described for cholesterol measurements. Lipid extracts were separated by thin-layer chromatography (TLC), the triglyceride band isolated, and radioactivity quantified by scintillation counting. All lipid measurements were normalized to lipid recovery and protein concentration. To determine protein concentration, proteins were extracted from cells remaining on the well after lipid extraction by incubation in 1mL of 0.1N NaOH for 10–15 minutes at RT, and subsequently quantified by the Bradford method (Bio-Rad, Hercules, CA). Experiments were performed in triplicate and statistical significance determined by the Student’s t-test.
Oil-Red-O staining
Control and P311 stable C3H10 cells were supplemented with 1mM Na oleate 1% BSA for 24 hours. Cells were then washed twice with PBS and fixed in 10% formalin in PBS at 37 °C for 1.5 hours. The cells were then washed three times with water and 2–3 mL of dye solution was added. The dye solution was prepared by dissolving 4.2g of Oil-Red-O (Sigma, St. Louis, MO) in 1200 mL of absolute isopropanol and left overnight without stirring at room temperature. The solution is vacuum-filtered through a Buchner funnel, diluted with 900 mL of double-distilled water, left overnight at 4 °C without stirring and then filtered twice. After incubation of the cells in the dye solution at 37 °C for 3 hours, excess dye was removed and the dish left to dry in the incubator for 10 minutes. The red stained lipid droplets are visualized under light microscopy using a Nikon TE100 microscope.
Adipogenic induction of NIH3T3-L1 cells
For infection, NIH3T3-L1 cells (ATCC, Manassas, VA) were incubated with recombinant lentivirus in the presence of 4 µg/mL polybrene and subjected to centrifugation at room temperature for 90 minutes at 2500 rpm. After 2–3 days at 37 °C infected cells were identified by the expression of the GFP co-marker signal. NIH3T3-L1 cells were subcultured at 3.3×103 cells/cm2 and grown to high confluency (no visible gaps between cells) in DMEM + 10% calf serum. Two days post-confluency, cells were stimulated with induction media (DMEM + 10% fetal bovine serum, 0.5mM IBMX, 1µg/mL insulin, 1µM dexamethasone) (Sigma, St. Louis, MO). After two days the induction media was changed to insulin-containing media (DMEM + 10% fetal bovine serum, 1µg/mL insulin). After two days this media was changed to DMEM + 10% fetal bovine serum which was then changed every two days. Full differentiation was achieved by 8 days. For lentivirus infection, cells were incubated with recombinant virus in the presence of 4 µg/mL polybrene and subjected to centrifugation at room temperature for 90 minutes at 2500 rpm. After 2–3 days, infected cells were subjected to induction of adipogenic differentiation as described above.
Immunocytochemistry
Cells were fixed for 6 minutes in 4% paraformaldehyde followed by permeabilization in 0.1% Triton X-100 for 6 minutes. Blocking was performed in PBS+5% bovine serum albumin (BSA). Cells were incubated with anti-adipophilin (Research Diagnostics Inc., Concord, MA) or anti-Tip47 antibody (a kind gift from Dr. Perry Bickel) for one hour in PBS+5% BSA, washed, and then incubated for 1 hour with either Alexa Fluor 594-conjuated goat anti-mouse antibody (Molecular Probes, Carlsabad, CA) or FITC-conjugated donkey anti-rabbit antibody (Southern Biotech, Birmingham, AL), washed, and mounted in Vectashield mounting media (Vector Laboratories, Burlingame, CA).
Isolation and culture of lung fibroblasts
Lung fibroblasts were isolated from one week old C57 mouse lungs as previously described (Bruce and Honaker, 1998). Briefly, following euthanasia lungs were promptly dissected, rinsed in Ca2+ and Mg2+ free Hank’s balanced salt solution (HBSS), minced into 1–2mm3 pieces, and placed into a solution of 0.3mg/mL Type IV collagenase and 0.5mg/mL trypsin in HBSS in a shaking 37 degree water bath for 60 minutes. Minced tissue was passed through a 25mL pipette at 20-minute intervals to dissociate the cells. Cells in suspension were removed from the lung homogenate and added to an equivalent volume of cold complete medium consisting of a 1:1 vol/vol of DMEM:Ham’s F12, 10% FBS, and glutamine and plated into a 100mm cell culture dish. For experiments involving the fatty acid exposure and retinoic acid treatment of lung fibroblasts, cells were seeded at 3.3×103 cells/cm2. The following day cells were treated with fatty acid complexes consisting of 1mM Na oleate and 1% BSA and retinoic acid (Sigma, St. Louis, MO) for the indicated times and dosage. Experiments were performed in triplicate and statistical significance determined by the Student’s t-test.
Quantitative real-time RT-PCR
2µg total RNA was treated with Dnase I (Invitrogen) and reverse transcribed using the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA). PCR was performed in the ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). Primers and probes were designed using the Primer Express program from the manufacturer. The sequences for the primers/probes used are as follows:P311: probe, 5’-AGA CTG GCG CTG CCT CGC TGA C-3’, forward primer, 5’-GAA GTG AAC CGA AAG AAG ATG AG-3’, reverse primer, 5’-GAA TTC ACG GCT GCC TGG-3’; GAPDH: probe, 5’-CCG CCT GGA GAA ACC TGC CAA GTA TG-3’, forward primer, 5’-TGT GTC CGT CGT GGA TCT GA-3’, reverse primer, 5’-CCT GCT TCA CCA CCT TCT TGA T-3’. The 5’ and 3’ modification of all the probes are FAM and BHQ, respectively. Thermal cycling conditions were 50°C for 2 min, 95 °C for 10 min, followed by 40 repetitive cycles of 95°C for 15 sec and 60°C for 1 min. Following PCR, quantification of transcript levels was done by determining the number of cycles to threshold (CT) of fluorescence detection within the geometric region of the semi-log plot of fluorescence detection. Relative gene expression was determined using the comparative CT method as described previously (Hettinger et al., 2001). Briefly, the ΔCT value was obtained by subtracting the GAPDH CT value from the P311 CT value in each sample. The ΔΔCT was obtained by subtracting the mean ΔCT of the control samples from the ΔCT of each experimental sample. The fold change in gene expression was determined by calculating 2-ΔΔCT and is expressed as fold change or as percentile change by multiplying by 100. Experiments were performed in triplicate and statistical significance determined by the Student’s t-test.
Lentivirus construction and infection
The lentiviral construct pSicoR and lentiviral packaging constructs VSV-g, REV, and MDL were provided by Dr. Tyler Jacks and Dr. Michael McManus. The 5’ phosphorylated, PAGE-purified oligos used for forming shRNA constructs targeting the knockdown of P311 transcripts were designed using the pSicoOligomaker program (http://web.mit.edu/ccr/labs/jacks/protocols/pSico.html). The shRNA construct was designed with the following pairs of oligos: Forward: 5’-TGA ATT CAC CTC TCC AGC TAT TCA AGA GAT AGC TGG AGA GGT GAA TTC TTT TTT C-3’, Reverse: 5’-TCG AGA AAA AAG AAT TCA CCT CTC CAG CTA TCT CTT GAA TAG CTG GAG AGG TGA ATT CA-3’. The oligos were checked by BLAST to verify their specificity. Oligos were resuspended to a final concentration of 100 µM and annealed in a reaction containing 23 µL ddH2O, 1uL sense oligo, 1uL antisense oligo, and 25 µL of 2x annealing buffer (200 mM potassium acetate, 60 mM HEPES-KOH pH 7.4, and 4 mM Mg-acetate) and incubated for 4 minutes at 95 °C, then at 10 minutes at 70 °C, and then slowly cooled down to 4 °C. To ligate the annealed oligos to the pSicoR vector, 1 µL of a 1:10 dilution of the annealed oligos was added to 50–100 ng of HpaI-XhoI digested pSicoR in a 10µl reaction mixture and incubated at ambient temperature for 3 hours. Transformation of 2µl of the ligation mixture yielded positive clones that were sequence verified. To produce lentiviral particles, the 293T packaging cell line (ATCC, Manassas, VA) was plated at 2×106/100mm and transfected with the plasmids encoding components of the lentiviral system (VSV-g, MDL, REV, SicoR). After 48–72 hours, cells were scraped and collected. Cells were disrupted by freeze-thaw 3 times, and cell debris removed by centrifugation at 3000 rpm for 10 minutes. The supernatant was filtered through a 0.45 µm-pore-size nitrocellulose membrane. Virus was aliquoted and frozen at −80 °C until use. For infection, cells were incubated with recombinant virus in the presence of 4 µg/mL polybrene and subjected to centrifugation at room temperature for 90 minutes at 2500 rpm. After 2–3 days at 37 °C infected cells were identified by the expression of the GFP co-marker signal.
Nile Red staining and flow cytometry
Lung fibroblast cells were detached with 0.05% trypsin/0.02% EDTA for five minutes and resuspended in complete medium and centrifuged at 1200 rpm for 10 minutes. Cells were then resuspended in normal saline, filtered through a nylon mesh and stained with Nile Red (Sigma, St.Louis, MO) at a final concentration of 0.1µg/mL. For flow cytometry, cells were subsequently washed three times, resuspended in saline, and analyzed for Nile Red fluorescence with a Becton Dickinson FACSort flow cytometer. Nile red fluorescence was detected through a 530/30-nm bandpass filter on FL2. To quantify the flow cytometry data we determined the mean fluorescent intensities (MFI) of each of the samples. The MFI is determined using the FCSPress analysis program (FCSPress, Cambridge, United Kingdom). For dual-color flow cytometric analysis, cells were washed three times, resuspended in saline, and analyzed using a Becton Dickinson FACSort flow cytometer. Uninfected and unstained cells were used as baseline controls for GFP and Nile Red signals and to adjust FL1 (GFP) and FL2 (Nile Red) PMT voltages. Separate control samples with GFP fluorescence or Nile red staining were used for instrument setup and compensation performed to ensure one fluorochrome didn't interfere with the other. All fluorescence data was collected on an arbitrary four-decade log scale and analyzed using FCSPress. FL1 versus FL2 cytograms were used to determine infected (shRNA-containing) cells with lipid droplets (FL1+/FL2+) and without lipid droplets (FL1+/FL2−).
ACKNOWLEDGMENTS
We thank Andrea Barczak and Rebecca Barbeau of the Sandler Microarray Core for their assistance with microarray hybridizations and data analysis. This study is supported by grants from the National Institute of Health (HL069925, HL075680, and HL073823), from the March of Dimes Research Foundation, and from the Sander Family Supporting Foundation to T.V. James K. Leung was supported by an NIH Ruth L. Kirschstein NRSA Individual Fellowship.
REFERENCES
- Abumrad NA, Park JH, Park CR. Permeation of long-chain fatty acid into adipocytes. Kinetics, specificity, and evidence for involvement of a membrane protein. J Biol Chem. 1984;259:8945–8953. [PubMed] [Google Scholar]
- Abumrad NA, Perkins RC, Park JH, Park CR. Mechanism of long chain fatty acid permeation in the isolated adipocyte. J Biol Chem. 1981;256:9183–9191. [PubMed] [Google Scholar]
- Badman MK, Flier JS. The adipocyte as an active participant in energy balance and metabolism. Gastroenterology. 2007;132:2103–2115. doi: 10.1053/j.gastro.2007.03.058. [DOI] [PubMed] [Google Scholar]
- Bruce MC, Honaker CE. Transcriptional regulation of tropoelastin expression in rat lung fibroblasts: changes with age and hyperoxia. Am J Physiol. 1998;274:L940–L950. doi: 10.1152/ajplung.1998.274.6.L940. [DOI] [PubMed] [Google Scholar]
- Cermelli S, Guo Y, Gross SP, Welte MA. The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr Biol. 2006;16:1783–1795. doi: 10.1016/j.cub.2006.07.062. [DOI] [PubMed] [Google Scholar]
- Chen H, Jackson S, Doro M, McGowan S. Perinatal expression of genes that may participate in lipid metabolism by lipid-laden lung fibroblasts. J Lipid Res. 1998;39:2483–2492. [PubMed] [Google Scholar]
- Chytil F. Retinoids in lung development. Faseb J. 1996;10:986–992. doi: 10.1096/fasebj.10.9.8801181. [DOI] [PubMed] [Google Scholar]
- Dani C, Smith AG, Dessolin S, Leroy P, Staccini L, Villageois P, Darimont C, Ailhaud G. Differentiation of embryonic stem cells into adipocytes in vitro. J Cell Sci. 1997;110(Pt 11):1279–1285. doi: 10.1242/jcs.110.11.1279. [DOI] [PubMed] [Google Scholar]
- Dutta-Roy AK. Cellular uptake of long-chain fatty acids: role of membrane-associated fatty-acid-binding/transport proteins. Cell Mol Life Sci. 2000;57:1360–1372. doi: 10.1007/PL00000621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak AM, Dvorak HF, Peters SP, Shulman ES, MacGlashan DW, Jr, Pyne K, Harvey VS, Galli SJ, Lichtenstein LM. Lipid bodies: cytoplasmic organelles important to arachidonate metabolism in macrophages and mast cells. J Immunol. 1983;131:2965–2976. [PubMed] [Google Scholar]
- Fujitani M, Yamagishi S, Che YH, Hata K, Kubo T, Ino H, Tohyama M, Yamashita T. P311 accelerates nerve regeneration of the axotomized facial nerve. J Neurochem. 2004;91:737–744. doi: 10.1111/j.1471-4159.2004.02738.x. [DOI] [PubMed] [Google Scholar]
- Green H, Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell. 1974;3:127–133. doi: 10.1016/0092-8674(74)90116-0. [DOI] [PubMed] [Google Scholar]
- Green S. PPAR: a mediator of peroxisome proliferator action. Mutat Res. 1995;333:101–109. doi: 10.1016/0027-5107(95)00136-0. [DOI] [PubMed] [Google Scholar]
- Hettinger AM, Allen MR, Zhang BR, Goad DW, Malayer JR, Geisert RD. Presence of the acute phase protein, bikunin, in the endometrium of gilts during estrous cycle and early pregnancy. Biol Reprod. 2001;65:507–513. doi: 10.1095/biolreprod65.2.507. [DOI] [PubMed] [Google Scholar]
- Hind M, Maden M. Retinoic acid induces alveolar regeneration in the adult mouse lung. Eur Respir J. 2004;23:20–27. doi: 10.1183/09031936.03.00119103. [DOI] [PubMed] [Google Scholar]
- Inazawa Y, Nakatsu M, Yasugi E, Saeki K, Yuo A. Lipid droplet formation in human myeloid NB4 cells stimulated by all trans retinoic acid and granulocyte colony-stimulating factor: possible involvement of peroxisome proliferator-activated receptor gamma. Cell Struct Funct. 2003;28:487–493. doi: 10.1247/csf.28.487. [DOI] [PubMed] [Google Scholar]
- Kaplan NB, Grant MM, Brody JS. The lipid interstitial cell of the pulmonary alveolus. Age and species differences. Am Rev Respir Dis. 1985;132:1307–1312. doi: 10.1164/arrd.1985.132.6.1307. [DOI] [PubMed] [Google Scholar]
- Liu B, Harvey CS, McGowan SE. Retinoic acid increases elastin in neonatal rat lung fibroblast cultures. Am J Physiol. 1993;265:L430–L437. doi: 10.1152/ajplung.1993.265.5.L430. [DOI] [PubMed] [Google Scholar]
- Maden M, Hind M. Retinoic acid, a regeneration-inducing molecule. Dev Dyn. 2003;226:237–244. doi: 10.1002/dvdy.10222. [DOI] [PubMed] [Google Scholar]
- Maden M, Hind M. Retinoic acid in alveolar development, maintenance and regeneration. Philos Trans R Soc Lond B Biol Sci. 2004;359:799–808. doi: 10.1098/rstb.2004.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani L, McDonough WS, Hoelzinger DB, Beaudry C, Kaczmarek E, Coons SW, Giese A, Moghaddam M, Seiler RW, Berens ME. Identification and validation of P311 as a glioblastoma invasion gene using laser capture microdissection. Cancer Res. 2001;61:4190–4196. [PubMed] [Google Scholar]
- Martin S, Parton RG. Caveolin, cholesterol, and lipid bodies. Semin Cell Dev Biol. 2005;16:163–174. doi: 10.1016/j.semcdb.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Martin S, Parton RG. Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol. 2006;7:373–378. doi: 10.1038/nrm1912. [DOI] [PubMed] [Google Scholar]
- Massaro GD, Massaro D. Retinoic acid treatment abrogates elastase-induced pulmonary emphysema in rats. Nat Med. 1997;3:675–677. doi: 10.1038/nm0697-675. [DOI] [PubMed] [Google Scholar]
- McGowan S, Jackson SK, Jenkins-Moore M, Dai HH, Chambon P, Snyder JM. Mice bearing deletions of retinoic acid receptors demonstrate reduced lung elastin and alveolar numbers. Am J Respir Cell Mol Biol. 2000;23:162–167. doi: 10.1165/ajrcmb.23.2.3904. [DOI] [PubMed] [Google Scholar]
- McGowan SE. Contributions of retinoids to the generation and repair of the pulmonary alveolus. Chest. 2002;121:206S–208S. doi: 10.1378/chest.121.5_suppl.206s. [DOI] [PubMed] [Google Scholar]
- McGowan SE, Torday JS. The pulmonary lipofibroblast (lipid interstitial cell) and its contributions to alveolar development. Annu Rev Physiol. 1997;59:43–62. doi: 10.1146/annurev.physiol.59.1.43. [DOI] [PubMed] [Google Scholar]
- Murphy DJ. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res. 2001;40:325–438. doi: 10.1016/s0163-7827(01)00013-3. [DOI] [PubMed] [Google Scholar]
- Pan D, Zhe X, Jakkaraju S, Taylor GA, Schuger L. P311 induces a TGF-beta1-independent, nonfibrogenic myofibroblast phenotype. J Clin Invest. 2002;110:1349–1358. doi: 10.1172/JCI15614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pol A, Martin S, Fernandez MA, Ferguson C, Carozzi A, Luetterforst R, Enrich C, Parton RG. Dynamic and regulated association of caveolin with lipid bodies: modulation of lipid body motility and function by a dominant negative mutant. Mol Biol Cell. 2004;15:99–110. doi: 10.1091/mbc.E03-06-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safonova I, Reichert U, Shroot B, Ailhaud G, Grimaldi P. Fatty acids and retinoids act synergistically on adipose cell differentiation. Biochem Biophys Res Commun. 1994;204:498–504. doi: 10.1006/bbrc.1994.2487. [DOI] [PubMed] [Google Scholar]
- Sato M, Suzuki S, Senoo H. Hepatic stellate cells: unique characteristics in cell biology and phenotype. Cell Struct Funct. 2003;28:105–112. doi: 10.1247/csf.28.105. [DOI] [PubMed] [Google Scholar]
- Schwarz EJ, Reginato MJ, Shao D, Krakow SL, Lazar MA. Retinoic acid blocks adipogenesis by inhibiting C/EBPbeta-mediated transcription. Mol Cell Biol. 1997;17:1552–1561. doi: 10.1128/mcb.17.3.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semeiks JR, Rizki A, Bissell MJ, Mian IS. Ensemble attribute profile clustering: discovering and characterizing groups of genes with similar patterns of biological features. BMC Bioinformatics. 2006;7:147. doi: 10.1186/1471-2105-7-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironen R, Elo M, Kaarniranta K, Helminen HJ, Lammi MJ. Transcriptional activation in chondrocytes submitted to hydrostatic pressure. Biorheology. 2000;37:85–93. [PubMed] [Google Scholar]
- Snyder JM, Jenkins-Moore M, Jackson SK, Goss KL, Dai HH, Bangsund PJ, Giguere V, McGowan SE. Alveolarization in retinoic acid receptor-beta-deficient mice. Pediatr Res. 2005;57:384–391. doi: 10.1203/01.PDR.0000151315.81106.D3. [DOI] [PubMed] [Google Scholar]
- Studler JM, Glowinski J, Levi-Strauss M. An abundant mRNA of the embryonic brain persists at a high level in cerebellum, hippocampus and olfactory bulb during adulthood. Eur J Neurosci. 1993;5:614–623. doi: 10.1111/j.1460-9568.1993.tb00527.x. [DOI] [PubMed] [Google Scholar]
- Toth IE, Szabo D, Bruckner GG. Lipoproteins, lipid droplets, lysosomes, and adrenocortical steroid hormone synthesis: morphological studies. Microsc Res Tech. 1997;36:480–492. doi: 10.1002/(SICI)1097-0029(19970315)36:6<480::AID-JEMT5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Vaccaro C, Brody JS. Ultrastructure of developing alveoli. I. The role of the interstitial fibroblast. Anat Rec. 1978;192:467–479. doi: 10.1002/ar.1091920402. [DOI] [PubMed] [Google Scholar]
- Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, Jacks T. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake K, Sato T. Intralobular heterogeneity of perisinusoidal stellate cells in porcine liver. Cell Tissue Res. 1993;273:227–237. doi: 10.1007/BF00312824. [DOI] [PubMed] [Google Scholar]
- Wan HC, Melo RC, Jin Z, Dvorak AM, Weller PF. Roles and origins of leukocyte lipid bodies: proteomic and ultrastructural studies. Faseb J. 2007;21:167–178. doi: 10.1096/fj.06-6711com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Lehto M, Rasilainen L, Metso J, Ehnholm C, Yla-Herttuala S, Jauhiainen M, Olkkonen VM. Oxysterol binding protein induces upregulation of SREBP-1c and enhances hepatic lipogenesis. Arterioscler Thromb Vasc Biol. 2007;27:1108–1114. doi: 10.1161/ATVBAHA.106.138545. [DOI] [PubMed] [Google Scholar]
- Yang L, Naltner A, Yan C. Overexpression of dominant negative retinoic acid receptor alpha causes alveolar abnormality in transgenic neonatal lungs. Endocrinology. 2003;144:3004–3011. doi: 10.1210/en.2002-0121. [DOI] [PubMed] [Google Scholar]
- Zhao L, Leung JK, Yamamoto H, Goswami S, Kheradmand F, Vu TH. Identification of P311 as a potential gene regulating alveolar generation. Am J Respir Cell Mol Biol. 2006;35:48–54. doi: 10.1165/rcmb.2005-0475OC. [DOI] [PMC free article] [PubMed] [Google Scholar]